

Dual Opioid–Neuropeptide FF Small Molecule Ligands Demonstrate Analgesia with Reduced Tolerance Liabilities

 ,
,  , ,
, ,  and
and
Abstract

1. Introduction
2. Results
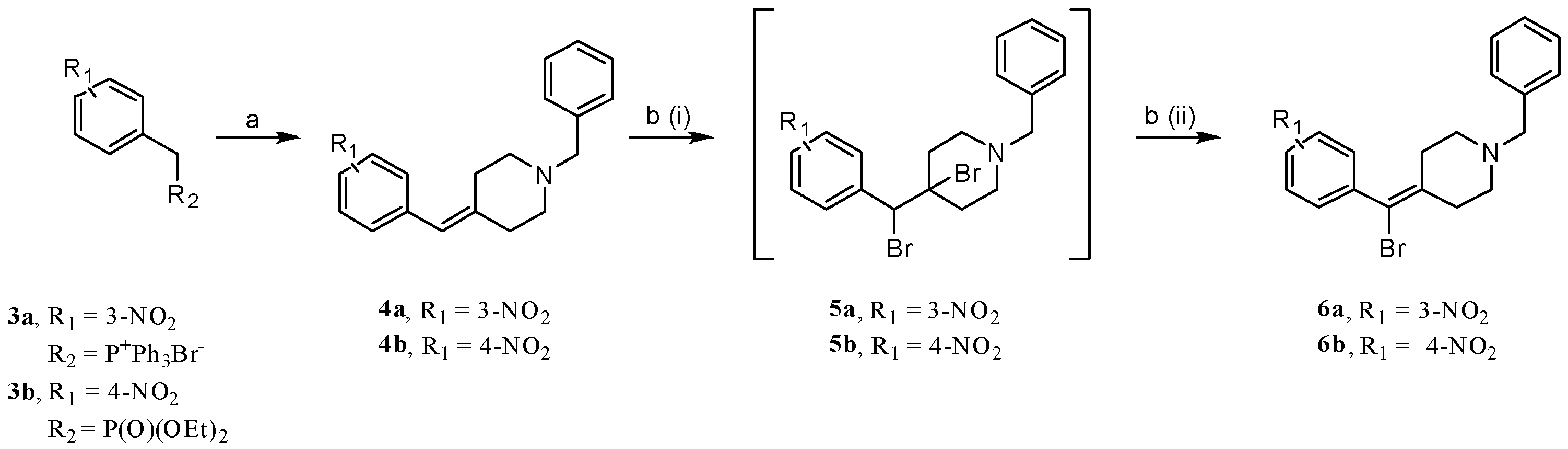
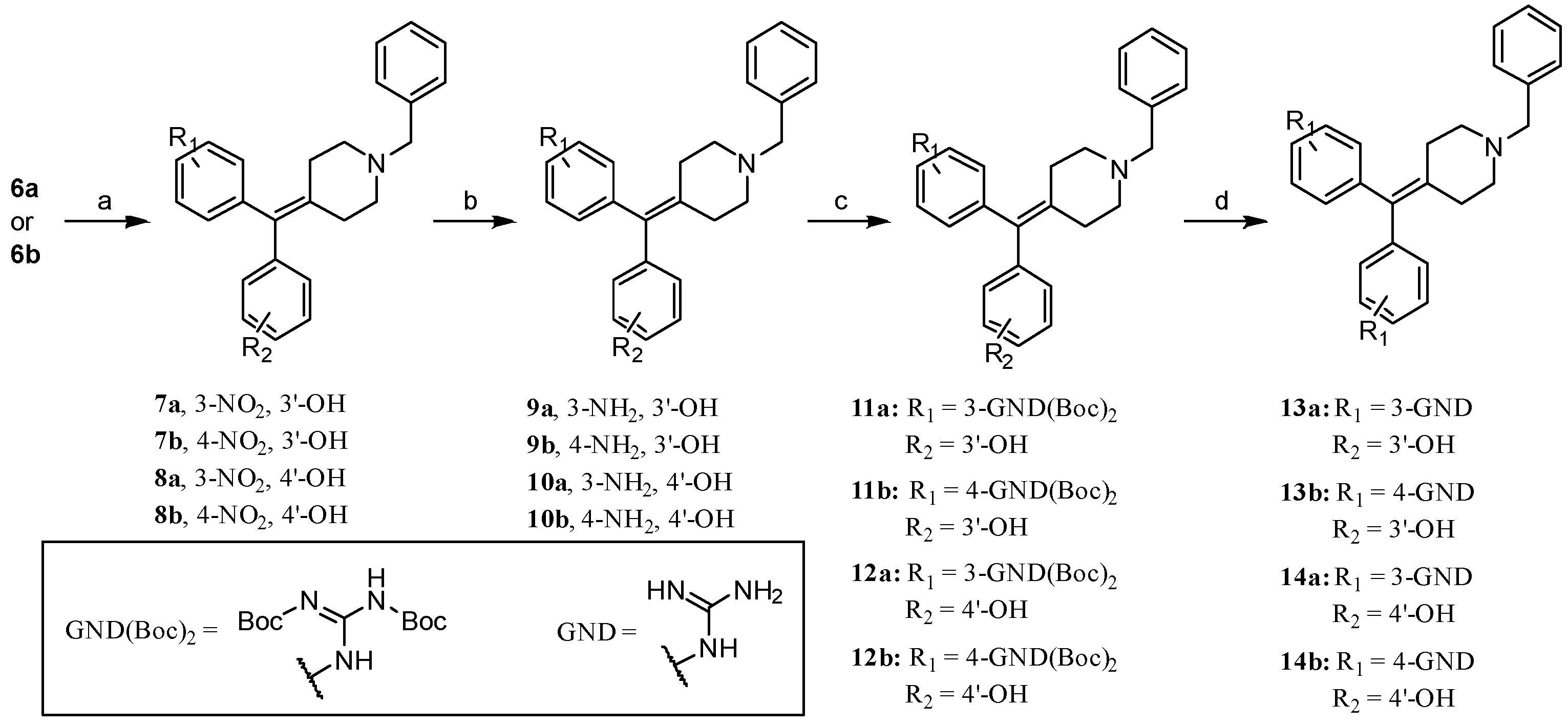
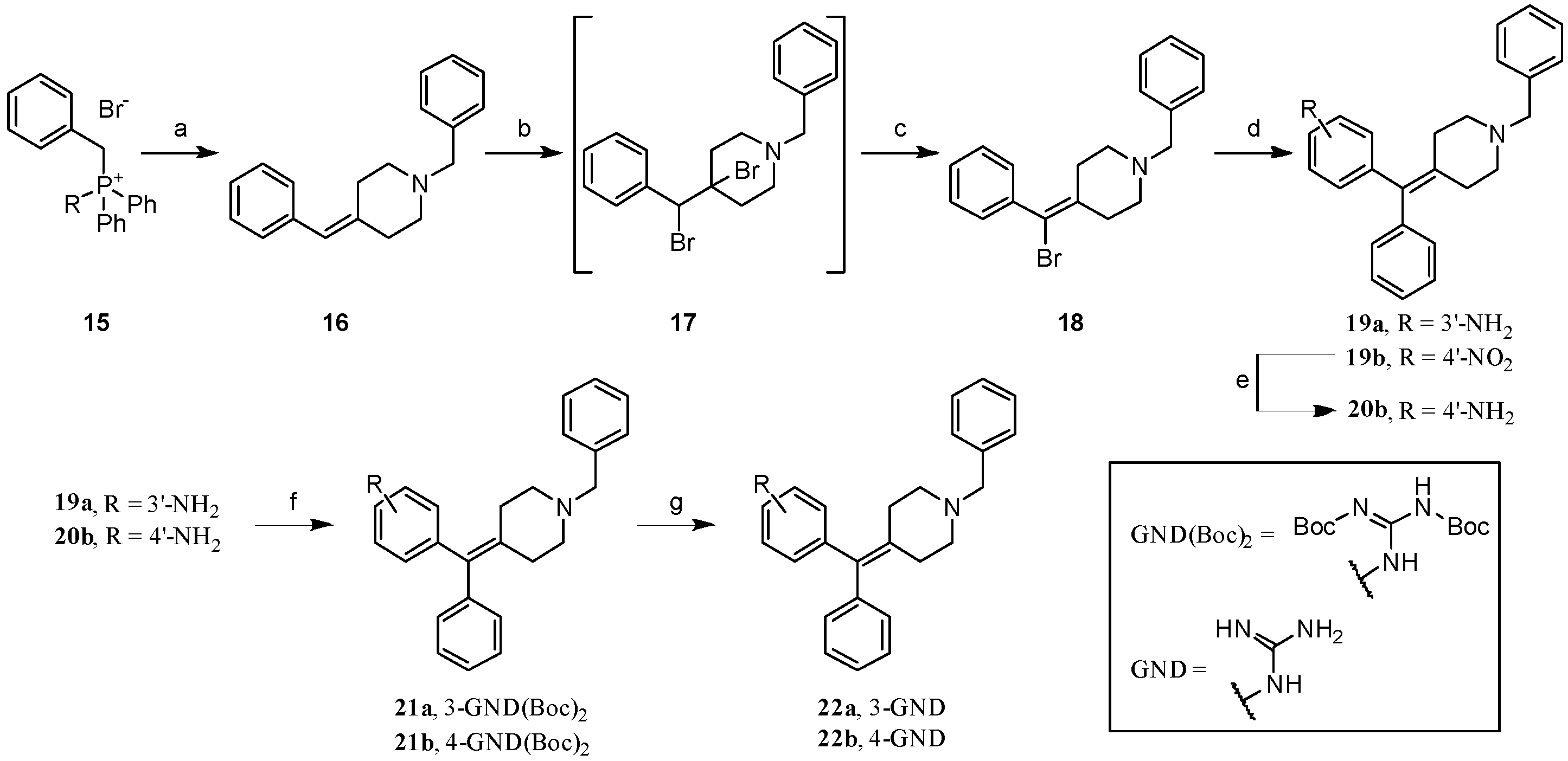
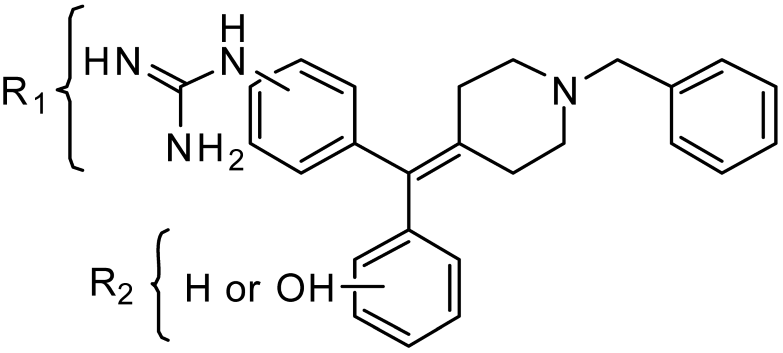
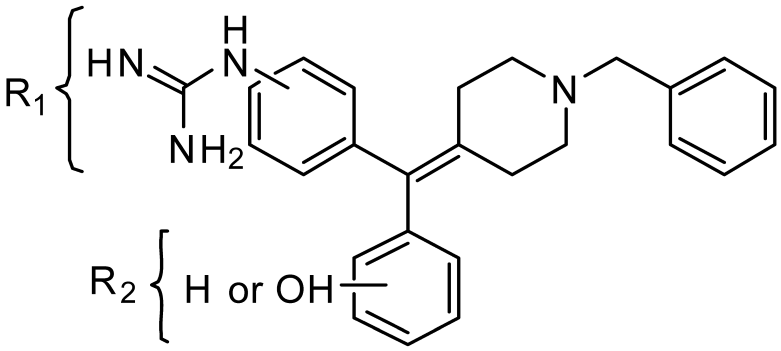
2.1. Chemistry: Synthesis of the Opioid Agonist/NPFF Antagonist DML
2.2. Determination of Binding Affinity of Lead Analogs at NPFF1-R, NPFF2-R, MOR, DOR, and KOR
2.3. Determination of Functional Efficacy of Lead Analogs at NPFF1-R, NPFF2-R, MOR, DOR, and KOR
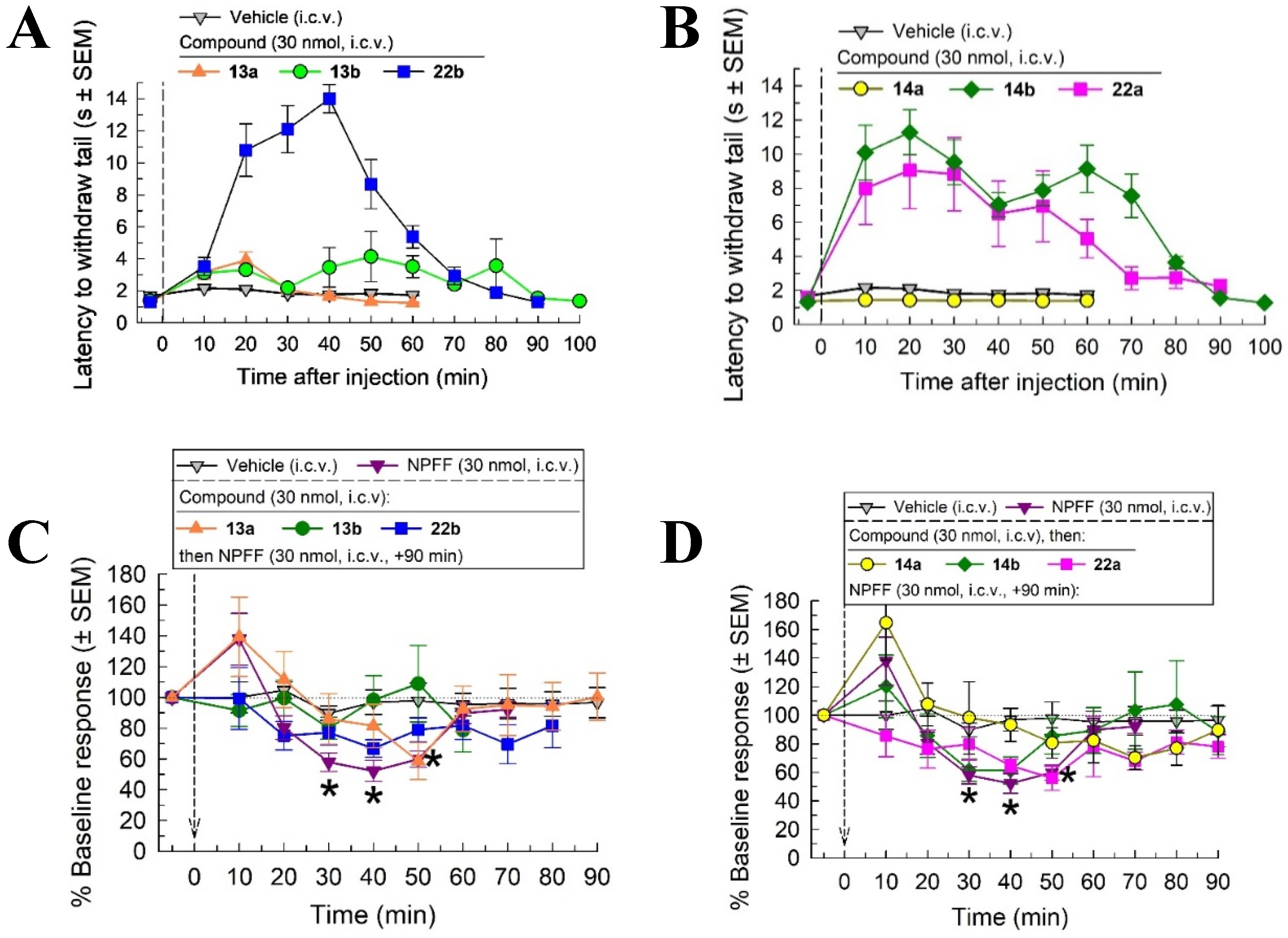
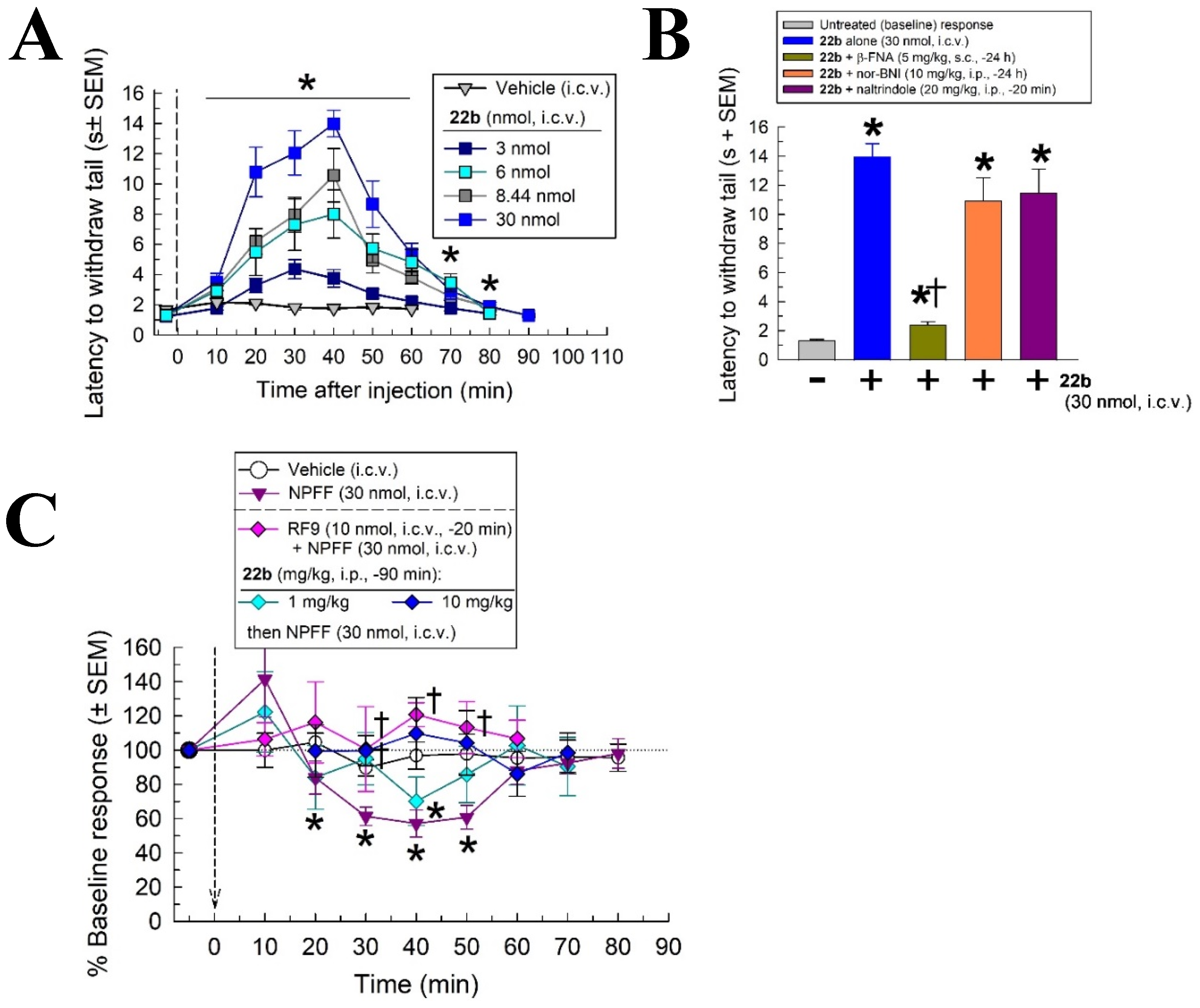
2.4. In Vivo Screening with the Warm Water Tail Withdrawal Assay
2.5. In Vivo Pharmacological Characterization of 22b
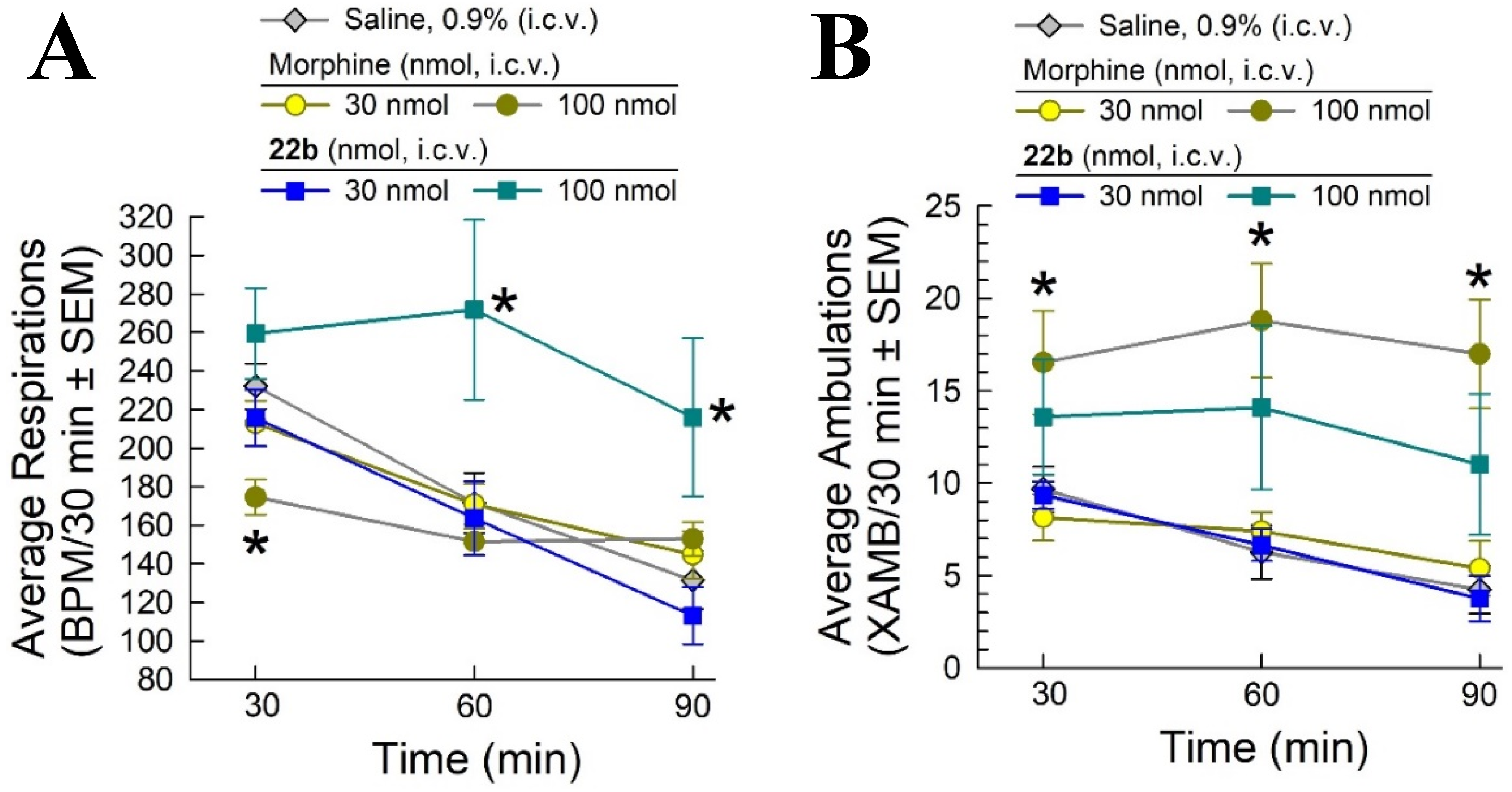
