3.2.7. Specific Procedures and Characterization
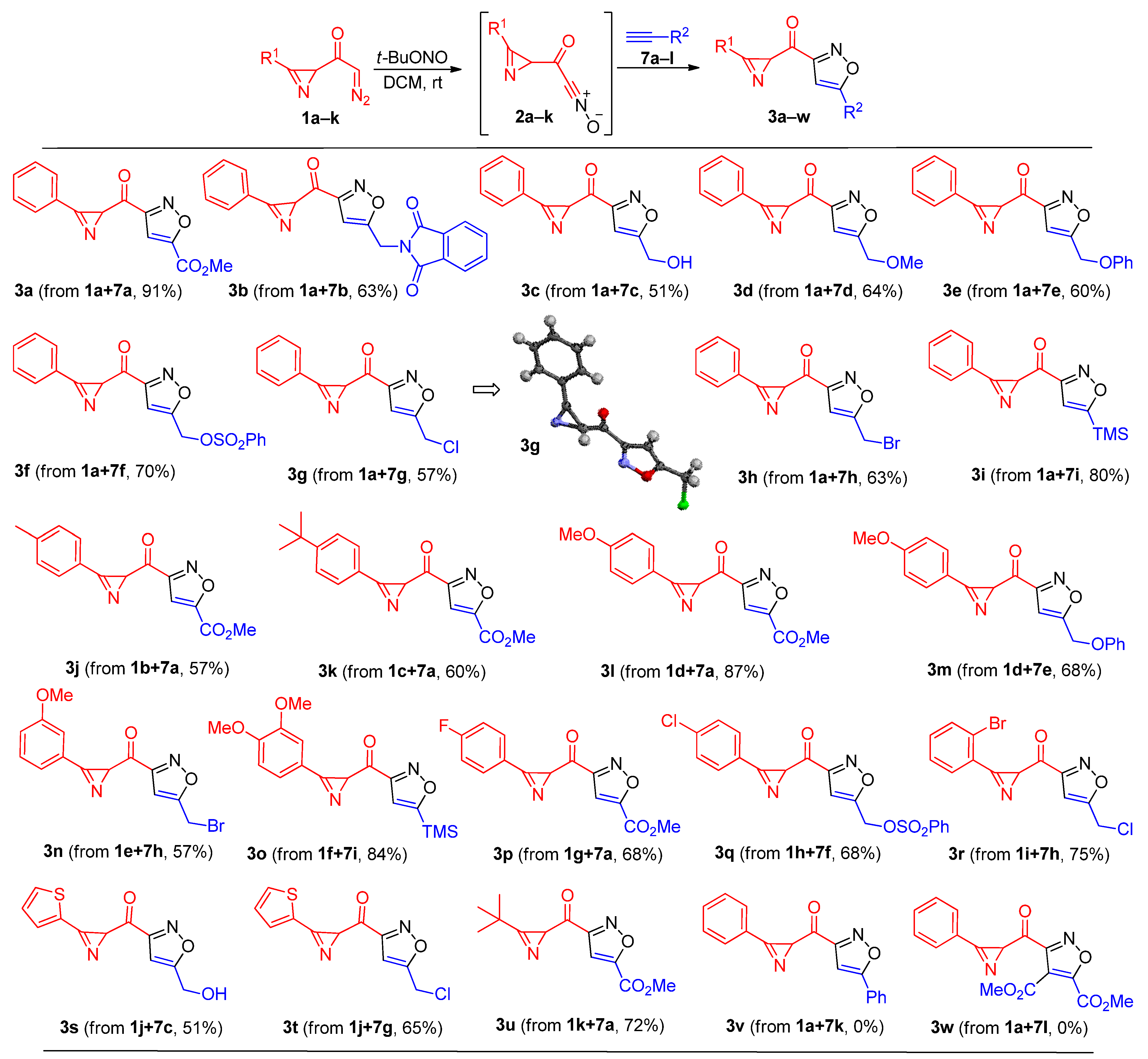
Methyl 3-(3-phenyl-2H-azirine-2-carbonyl)isoxazole-5-carboxylate (3a). Compound 3a was prepared following the general procedure GP-A from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (50 mg, 0.27 mmol), tert-butyl nitrite (175 μL, 1.35 mmol), and methyl propiolate 7a (114 μL, 1.35 mmol) in DCM (5 mL) to produce pure product in 69 mg (91% yield), after column chromatography on silica (light petroleum/ethyl acetate, 4:1, (v/v)).