2.6. Evaluation of Potential Opioid-like Liabilities of 22b
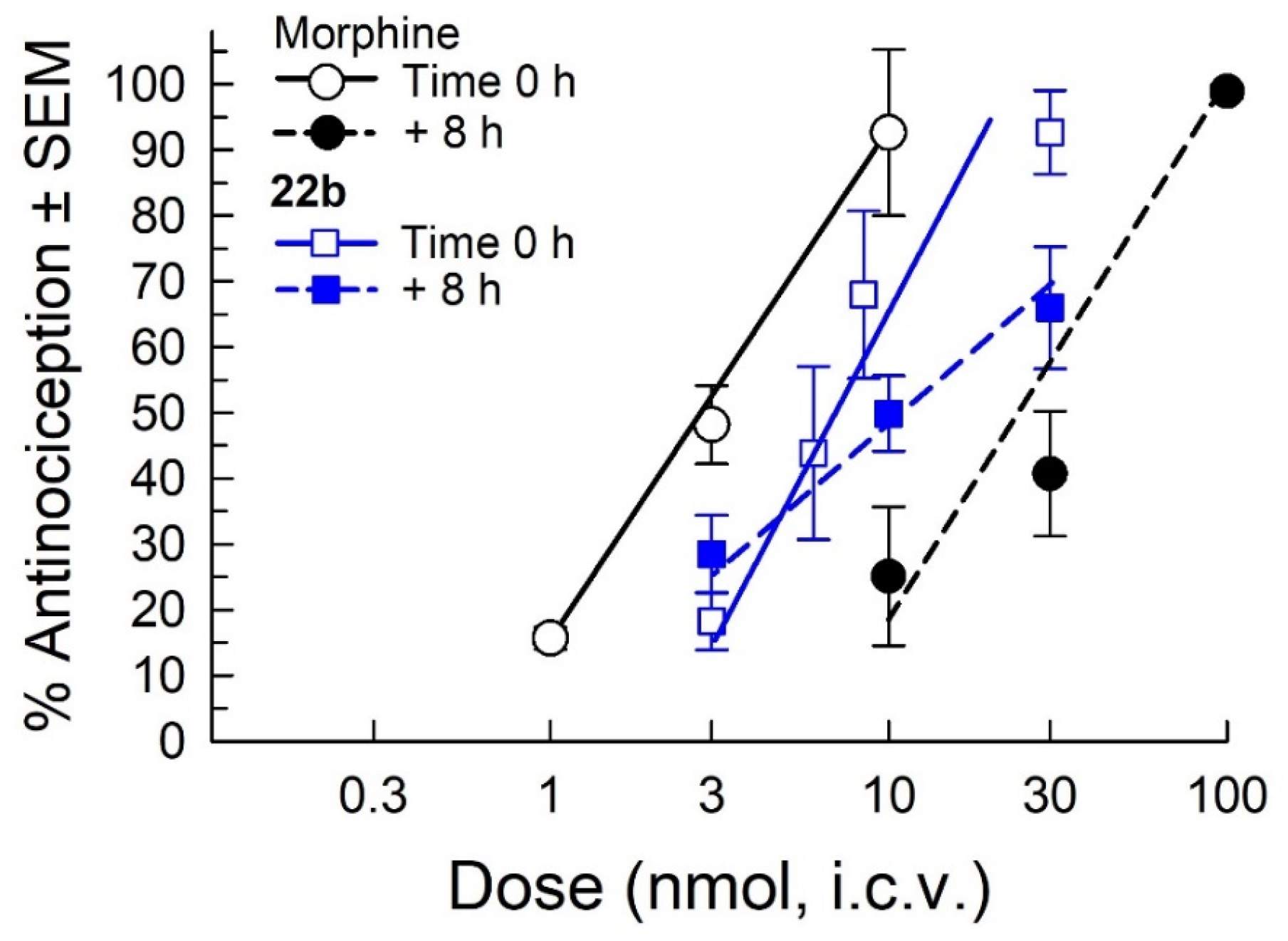
2.7. Assessment of Antinociceptive Tolerance
2.8. Pharmacokinetic (PK) Studies
3. Discussion
4. Materials and Methods
4.1. Chemistry
- (3-Nitrobenzyl)triphenylphosphonium bromide (3a). 3-Nitrobenzyl bromide (30.00 g, 138.56 mmol) and PPh3 (36.42 g, 138.56 mmol) were dissolved in toluene (500 mL) and heated to reflux overnight. The suspension obtained was cooled to rt and filtered, and the precipitate was washed with toluene to afford the desired compound (63.24 g, 95% yield). 1H NMR: (400 MHz, DMSO-d6) δ 8.15–8.13 (m, 1H), 7.92–7.89 (m, 3H), 7.76–7.73 (m, 13H), 7.58–7.55 (m, 2H), 5.48 (d, JPH = 15.8 Hz, 2H). 13C NMR: (101 MHz, DMSO- d6) δ 147.9 (d, JPC = 3 Hz), 137.7, 135.8 (d, JPC = 2 Hz), 134.6 (d, JPC = 10 Hz), 130.8 (d, JPC = 31 Hz), 128.8, 126.0 (d, JPC = 5 Hz), 126.0, 123.7, 117.7 (d, JPC = 86 Hz), 28.1 (d, JPC = 46 Hz). MS(ESI+): 398.0 m/z [M]+.
- Diethyl (4-nitrobenzyl)phosphonate (3b). 4-Nitrobenzyl bromide (5.00 g, 23.14 mmol) was suspended in triethylphosphite (16.0 mL, 92.57 mmol), heated to ca. 100 °C for 2 h, and then concentrated. The residue was purified via flash column chromatography using a gradient of hexanes/EtOAc to afford the desired product (4.98 g, 79% yield), which was spectroscopically equivalent to what was reported in reference [41]. 1H NMR: (400 MHz, CDCl3) δ 8.01 (d, J = 8.5 Hz, 2H), 7.39–7.33 (m, 2H), 3.97–3.87 (m, 4H), 3.13 (d, JPH = 22.3 Hz, 2H), 1.12 (t, J = 7.1 Hz, 6H). 13C NMR: (101 MHz, CDCl3) δ 146.8 (d, JPC = 4 Hz, C), 139.8 (d, JPC = 9 Hz, C), 130.5 (d, JPC = 6 Hz, CH), 123.5 (d, JPC = 3 Hz, CH), 62. 3 (d, JPC = 7 Hz, OCH2), 33.7 (d, JPC = 137 Hz, PCH2), 16.2 (d, JPC = 6 Hz, CH3).
- 1-Benzyl-4-(3-nitrobenzylidene)piperidine (4a). Prepared from 3a according to general procedure A using (3-nitrobenzyl)(triphenyl)phosphonium bromide to afford the title compound with 19% yield. 1H NMR: (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.53–7.43 (m, 2H), 7.40–7.23 (m, 5H), 6.31 (s, 1H), 3.56 (s, 2H), 2.59–2.43 (m, 8H). 13C NMR: (101 MHz, CDCl3) δ 148.2, 142.8, 139.5, 138.2, 134.9, 129.1, 128.9, 128.2, 127.1, 123.6, 121.1, 120.9, 62.8, 54.8, 54.1, 36.4, 29.1. MS(ESI+): 309.0 m/z (M+H)+.
- 1-Benzyl-4-(4-nitrobenzylidene)piperidine (4b). Prepared from 3b according to general procedure A using diethyl(4-nitrobenzyl)phosphonate to afford the title compound with 67% yield. 1H NMR: (400 MHz, CDCl3) δ 8.17 (d, 2H, J = 8.7 Hz), 7.45–7.22 (m, 7H), 6.33 (s, 1H), 3.56 (s, 2H), 2.59–2.44 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 145.9, 144.8, 144.2, 138.3, 129.5, 129.1, 128.3, 127.1, 123.5, 121.6, 62.8, 54.8, 54.1, 36.7, 29.4. MS(ESI+): 309.1 m/z (M+H)+.
- 1-Benzyl-4-(bromo(3-nitrophenyl)methylene)piperidine (6a). Prepared from 4a according to general procedure B to afford the title material with 40% yield. 1H NMR (400 MHz, CDCl3) δ 8.15–8.13 (m, 2H), 7.61–7.59 (m, 1H), 7.54–7.49 (m, 1H), 7.32–7.31 (m, 4H), 7.28–7.26 (s, 1H), 3.54 (s, 2H), 2.72–2.70 (m, 2H), 2.59–2.56 (m, 2H), 2.45–2.37 (m, 2H), 2.30–2.22 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 148.1, 141.9, 140.2, 138.1, 135.5, 129.3, 129.1, 128.3, 127.2, 124.5, 122.8, 112.2, 62.6, 53.9, 53.6, 34.5, 31.9. MS(ESI+): 388.9 m/z (M+H)+ for 81Br.
- 1-Benzyl-4-(bromo(4-nitrophenyl)methylene)piperidine (6b). Prepared from 4b according to general procedure B to afford the title material with 27% yield. 1H NMR: (400 MHz, CDCl3) δ 8.20 (d, 2H, J = 8.6 Hz), 7.46 (d, 2H, J = 8.6 Hz), 7.40–7.19 (m, 5H), 3.56 (s, 2H), 2.77–2.68 (m, 2H), 2.65–2.56 (m, 2H), 2.45–2.37 (m, 2H), 2.34–2.25 (m, 2H). 13C NMR: (101 MHz, CDCl3) δ 147.1, 146.7, 140.3, 138.0, 130.5, 129.1, 128.3, 127.2, 123.6, 112.4, 62.6, 53.9, 53.6, 34.6, 32.1. MS(ESI+): 388.9 m/z (M+H)+ for 81Br.
- 3-((1-Benzylpiperidin-4-ylidene)(3-nitrophenyl)methyl)phenol (7a). Prepared from 6a according to general procedure C using 3-hydroxyphenylboronic acid to give the title material with 80% yield. 1H NMR: (400 MHz, CDCl3) δ 8.07–8.00 (m, 1H), 7.94 (s, 1H), 7.42–7.41 (m, 2H), 7.35–7.22 (m, 6H), 7.16 (t, 1H, J = 7.8 Hz), 6.73–6.60 (m, 2H), 6.50 (s, 1H), 3.56 (s, 2H), 2.56–2.46 (s, 4H), 2.44–2.36 (m, 2H), 2.36–2.29 (m, 2H). 13C NMR: (101 MHz, CDCl3) δ 156.1, 148.0, 143.8, 142.5, 137.4, 136.9, 135.9, 133.7, 129.6, 129.5, 128.9, 128.3, 127.4, 124.4, 121.6, 121.4, 116.9, 114.3, 62.9, 54.7, 54.7, 31.2, 31.1. MS(ESI+): 401.0 m/z (M+H)+.
- 3-((1-Benzylpiperidin-4-ylidene)(4-nitrophenyl)methyl)phenol (7b). Prepared from 6b according to general procedure C using 3-hydroxyphenylboronic acid to give the title material with 81% yield. 1H NMR: (400 MHz, DMSO-d6) δ 9.46 (bs, 1H), 8.14 (d, 2H, J = 8.4 Hz), 7.39–7.17 (m, 6H), 7.11 (t, 1H, J = 7.7 Hz), 6.66 (d, 1H, J = 7.3 Hz), 6.57–6.44 (m, 2H), 3.45 (s, 2H), 2.49–2.23 (m, 8H). 13C NMR: (101 MHz, DMSO-d6) δ 157.7, 149.5, 146.2, 142.6, 138.7, 138.1, 136.4, 134.0, 131.1, 129.8, 129.2, 128.6, 127.3, 123.8, 120.7, 116.8, 114.4, 62.3, 54.8, 31.9, 31.7. MS(ESI+): 401.1 m/z (M+H)+.
- 4-((1-Benzylpiperidin-4-ylidene)(3-nitrophenyl)methyl)phenol (8a). Prepared from 7a according to general procedure C using 4-hydroxyphenylboronic acid to give the title material with 50% yield. 1H NMR: (400 MHz, DMSO-d6) δ 8.07–8.05 (d, 1H, J = 7.7 Hz), 7.79 (s, 1H), 7.59 (t, 1H, J = 7.9 Hz), 7.53–7.51 (m, 1H), 7.30–7.21 (m, 5H), 6.90–6.88 (m, 2H), 6.72–6.69 (m, 2H), 3.48 (s, 2H), 2.45–2.41 (m, 4H), 2.32–2.30 (m, 2H), 2.23–2.21 (m, 2H). 13C NMR: (101 MHz, DMSO--d6) δ 156.7, 148.1, 144.6, 138.6, 137.0, 136.6, 133.5, 132.0, 131.2, 130.1, 129.3, 128.6, 127.4, 124.1, 121.8, 115.6, 62.2, 54.8, 31.7. MS(ESI+): 401.1 m/z (M+H)+.
- 4-((1-Benzylpiperidin-4-ylidene)(4-nitrophenyl)methyl)phenol (8b). Prepared from 7b according to general procedure C using 4-hydroxyphenylboronic acid to give the title material with 93% yield. 1H NMR: (500 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.15 (d, 2H, J = 8.1 Hz), 7.32–7.24 (m, 7H), 6.88 (d, 2H, J = 7.8 Hz), 6.72 (d, 2H, J = 8.3 Hz), 3.48 (s, 2H), 2.49–2.12 (m, 8H). 13C NMR: (126 MHz, DMSO--d6) δ 156.8, 150.2, 146.2, 138.8, 137.6, 133.9, 131.9, 131.2, 129.3, 128.6, 127.4, 123.8, 115.6, 62.3, 54.9, 31.9. MS(ESI+): m/z 401.1 (M+H)+.
- 3-((3-Aminophenyl)(1-benzylpiperidin-4-ylidene)methyl)phenol (9a). Prepared from 7a according to general procedure D to give the title material, which was used directly in the next reaction.
- 3-((4-Aminophenyl)(1-benzylpiperidin-4-ylidene)methyl)phenol (9b). Prepared from 7b according to general procedure D to give the title material with 56% yield. 1H NMR: (500 MHz, CDCl3) δ 7.43–7.20 (m, 5H), 7.12 (t, 1H, J = 7.7 Hz), 6.84 (d, 2H, J = 7.9 Hz), 6.66 (d, 2H, J = 7.2 Hz), 6.56 (d, 2H, J = 7.9 Hz), 6.44 (s, 1H), 3.55 (bs, 4H), 2.35–2.50 (m, 8H). 13C NMR: (126 MHz, CDCl3) δ 155.8, 144.5, 144.5, 137.2, 135.5, 133.9, 132.8, 130.8, 129.7, 128.9, 128.2, 127.3, 121.8, 117.1, 114.7, 113.5, 63.0, 55.0, 54.9, 31.3, 31.2.
- 4-((3-Aminophenyl)(1-benzylpiperidin-4-ylidene)methyl)phenol (10a). Prepared from 8a according to general procedure D to give the title material with 43% yield. 13C NMR: (126 MHz, DMSO-d6) δ 155.9, 148.5, 143.7, 138.4, 136.1, 133.6, 133.4, 130.8, 129.5, 128.9, 128.6, 127.4, 117.7, 115.5, 115.2, 112.6, 62.4, 55.0, 54.9, 31.7, 31.4. MS (ESI+): 371.2 m/z (M+H)+.
- 4-((4-Aminophenyl)(1-benzylpiperidin-4-ylidene)methyl)phenol (10b). Prepared from 7b according to general procedure D to give the title material with 52% yield. 1H NMR: (500 MHz, CDCl3) δ 9.30 (s, 1H), 7.31–7.24 (m, 5H), 6.83–6.81 (m, 2H), 6.69–6.65 (m, 3H), 6.47–6.45 (m, 2H), 4.99 (s, 2H), 3.46 (s, 2H), 2.39–2.25 (m, 8H). 13C NMR: (126 MHz, CDCl3) δ 156.1, 147.4, 138.9, 135.9, 134.1, 132.6, 131.1, 130.7, 130.5, 130.5, 129.3, 128.6, 127.3, 115.1, 113.8, 62.6, 55.3, 55.2, 31.9.