Compound 3a was also prepared following the general procedure GP-B from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (300 mg, 1.62 mmol), tert-butyl nitrite (1.02 mL, 8.1 mmol), methyl propiolate 7a (811 μL, 8.1 mmol), and boron trifluoride etherate (10 μL, 81 μmol) in DMF (10 mL) to produce pure product in 348 mg (79% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 2:1, (v/v)) as a yellow solid: mp 85–87 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.91–7.88 (m, 2H), 7.68–7.64 (m, 1H), 7.60–7.56 (m, 2H), 7.32 (s, 1H), 4.05 (s, 1H), 4.01 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.5 (C), 162.1 (C), 161.4 (C), 156.6 (C), 156.0 (C), 134.2 (CH), 130.8 (CH), 129.4 (CH), 121.6 (C), 107.9 (C), 53.2 (CH), 34.5 (CH3); IR (KBr, cm–1): 3134, 2956, 1741, 1680, 1586, 1450, 1313, 1229, 1130, 998, 979, 931, 756, 687, 554; HRMS (ESI) m/z [M + H]+ calcd for C14H11N2O4+ 271.0713, found 271.0711.
2-((3-(3-Phenyl-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl)isoindoline-1,3-dione (3b). Compound 3b was prepared following the general procedure GP-A from 2-diazo-1-(3-(4-fluorophenyl)-2H-azirin-2-yl)ethan-1-one 1a (50 mg, 0.27 mmol), tert-butyl nitrite (175 μL, 1.35 mmol), and 2-(prop-2-yn-1-yl)isoindoline-1,3-dione 7b (250 mg, 1.35 mmol) in DCM (7 mL) to produce pure product in 63 mg (63% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 93–94 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.92–7.90 (m, 2H), 7.87–7.85 (m, 2H), 7.78–7.76 (m, 2H), 7.65–7.61 (m, 1H), 7.57–7.53 (m, 2H), 6.67 (s, 1H), 5.07 (s, 2H), 3.99 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.1 (C), 168.2 (C), 167.0 (C), 162.0 (C), 156.2 (C), 134.5 (CH), 134.0 (CH), 131.7 (C), 130.8 (CH), 129.4 (CH), 123.8 (CH), 121.7 (C), 101.9 (CH), 34.4 (CH), 33.0 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H14N3O4+ 372.0979, found 372.0971.
(5-(Hydroxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone (3c). Compound 3c was prepared following the general procedure GP-A from 2-diazo-1-(3-(4-fluorophenyl)-2H-azirin-2-yl)ethan-1-one 1a (50 mg, 0.27 mmol), tert-butyl nitrite (175 μL, 1.35 mmol), and prop-2-yn-1-ol 7c (80 μL, 1.35 mmol) in DCM (5 mL) to produce pure product in 33 mg (51% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 2:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.90–7.88 (m, 2H), 7.67–7.63 (m, 1H), 7.59–7.55 (m, 2H), 6.69 (s, 1H), 4.87–4.86 (m, 2H), 4.03 (s, 1H), 2.11 (br. s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.5 (C), 173.1 (C), 161.8 (C), 156.3 (C), 134.1 (CH), 130.8 (CH), 129.4 (CH), 121.7 (C), 100.7 (CH), 56.3 (CH2), 34.4 (CH); HRMS (ESI) m/z [M + H]+ calcd for C13H11N2O3+ 243.0764, found 243.0760.
(5-(Methoxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone (3d). Compound 3d was prepared following the general procedure GP-A from 2-diazo-1-(3-(4-fluorophenyl)-2H-azirin-2-yl)ethan-1-one 1a (200 mg, 1.08 mmol), tert-butyl nitrite (465 μL, 5.4 mmol), and 3-methoxyprop-1-yne (560 mg, 5.4 mmol) in DCM (10 mL) to produce pure product in 164 mg (64% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.91–7.88 (m, 2H), 7.67–7.62 (m, 1H), 7.59–7.55 (m, 2H), 6.70 (s, 1H), 4.62 (s, 2H), 4.04 (s, 1H), 3.46 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.3 (C), 170.8 (C), 161.9 (C), 156.3 (C), 134.1 (CH), 130.8 (CH), 129.4 (CH), 121.8 (C), 101.8 (CH), 65.0 (CH2), 59.0 (CH), 34.3 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C14H13N2O3+ 257.0921, found 257.0918.
(5-(Phenoxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone (3e). Compound 3e was prepared following the general procedure GP-A from 2-diazo-1-(3-(4-fluorophenyl)-2H-azirin-2-yl)ethan-1-one 1a (150 mg, 0.81 mmol), tert-butyl nitrite (530 μL, 4.05 mmol), and (prop-2-yn-1-yloxy)benzene 7e (510 μL, 4.05 mmol) in DCM (10 mL) to produce pure product in 258 mg (60% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 70–72 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.90–7.88 (m, 2H), 7.67–7.63 (m, 1H), 7.59–7.55 (m, 2H), 7.35–7.31 (m, 2H), 7.05–7.01 (m, 1H), 6.98–6.96 (m, 2H), 6.78 (s, 1H), 5.24 (s, 2H), 4.04 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.2 (C), 169.7 (C), 161.9 (C), 157.6 (C), 156.2 (C), 134.1 (CH), 130.8 (CH), 129.8 (CH), 129.4 CH), 122.1 (CH), 121.8 (C), 114.8 (CH), 102.2 (CH), 61.0 (CH2), 34.5 (CH); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O3+ 319.1077, found 319.1079.