- tert-Butyl N-[({3-[(1-benzylpiperidin-4-ylidene)(3-hydroxyphenyl)methyl]phenyl}amino)({[(tert-butoxy)carbonyl]imino})methyl]carbamate (11a). Prepared from 9a according to general procedure E to give the title material with 27% yield. 1H NMR: (400 MHz, CDCl3) δ 11.60 (bs, 1H), 10.20 (s, 1H), 7.55 (d, 1H, J = 7.9 Hz), 7.29 (dd, 5H, J = 12.6, 4.3 Hz), 7.19–7.05 (m, 3H), 6.79 (d, 1H, J = 7.6 Hz), 6.67–6.62 (m, 2H), 6.47 (s, 1H), 3.56 (s, 2H), 2.52–2.48 (m, 4H), 2.37 (dd, 4H, J = 22.2, 4.6 Hz), 1.61–1.38 (m, 18H). 13C NMR: (126 MHz, CDCl3) δ 163.5, 155.9, 153.7, 153.3, 143.4, 142.7, 136.2, 135.1, 129.6, 129.1, 128.6, 128.3, 127.3, 126.5, 123.7, 121.7, 120.9, 117.0, 113.8, 83.7, 79.7, 62.9, 55.0, 54.9, 31.2, 31.2, 28.2, 28.1. MS(ESI+): 613.2 m/z (M+H)+.
- tert-Butyl N-[({4-[(1-benzylpiperidin-4-ylidene)(3-hydroxyphenyl)methyl]phenyl}amino)({[(tert-butoxy)carbonyl]imino})methyl]carbamate (11b). Prepared from 9b according to general procedure E to give the title material with 74% yield. 1H NMR: (500 MHz, CDCl3) δ 11.64 (s, 1H), 10.29 (s, 1H), 7.48 (d, 2H, J = 8.4 Hz), 7.40–7.21 (m, 5H), 7.10 (t, 1H, J = 7.8 Hz), 7.01 (d, 2H, J = 8.4 Hz), 6.65–6.60 (m, 2H), 6.48 (s, 1H), 3.56 (s, 2H), 2.66–2.24 (m, 8H), 1.51 (s, 9H), 1.55 (s, 9H). 13C NMR: (126 MHz, CDCl3) δ 163.5, 155.9, 153.6, 153.3, 143.7, 138.9, 137.2, 135.2, 134.9, 130.3, 129.7, 129.1, 128.3, 127.3, 121.8, 117.1, 113.7, 83.7, 79.8, 62.9, 55.0, 54.9, 31.4, 31.2, 28.2, 28.1. MS(ESI+): 613.1 m/z (M+H)+.
- tert-Butyl N-[({3-[(1-benzylpiperidin-4-ylidene)(4-hydroxyphenyl)methyl]phenyl}amino)({[(tert-butoxy)carbonyl]imino})methyl]carbamate (12a). Prepared from 10a according to general procedure E to give the title material with 27% yield. 1H NMR: (500 MHz, CDCl3) δ 11.35 (s, 1H), 9.93 (s, 1H), 9.35 (s, 1H), 7.31–7.25 (m, 7H), 6.89–6.88 (m, 2H), 6.80 (d, 1H, J = 5.2 Hz), 6.69 (d, 2H, J = 7.7 Hz), 3.48 (s, 2H), 2.41–2.29 (m, 8H), 1.48–1.37 (m, 18H). 13C NMR: (126 MHz, CDCl3) δ 156.4, 153.2, 143.4, 139.0, 137.8, 136.7, 135.3, 135.0, 132.9, 131.0, 129.2, 128.7, 128.6, 127.3, 126.4, 124.5, 121.0, 115.3, 83.7, 62.5, 55.2, 54.9, 31.8, 31.7, 28.2, 28.2. MS(ESI+): 613.1 m/z (M+H)+.
- tert-Butyl N-[({4-[(1-benzylpiperidin-4-ylidene)(4-hydroxyphenyl)methyl]phenyl}amino)({[(tert-butoxy)carbonyl]imino})methyl]carbamate (12b). Prepared from 10b according to general procedure E to give the title material with 69% yield. 1H NMR: (500 MHz, DMSO-d6) δ 11.42 (s, 1H), 9.99 (s, 1H), 9.39 (s, 1H), 7.47 (d, 1H, J = 7.6 Hz), 7.31–7.25 (m, 5H), 7.02 (d, 2H, J = 7.8 Hz), 6.87–6.85 (m, 2H), 6.70–6.68 (m, 2H), 3.48 (s, 2H), 2.47–2.15 (m, 8H), 1.55–1.30 (m, 18H). 13C NMR: (126 MHz, DMSO-d6) δ 156.4, 153.1, 139.6, 138.9, 135.3, 135.3, 134.9, 133.1, 131.1, 130.2, 129.3, 128.6, 127.3, 122.6, 115.3, 62.5, 55.1, 31.8, 31.8, 28.2. MS(ESI+): 613.2 m/z [M+H]+.
- 1-(3-((1-Benzylpiperidin-4-ylidene)(3-hydroxyphenyl)methyl)phenyl)guanidine (13a). Prepared from 11a according to general procedure F to give the title material as a hydrochloride salt with 40% yield. 1H NMR: (600 MHz, Methanol-d4) δ 7.49 (dd, J = 6.6, 3.0 Hz, 2H), 7.42–7.35 (m, 3H), 7.34 (t, J = 7.9 Hz, 1H), 7.10 (dd, J = 7.7, 2.0 Hz, 1H), 7.08–7.01 (m, 2H), 6.96 (t, J = 1.9 Hz, 1H), 6.60 (ddd, J = 8.2, 2.5, 0.8 Hz, 1H), 6.54 (dt, J = 7.4, 1.1 Hz, 1H), 6.49 (t, J = 2.0 Hz, 1H), 4.27 (s, 2H), 3.52–3.25 (m, 2H), 3.14–2.89 (m, 2H), 2.81–2.33 (m, 4H). 13C NMR: (151 MHz, Methanol-d4) δ 158.7, 158.0, 144.4, 143.3, 140.0, 136.3, 132.5, 131.2, 131.1, 130.6, 130.5, 130.3, 129.5, 127.2, 125.0, 121.5, 117.2, 115.4, 61.4, 54.2, 54.2, 29.4, 29.2. HRMS (ESI+): m/z calcd. for C26H29N4O [M+H]+ 413.2341, found 413.2348. Purity by HPLC: 94.0%, tR = 3.2 min.
- 1-(4-((1-Benzylpiperidin-4-ylidene)(3-hydroxyphenyl)methyl)phenyl)guanidine (13b). Prepared from 11b according to general procedure F to give the title material as a hydrochloride salt with 36% yield. 1H NMR: (500 MHz, Methanol-d4) δ 7.52–7.43 (m, 2H), 7.43–7.36 (m, 3H), 7.26–7.13 (m, 4H), 7.05 (td, J =7.9, 2.0 Hz, 1H), 6.60 (d, J =8.1 Hz, 1H), 6.54 (d, J =7.7 Hz, 1H), 6.51–6.43 (m, 1H), 4.26 (s, 2H), 3.42 (br s, 2H), 3.03 (br s, 2H), 2.69 (br s, 2H), 2.45 (br s, 2H). 13C NMR: (126 MHz, Methanol-d4) δ 158.7, 143.4, 141.8, 140.3, 135.1, 132.4, 132.0, 131.3, 130.6, 130.4, 126.3, 121.4, 117.2, 115.3, 61.5, 54.3, 54.2, 49.0, 29.4, 29.2. HRMS (ESI+) m/z calcd. for C26H29N4O [M+H]+ 413.2341, found 413.2333. Purity by HPLC: >99%, tR = 3.84 min.
- 1-(3-((1-Benzylpiperidin-4-ylidene)(4-hydroxyphenyl)methyl)phenyl)guanidine (14a). Prepared from 12a according to general procedure F to give the title material as a hydrochloride salt with 79% yield. 1H NMR: (600 MHz, Methanol-d4) δ 7.60 (dd, J = 6.6, 2.9 Hz, 2H), 7.53–7.48 (m, 3H), 7.46 (t, J = 7.8 Hz, 1H), 7.25–7.19 (m, 1H), 7.17 (dt, J = 7.7, 1.3 Hz, 1H), 7.06 (t, J = 1.9 Hz, 1H), 7.00 (d, J = 8.5 Hz, 2H), 6.77 (d, J = 8.5 Hz, 2H), 4.38 (s, 2H), 3.69–3.40 (m, 2H), 3.27–3.01 (m, 2H), 2.97–2.42 (m, 4H). 13C NMR: (151 MHz, Methanol-d4) δ 156.7, 156.6, 143.6, 138.6, 134.9, 131.6, 131.0, 130.4, 129.8, 129.6, 129.1, 129.0, 128.3, 128.2, 126.0, 123.6, 114.8, 60.1, 52.9, 28.1, 28.0. HRMS (ESI+) m/z calcd. for C26H29N4O [M+H]+ 413.2341, found 413.2325. Purity by HPLC: >99%, tR = 2.21 min.
- 1-(4-((1-Benzylpiperidin-4-ylidene)(4-hydroxyphenyl)methyl)phenyl)guanidine (14b). Prepared from 12b according to general procedure F to give the title material as a hydrochloride salt with 55% yield. 1H NMR: (500 MHz, Methanol-d4) δ 7.56 (dd, J = 6.4, 3.0 Hz, 2H), 7.52–7.43 (m, 3H), 7.24 (s, 4H), 6.96 (d, J = 8.1 Hz, 2H), 6.74 (d, J = 8.1 Hz, 2H), 4.35 (s, 2H), 3.48 (br s, 2H), 3.13 (br s, 2H), 2.95–2.34 (m, 4H). 13C NMR: (126 MHz, Methanol-d4) δ 156.6, 154.2, 141.0, 138.8, 133.5, 131.7, 131.0, 130.7, 130.3, 129.8, 129.1, 129.0, 128.0, 124.9, 114.8, 60.1, 52.9, 28.1, 28.0. HRMS (ESI+) m/z calcd. for C26H29N4O [M+H]+ 413.2341, found 413.2376. Purity by HPLC: >99%, tR =3.80 min.
- Benzyltriphenylphosphonium bromide (15). Benzyl bromide (10.4 mL, 87.7 mmol) and PPh3 (23.0 g, 87.7 mmol) were dissolved in toluene (500 mL) and heated to reflux overnight. The suspension obtained was cooled to rt and filtered. The precipitate was washed with toluene and dried to afford the desired compound (38.0 g, >99% yield). 1H NMR: (400 MHz, DMSO-d6) δ 7.94–7.85 (m, 3H), 7.79–7.65 (m, 12H), 7.29–7.26 (m, 1H), 7.20 (t, J = 7.5 Hz, 2H), 7.04–6.96 (m, 2H), 5.28 (d, JPH = 15.7 Hz, 2H). 13C NMR: (101 MHz, DMSO-d6) δ 135.6 (d, JPC = 3 Hz),134.5 (d, JPC = 10 Hz), 131.4 (d, JPC = 6 Hz), 130.5 (d, JPC = 12 Hz),129.2 (d, JPC = 3 Hz), 128.8 (d, JPC = 3 Hz), 128.5 (d, JPC = 9 Hz), 118.3 (d, JPC = 86 Hz), 28.6 (d, JPC = 46 Hz). MS(ESI+): m/z 353.99 [M]+.
- 1-Benzyl-4-benzylidenepiperidine (16). Prepared from 15 according to general procedure A to afford the title compound with 52% yield. 1H NMR: (400 MHz, CDCl3) δ 7.52–7.13 (m, 10H), 6.31 (s, 1H), 3.56 (s, 2H), 2.71–2.31 (m, 8H). 13C NMR: (101 MHz, CDCl3) δ 139.8, 138.6, 137.9, 129.2, 129.0, 128.2, 128.1, 127.0, 126.1, 123.2, 63.1, 55.1, 54.5, 36.6, 29.2. MS(ESI+): 264.2 m/z (M+H)+.
- 1-Benzyl-4-(bromo(phenyl)methylene)piperidine (18). Prepared from 16 according to general procedure B to afford the title compound with 29% yield. 1H NMR: (CDCl3) δ 7.36–7.29 (m, 10H), 3.56 (s, 2H), 2.73 (t, 2H, J = 5.4 Hz), 2.59 (t, 2H, J = 5.9 Hz), 2.40 (t, 2H, J = 5.1 Hz), 2.31 (t, 2H, J = 5.8 Hz). 13C NMR: (CDCl3) δ 140.6, 138.4, 137.8, 129.6, 129.3, 128.4, 128.4, 128.1, 127.2, 115.7, 62.9, 54.3, 54.0, 34.7, 32.1. MS (ESI+): 342.3 m/z (M+H)+ for 79Br, 344.9 m/z (M+H)+ for 81Br.
- 3-((1-Benzylpiperidin-4-ylidene)(phenyl)methyl)aniline (19a). Prepared from 18 according to general procedure C using 3-aminophenylboronic acid hydrochloride to give the title material with 98% yield. 1H NMR: (DMSO-d6) δ 7.30–7.15 (m, 8H), 7.06 (d, 2H, J = 7.9 Hz), 6.93 (t, 1H, J = 7.7 Hz), 6.40 (d, 1H, J = 8.0 Hz), 6.29–6.25 (m, 2H), 3.46 (s, 2H), 2.42–2.39 (m, 4H), 2.29–2.26 (m, 2H), 2.24–2.23 (m, 2H). 13C NMR: (DMSO- d6) δ 148.3, 142.7, 142.2, 138.4, 135.9, 134.2, 129.2, 128.8, 128.4, 128.1, 127.9, 126.8, 126.1, 116.9, 114.9, 112.1, 61.9, 54.6, 54.6, 31.3, 31.1. MS (ESI+): 355 m/z (M+H)+.
- 1-Benzyl-4-((4-nitrophenyl)(phenyl)methylene)piperidine (19b). Prepared from 18 according to general procedure C using 4-nitrophenylboronic acid to give the title material with 33% yield. 1H NMR: (400 MHz, CDCl3) δ 8.16 (d, 2H, J = 8.4 Hz), 7.45–7.22 (m, 10H), 7.15–7.13 (m, 2H), 3.60 (s, 2H), 2.66–2.52 (m, 4H), 2.51–2.37 (m, 4H). 13C NMR: (101 MHz, CDCl3) δ 149.5, 146.3, 138.5, 134.2, 130.8, 129.9, 129.3, 128.4, 128.3, 127.2, 127.0, 126.3, 123.4, 115.9, 62.9, 54.9, 54.9, 31.8, 31.8. MS(ESI+): 385.0 m/z (M+H)+.
- 4-((1-Benzylpiperidin-4-ylidene)(phenyl)methyl)aniline (20b). Prepared from 19b according to general procedure D to give the title material with 95% yield. 1H NMR: (400 MHz, CDCl3) δ 7.44–7.08 (m, 10H), 6.92 (d, 2H, J = 8.3 Hz), 6.61 (d, 2H, J = 8.3 Hz), 3.61 (bs, 2H), 3.55 (s, 2H), 2.59–2.42 (m, 6H), 2.42–2.35 (m, 2H). 13C NMR: (101 MHz, CDCl3) δ 144.7, 143.1, 138.5, 135.6, 134.6, 133.0, 130.9, 129.9, 129.2, 128.2, 127.8, 127.0, 126.1, 114.6, 63.1, 55.3, 55.3, 31.8, 31.8. MS(ESI+): 355.1 m/z (M+H)+.
- tert-Butyl(3-((1-benzylpiperidin-4-ylidene)(phenyl)methyl)phenylamino)(tert-butoxycarbonylamino)methylenecarbamate (21a). Prepared from 19a according to general procedure E to give the title material with 75% yield. 1H NMR: (CDCl3) δ 11.61 (s, 1H), 10.21 (s, 1H), 7.57 (d, 1H, J = 8.1 Hz), 7.34–7.18 (m, 10H), 7.12 (d, 2H, J = 7.8 Hz), 6.87 (d, 1H, J = 7.4 Hz), 3.53 (s, 2H), 2.50–2.49 (m, 4H), 2.45–2.44 (m, 2H), 2.39–2.38 (m, 2H), 1.53 (s, 9H), 1.48 (s, 9H). 13C NMR: (CDCl3) δ 163.8, 153.7, 153.5, 143.2, 142.5, 138.7, 136.5, 136.4, 135.3, 130.0, 129.3, 128.7, 128.4, 128.1, 127.1, 126.7, 126.5, 123.9, 120.8, 83.8, 79.7, 63.2, 55.5, 55.3, 31.9, 28.4, 28.3. MS (ESI+): 597 m/z (M+H)+.
- Tert-butyl(4-((1-benzylpiperidin-4-ylidene)(phenyl)methyl)phenylamino)(tert-butoxycarbonylamino)methylenecarbamate (21b). Prepared from 20b according to general procedure E to give the title material with 71% yield. 1H NMR: (600 MHz, CDCl3) δ 11.65 (s, 1H), 10.33 (s, 1H), 7.54 (d, J = 8.5 Hz, 2H), 7.42–7.36 (m, 2H), 7.35 (t, J = 7.4 Hz, 2H), 7.32–7.25 (m, 3H), 7.21 (t, J = 7.4 Hz, 1H), 7.11 (d, J = 7.1 Hz, 2H), 7.08 (d, J = 8.6 Hz, 2H), 3.63 (s, 2H), 2.70–2.36 (m, 8H), 1.55 (s, 9H), 1.52 (s, 9H). 13C NMR: (151 MHz, CDCl3) δ 163.7, 153.5, 153.4, 142.4, 138.9, 135.8, 135.2, 130.5, 130.0, 129.6, 128.4, 128.1, 127.5, 126.5, 121.7, 83.8, 79.7, 62.9, 55.1, 31.5, 31.4, 28.3, 28.2. MS(ESI+): 597.1 m/z (M+H)+.
- 1-(3-((1-Benzylpiperidin-4-ylidene)(phenyl)methyl)phenyl)guanidine (22a). To a solution of intermediate 21a (0.36 g, 0.587 mmol) in CH2Cl2 (20 mL) was added 4N HCl/dioxane (1.5 mL, 5.87 mmol), and the reaction mixture was stirred for 3 days at rt. The solvent was evaporated and the residue re-dissolved in 5% NaOH (aq) and then extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried and evaporated, and the residue was purified via flash column chromatography using a gradient of CH2Cl2/MeOH satd. with NH3 90:10→50:50 as the eluent to afford the title material, which was converted to the HCl salt through addition of HCl/dioxane (with presence of 1/2 molecule of dioxane, white solid, 0.21 g, 74%). 1H NMR: (500 MHz, Methanol-d4) δ 8.13 (s, 1H), 7.59–7.36 (m, 5H), 7.26 (t, J = 7.4 Hz, 2H), 7.23–7.15 (m, 5H), 7.09 (d, J = 7.3 Hz, 2H), 4.23 (s, 2H), 3.33–3.12 (m, 4H), 2.57 (dt, J = 16.5, 5.9 Hz, 4H). 13C NMR: (126 MHz, Methanol--d4) δ 156.6, 140.8, 140.5, 138.7, 133.7, 130.9, 130.7, 129.7, 129.5, 129.3, 129.0, 128.9, 128.2, 127.1, 124.9, 60.2, 52.9, 52.9, 28.1, 28.0. HRMS (ESI+) m/z calcd. for C26H29N4 [M+H]+ 397.2392, found 397.2728. Purity by HPLC: >99%, tR =1.415 min.
- 1-(4-((1-Benzylpiperidin-4-ylidene)(phenyl)methyl)phenyl)guanidine (22b). Prepared from 21b according to general procedure F to give the title material as a hydrochloride salt with 49% yield. 1H NMR: (500 MHz, Methanol-d4) δ 7.59–7.46 (m, 2H), 7.47–7.38 (m, 3H), 7.37 (t, J = 7.8 Hz, 1H), 7.26 (t, J = 7.4 Hz, 2H), 7.19 (t, J = 7.4 Hz, 1H), 7.17–7.08 (m, 4H), 7.03–6.96 (m, 1H), 4.29 (s, 2H), 3.54–3.39 (m, 2H), 3.16–2.99 (m, 2H), 2.71 (dd, J = 29.5, 15.1 Hz, 2H), 2.60–2.43 (m, 2H). 13C NMR: (126 MHz, DMSO-d6) δ 156.6, 141.4, 139.5, 137.5, 134.5, 131.9, 130.8, 130.7, 130.4, 129.8, 129.6, 129.2, 128.9, 127.5, 124.4, 59.0, 52.2, 52.2, 28.1, 28.0. HRMS (ESI+), m/z calc. for C26H29N4 [M+H]+ 397.2392, found 397.2412. Purity by HPLC: >99%, tR = 3.89 min.
4.2. In Vitro Pharmacology
4.3. In Vivo Behavioral Pharmacology
4.4. Metabolic Stability in Rat Liver Microsomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- National Institute on Drug Abuse (NIDA). Prescription Opioids DrugFacts. Available online: https://nida.nih.gov/publications/drugfacts/prescription-opioids (accessed on 27 March 2025).
- Centers for Disease Control and Prevention (CDC). U.S. Opioid Dispensing Rate Maps. Available online: https://www.cdc.gov/overdose-prevention/data-research/facts-stats/opioid-dispensing-rate-maps.html (accessed on 27 September 2024).
- Lee, M.; Silverman, S.M.; Hansen, H.; Patel, V.B.; Manchikanti, L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician 2011, 14, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Moal, M.L. Drug Abuse: Hedonic Homeostatic Dysregulation. Science 1997, 278, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Aghajanian, G.K. Molecular and Cellular Basis of Addiction. Science 1997, 278, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). U.S. Overdose Deaths Decrease Almost 27% in 2024. Available online: https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2025/20250514.htm#:~:text=27%25%20in%202024-,U.S.%20Overdose%20Deaths%20Decrease%20Almost%2027%25%20in%202024,like%20methamphetamine)%20decreased%20as%20well (accessed on 5 June 2025).
- McLaughlin, J.P.; Myers, L.C.; Zarek, P.E.; Caron, M.G.; Lefkowitz, R.J.; Czyzyk, T.A.; Pintar, J.E.; Chavkin, C. Prolonged Kappa Opioid Receptor Phosphorylation Mediated by G-protein Receptor Kinase Underlies Sustained Analgesic Tolerance. J. Biol. Chem. 2004, 279, 1810–1818. [Google Scholar] [CrossRef]
- Allouche, S.; Noble, F.; Marie, N. Opioid receptor desensitization: Mechanisms and its link to tolerance. Front. Pharmacol. 2014, 5, 280. [Google Scholar] [CrossRef]
- Catherine, M.; Michel, R.; Jean-Marie, Z. Opioid-modulating Peptides: Mechanisms of Action. Curr. Top. Med. Chem. 2005, 5, 341–355. [Google Scholar]
- Rothman, R.B. A review of the role of anti-opioid peptides in morphine tolerance and dependence. Synapse 1992, 12, 129–138. [Google Scholar] [CrossRef]
- Kim, S.H.; Stoicea, N.; Soghomonyan, S.; Bergese, S.D. Intraoperative use of remifentanil and opioid induced hyperalgesia/acute opioid tolerance: Systematic review. Front. Pharmacol. 2014, 5, 108. [Google Scholar] [CrossRef]
- Chu, L.F.; Angst, M.S.; Clark, D. Opioid-induced hyperalgesia in humans: Molecular mechanisms and clinical considerations. Clin. J. Pain 2008, 24, 479–496. [Google Scholar] [CrossRef]
- Chu, L.F.; Clark, D.J.; Angst, M.S. Opioid Tolerance and Hyperalgesia in Chronic Pain Patients After One Month of Oral Morphine Therapy: A Preliminary Prospective Study. J. Pain 2006, 7, 43–48. [Google Scholar] [CrossRef]
- Mao, J. Opioid-induced abnormal pain sensitivity: Implications in clinical opioid therapy. Pain 2002, 100, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Kaczynska, K.; Wojciechowski, P. Non-Opioid Peptides Targeting Opioid Effects. Int. J. Mol. Sci. 2021, 22, 13619. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Marusich, J.; Li, J.-X.; Zhang, Y. Neuropeptide FF and Its Receptors: Therapeutic Applications and Ligand Development. J. Med. Chem. 2020, 63, 12387–12402. [Google Scholar] [CrossRef] [PubMed]
- Simonin, F.; Schmitt, M.; Laulin, J.-P.; Laboureyras, E.; Jhamandas, J.H.; MacTavish, D.; Matifas, A.; Mollereau, C.; Laurent, P.; Parmentier, M.; et al. RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc. Natl. Acad. Sci. USA 2006, 103, 466–471. [Google Scholar] [CrossRef]
- Hammoud, H.; Elhabazi, K.; Quillet, R.; Bertin, I.; Utard, V.; Laboureyras, E.; Bourguignon, J.-J.; Bihel, F.; Simonnet, G.; Simonin, F.; et al. Aminoguanidine Hydrazone Derivatives as Nonpeptide NPFF1 Receptor Antagonists Reverse Opioid Induced Hyperalgesia. ACS Chem. Neurosci. 2018, 9, 2599–2609. [Google Scholar] [CrossRef]
- Quillet, R.; Schneider, S.; Utard, V.; Drieu la Rochelle, A.; Elhabazi, K.; Henningsen, J.B.; Gizzi, P.; Schmitt, M.; Kugler, V.; Simonneaux, V.; et al. Identification of an N-acylated-DArg-Leu-NH2 Dipeptide as a Highly Selective Neuropeptide FF1 Receptor Antagonist That Potently Prevents Opioid-Induced Hyperalgesia. J. Med. Chem. 2021, 64, 7555–7564. [Google Scholar] [CrossRef]
- Gealageas, R.; Schneider, S.; Humbert, J.-P.; Bertin, I.; Schmitt, M.; Laboureyras, E.; Dugave, C.; Mollereau, C.; Simonnet, G.; Bourguignon, J.-J.; et al. Development of sub-nanomolar dipeptidic ligands of neuropeptide FF receptors. Bioorganic Med. Chem. Lett. 2012, 22, 7471–7474. [Google Scholar] [CrossRef]
- Zhang, T.; Han, Z.; Shi, X.; Zhao, W.; Wang, Z.; Zhang, R.; Xu, B.; Zhang, M.; Zhang, Q.; Xiao, J.; et al. Discovery of two novel branched peptidomimetics containing endomorphin-2 and RF9 pharmacophores: Synthesis and neuropharmacological evaluation. Bioorganic Med. Chem. 2019, 27, 630–643. [Google Scholar] [CrossRef]
- Elhabazi, K.; Trigo, J.M.; Mollereau, C.; Moulédous, L.; Zajac, J.M.; Bihel, F.; Schmitt, M.; Bourguignon, J.J.; Meziane, H.; Petit-demoulière, B.; et al. Involvement of neuropeptide FF receptors in neuroadaptive responses to acute and chronic opiate treatments. Br. J. Pharmacol. 2012, 165, 424–435. [Google Scholar] [CrossRef]
- Elhabazi, K.; Humbert, J.-P.; Bertin, I.; Quillet, R.; Utard, V.; Schneider, S.; Schmitt, M.; Bourguignon, J.-J.; Laboureyras, E.; Ben Boujema, M.; et al. RF313, an orally bioavailable neuropeptide FF receptor antagonist, opposes effects of RF-amide-related peptide-3 and opioid-induced hyperalgesia in rodents. Neuropharmacology 2017, 118, 188–198. [Google Scholar] [CrossRef]
- Bihel, F.; Humbert, J.-P.; Schneider, S.; Bertin, I.; Wagner, P.; Schmitt, M.; Laboureyras, E.; Petit-Demoulière, B.; Schneider, E.; Mollereau, C.; et al. Development of a Peptidomimetic Antagonist of Neuropeptide FF Receptors for the Prevention of Opioid-Induced Hyperalgesia. ACS Chem. Neurosci. 2015, 6, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, B.; Li, N.; Zhang, R.; Zhang, Q.; Shi, X.; Xu, K.; Xiao, J.; Chen, D.; Niu, J.; et al. Development of Multifunctional and Orally Active Cyclic Peptide Agonists of Opioid/Neuropeptide FF Receptors that Produce Potent, Long-Lasting, and Peripherally Restricted Antinociception with Diminished Side Effects. J. Med. Chem. 2021, 64, 13394–13409. [Google Scholar] [CrossRef]
- He, C.; Wang, X.; Zhang, J.; Wang, H.; Zhao, Y.; Shah, J.N.; Ma, C.; Li, H.; Su, W.; Zhang, Z.; et al. MCRT, a multifunctional ligand of opioid and neuropeptide FF receptors, attenuates neuropathic pain in spared nerve injury model. Basic Clin. Pharmacol. Toxicol. 2021, 128, 731–740. [Google Scholar] [CrossRef]
- Li, N.; Han, Z.L.; Wang, Z.L.; Xing, Y.H.; Sun, Y.L.; Li, X.H.; Song, J.J.; Zhang, T.; Zhang, R.; Zhang, M.N.; et al. BN-9, a chimeric peptide with mixed opioid and neuropeptide FF receptor agonistic properties, produces nontolerance-forming antinociception in mice. Br. J. Pharmacol. 2016, 173, 1864–1880. [Google Scholar] [CrossRef]
- Journigan, V.B.; Mésangeau, C.; Vyas, N.; Eans, S.O.; Cutler, S.J.; McLaughlin, J.P.; Mollereau, C.; McCurdy, C.R. Nonpeptide Small Molecule Agonist and Antagonist Original Leads for Neuropeptide FF1 and FF2 Receptors. J. Med. Chem. 2014, 57, 8903–8927. [Google Scholar] [CrossRef]
- Dantzman, C.L.; King, M.M.; Ernst, G.E.; Wang, X.; McCauley, J.P.; Andisik, D.W.; Brush, K.; Bui, K.H.; Frietze, W.; Hoesch, V.; et al. 4-Piperidin-4-ylidenemethyl-benzamides as δ-opioid receptor agonists for CNS indications: Identifying clinical candidates. Bioorg. Med. Chem. Lett. 2012, 22, 1174–1178. [Google Scholar] [CrossRef]
- Wei, Z.-Y.; Brown, W.; Takasaki, B.; Plobeck, N.; Delorme, D.; Zhou, F.; Yang, H.; Jones, P.; Gawell, L.; Gagnon, H.; et al. N,N-Diethyl-4-(phenylpiperidin-4-ylidenemethyl)benzamide: A Novel, Exceptionally Selective, Potent δ Opioid Receptor Agonist with Oral Bioavailability and Its Analogues. J. Med. Chem. 2000, 43, 3895–3905. [Google Scholar] [CrossRef]
- Chang, K.J.; Rigdon, G.C.; Howard, J.L.; McNutt, R.W. A novel, potent and selective nonpeptidic delta opioid receptor agonist BW373U86. J. Pharmacol. Exp. Ther. 1993, 267, 852–857. [Google Scholar] [CrossRef]
- Calderon, S.N.; Rothman, R.B.; Porreca, F.; Flippen-Anderson, J.L.; McNutt, R.W.; Xu, H.; Smith, L.E.; Bilsky, E.J.; Davis, P.; Rice, K.C. Probes for Narcotic Receptor Mediated Phenomena. 19. Synthesis of (+)-4-[(αR)-α-((2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC 80): A Highly Selective, Nonpeptide .delta. Opioid Receptor Agonist. J. Med. Chem. 1994, 37, 2125–2128. [Google Scholar] [CrossRef]
- Coats, S.J.; Schulz, M.J.; Carson, J.R.; Codd, E.E.; Hlasta, D.J.; Pitis, P.M.; Stone, D.J.; Zhang, S.-P.; Colburn, R.W.; Dax, S.L. Parallel methods for the preparation and SAR exploration of N-ethyl-4-[(8-alkyl-8-aza-bicyclo[3.2.1]oct-3-ylidene)-aryl-methyl]-benzamides, powerful mu and delta opioid agonists. Bioorg. Med. Chem. Lett. 2004, 14, 5493–5498. [Google Scholar] [CrossRef] [PubMed]
- Maryanoff, B.E.; Reitz, A.B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Sharma, S.K.; Jones, R.M.; Metzger, T.G.; Ferguson, D.M.; Portoghese, P.S. Transformation of a κ-Opioid Receptor Antagonist to a κ-Agonist by Transfer of a Guanidinium Group from the 5′- to 6′-Position of Naltrindole. J. Med. Chem. 2001, 44, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Talmont, F.; Garcia, L.P.; Mazarguil, H.; Zajac, J.-M.; Mollereau, C. Characterization of two novel tritiated radioligands for labelling Neuropeptide FF (NPFF1 and NPFF2) receptors. Neurochem. Int. 2009, 55, 815–819. [Google Scholar] [CrossRef]
- Loewa, A.; Feng, J.J.; Hedtrich, S. Human disease models in drug development. Nat. Rev Bioeng. 2023, 1, 545–559. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, B.; Zhang, Q.; Chen, D.; Zhang, M.; Zhao, G.; Xu, K.; Xiao, J.; Zhu, H.; Niu, J.; et al. Spinal administration of the multi-functional opioid/neuropeptide FF agonist BN-9 produced potent antinociception without development of tolerance and opioid-induced hyperalgesia. Eur. J. Pharmacol. 2020, 880, 173169. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, B.; Jiang, C.; Zhang, T.; Zhang, M.; Li, N.; Zhang, Q.; Xu, K.; Chen, D.; Xiao, J.; et al. Spinal DN-9, a Peptidic Multifunctional Opioid/Neuropeptide FF Agonist Produced Potent Nontolerance Forming Analgesia with Limited Side Effects. J. Pain 2020, 21, 477–493. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Hoot, M.R.; Sypek, E.I.; Reilley, K.J.; Carey, A.N.; Bidlack, J.M.; McLaughlin, J.P. Inhibition of Gβγ-subunit signaling potentiates morphine-induced antinociception but not respiratory depression, constipation, locomotion, and reward. Behav. Pharmacol. 2013, 24, 144–152. [Google Scholar] [CrossRef]
- Gavériaux-Ruff, C.; Kieffer, B.L. Delta opioid receptor analgesia: Recent contributions from pharmacology and molecular approaches. Behav. Pharmacol. 2011, 22, 405–414. [Google Scholar] [CrossRef]
- Crain, S.M.; Shen, K.-F. Acute thermal hyperalgesia elicited by low-dose morphine in normal mice is blocked by ultra-low-dose naltrexone, unmasking potent opioid analgesia. Brain Res. 2001, 888, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Way, E.L.; Loh, H.H.; Shen, F.H. Simultaneous quantitative assessment of morphine tolerance and physical dependence. J. Pharmacol. Exp. Ther. 1969, 167, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, C.; Mazarguil, H.; Marcus, D.; Quelven, I.; Kotani, M.; Lannoy, V.; Dumont, Y.; Quirion, R.; Detheux, M.; Parmentier, M.; et al. Pharmacological characterization of human NPFF1 and NPFF2 receptors expressed in CHO cells by using NPY Y1 receptor antagonists. Eur. J. Pharmacol. 2002, 451, 245–256. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Binding, Ki (nM) ± SEM | |||||||
|---|---|---|---|---|---|---|---|
| Cpd. | R1 | R2 | NPFF1-R | NPFF2-R | MOR | DOR | KOR |
| NPFF | 1.39 ± 0.08 | 6.09 ± 1.59 | >10,000 | >10,000 | >10,000 | ||
| NPAF | 99.6 ± 5.0 | 3.54 ± 0.82 | >10,000 | >10,000 | >10,000 | ||
| RF9 | 163 ± 26 | 158 ± 66 | 1920 ± 161 | >10,000 | 6710 ± 639 | ||
| DAMGO | 4.15 ± 0.34 | 857 ± 61 | 1200 ± 354 | ||||
| SNC80 | 2761 ± 322 | 34.6 ± 3.8 | 2020 ± 617 | ||||
| U69,593 | 4050 ± 534 | 6700 ± 739 | 1.62 ± 0.11 | ||||
| 13a | 3- | 3′-OH | 2180 ± 280 | 7080 ± 1260 | 5.06 ± 0.21 | 0.481 ± 0.192 | 2.82 ± 0.39 |
| 13b | 4- | 3′-OH | 605 ± 136 | 6020 ± 1150 | 56.2 ± 7.5 | 1.67 ± 0.61 | 30.6 ± 1.1 |
| 14a | 3- | 4′-OH | 1720 ± 308 | >10,000 | 33.5 ± 4.7 | 2.33 ± 0.60 | 32.3 ± 3.5 |
| 14b | 4- | 4′-OH | 1980 ± 288 | 4825 ± 1010 | 313 ± 11 | 6.46 ± 1.11 | 56.6 ± 3.8 |
| 22a | 3- | H | 1400 ± 61 | 2570 ± 423 | 6.45 ± 1.23 | 3.37 ± 0.59 | 91.0 ± 9.9 |
| 22b | 4- | H | 929 ± 183 | 272 ± 1 | 428 ± 33 | 11.4 ± 4.3 | 270 ± 43 |
 | |||||||
| Functional Activity (Agonist Mode) (nM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| [35S]GTPγS MOR | [35S]GTPγS DOR | [35S]GTPγS KOR | ||||||
| Cpd. | R1 | R2 | EC50 ± SEM | Type | EC50 ± SEM | Type | EC50 ± SEM | Type |
| DAMGO | 27.7 ± 2.9 | C | ||||||
| DPDPE | 16.3 ± 1.9 | C | ||||||
| U69,593 | 6.07 ± 0.65 | C | ||||||
| 13a | 3- | 3′-OH | 28.3 ± 2.5 | F | 24.1 ± 2.5 | F | 41.8 ± 4.9 | P |
| 13b | 4- | 3′-OH | 1442 ± 226 | P | 9.62 ±1.27 | F | 267.9 ± 31.9 | P |
| 14a | 3- | 4′-OH | 419.8 ± 54.5 | P | 455.0 ± 61.7 | F | 481.9 ± 81.6 | P |
| 14b | 4- | 4′-OH | n/a | A | 227.0 ± 12.5 | F | 135.8 ± 23.7 | P |
| 22a | 3- | H | 101.9 ± 21.3 | P | 255.9 ± 24.9 | F | 254.7 ± 57.7 | P |
| 22b | 4- | H | 576.2 ± 311.3 | P | 539.4 ± 93.2 | F | 2711 ± 398 | P |
 | ||||||||
| Parameters | |
|---|---|
| C0 (ng/mL) | 9152.0 ± 3736.3 |
| AUC/dose0_inf (ng × h/mL/mg) | 3004.7 ± 108.1 |
| CL (L/h) | 0.3 ± 0.0 |
| k (1/h) | 0.2 ± 0.0 |
| T1/2 (h) | 4.1 ± 0.5 |
| Vd (L) | 2.0 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mottinelli, M.; Journigan, V.B.; Obeng, S.; Pallares, V.L.C.; Mѐsangeau, C.; Kapanda, C.N.; Cutler, S.J.; Lambert, J.A.; Eans, S.O.; Ganno, M.L.; et al. Dual Opioid–Neuropeptide FF Small Molecule Ligands Demonstrate Analgesia with Reduced Tolerance Liabilities. Molecules 2025, 30, 2851. https://doi.org/10.3390/molecules30132851
Mottinelli M, Journigan VB, Obeng S, Pallares VLC, Mѐsangeau C, Kapanda CN, Cutler SJ, Lambert JA, Eans SO, Ganno ML, et al. Dual Opioid–Neuropeptide FF Small Molecule Ligands Demonstrate Analgesia with Reduced Tolerance Liabilities. Molecules. 2025; 30(13):2851. https://doi.org/10.3390/molecules30132851
Chicago/Turabian StyleMottinelli, Marco, V. Blair Journigan, Samuel Obeng, Victoria L. C. Pallares, Christophe Mѐsangeau, Coco N. Kapanda, Stephen J. Cutler, Janet A. Lambert, Shainnel O. Eans, Michelle L. Ganno, and et al. 2025. "Dual Opioid–Neuropeptide FF Small Molecule Ligands Demonstrate Analgesia with Reduced Tolerance Liabilities" Molecules 30, no. 13: 2851. https://doi.org/10.3390/molecules30132851
APA StyleMottinelli, M., Journigan, V. B., Obeng, S., Pallares, V. L. C., Mѐsangeau, C., Kapanda, C. N., Cutler, S. J., Lambert, J. A., Eans, S. O., Ganno, M. L., Sheng, W., King, T., Sharma, A., Mollereau, C., Avery, B. A., McLaughlin, J. P., & McCurdy, C. R. (2025). Dual Opioid–Neuropeptide FF Small Molecule Ligands Demonstrate Analgesia with Reduced Tolerance Liabilities. Molecules, 30(13), 2851. https://doi.org/10.3390/molecules30132851