(3-(3-Phenyl-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl benzenesulfonate (3f). Compound 3f was prepared following the general procedure GP-A from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (50 mg, 0.27 mmol), tert-butyl nitrite (175 μL, 1.35 mmol), and prop-2-yn-1-yl benzenesulfonate 7f (215 μL, 1.35) in DCM (5 mL) to produce pure product in 72 mg (70% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.95–7.92 (m, 2H), 7.88–7.86 (m, 2H), 7.72–7.63 (m, 2H), 7.60–7.55 (m, 4H), 6.68 (s, 1H), 5.24 (s, 2H), 3.96 (m, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.7 (C), 165.9 (C), 161.8 (C), 156.1 (C), 135.5 (C), 134.4 (CH), 134.2 (CH), 130.8 (CH), 129.5 (CH), 129.4 (CH), 128.0 (CH), 121.6 (C), 103.9 (CH), 60.6 (CH2), 34.4 (CH); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O5S+ 383.0696, found 383.0697.
(5-(Chloromethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanoneole (3g). Compound 3g was prepared following the general procedure GP-A from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (200 mg, 1.08 mmol), tert-butyl nitrite (700 μL, 5.4 mmol), and 3-chloroprop-1-yne 7g (570 μL, 5.4 mmol) in DCM (10 mL) to produce pure product in 162 mg (57% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-yellow solid: mp 92–94 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.91–7.88 (m, 2H), 7.68–7.63 (m, 1H), 7.59–7.55 (m, 2H), 6.76 (s, 1H), 4.69 (s, 2H), 4.03 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.0 (C), 169.1 (C), 162.0 (C), 156.2 (C), 134.1 (CH), 130.8 (CH), 129.4 (CH), 121.7 (C), 102.6 (CH), 34.4 (CH), 34.0 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C13H10ClN2O2+ 261.0424, found 261.0425.
(5-(Bromomethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone (3h). Compound 3h was prepared following the general procedure GP-A from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (300 mg, 1.62 mmol), tert-butyl nitrite (1050 μL, 8.1 mmol), and 3-bromoprop-1-yne 7h (900 μL, 8.1 mmol) in DCM (10 mL) to produce pure product in 311 mg (63% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-red–brown solid: mp 73–74 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.90–7.88 (m, 2H), 7.67–7.63 (m, 1H), 7.59–7.55 (m, 2H), 6.75 (s, 1H), 4.53 (s, 2H), 4.02 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.0 (C), 169.1 (C), 162.1 (C), 156.2 (C), 134.1 (CH), 130.8 (CH), 129.4 (CH), 121.7 (C), 102.6 (CH), 34.4 (CH), 17.8 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C13H10BrN2O2+ 304.9927, found 304.9920.
(3-Phenyl-2H-azirin-2-yl)(5-(trimethylsilyl)isoxazol-3-yl)methanone (3i). Compound 3i was prepared following the general procedure GP-A from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (300 mg, 1.62 mmol), tert-butyl nitrite (1050 μL, 8.1 mmol), and ethynyltrimethylsilane 7i (1150 μL, 8.1) in DCM (10 mL) to produce pure product in 370 mg (80% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow–green solid: mp 47–48 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.90–7.88 (m, 2H), 7.66–7.62 (m, 1H), 7.58–7.54 (m, 2H), 6.88 (s, 1H), 4.06 (s, 1H), 0.39 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.8 (C), 180.2 (C), 160.4 (C), 156.5 (C), 134.0 (CH), 130.8 (CH), 129.4 (CH), 122.0 (C), 111.3 (CH), 34.9 (CH), –1.99 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H17N2O2Si+ 285.1054, found 285.1050.
Methyl 3-(3-(p-tolyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate (3j). Compound 3j was prepared following the general procedure GP-A from 2-diazo-1-(3-(p-tolyl)-2H-azirin-2-yl)ethan-1-one 1b (150 mg, 0.75 mmol), tert-butyl nitrite (490 μL, 3.75 mmol), and methyl propiolate 7a (360 μL, 3.75 mmol) in DCM (10 mL) to produce pure product in 120 mg (57% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-red–brown solid: mp 111–113 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.77 (d, 2H, J = 8.1 Hz), 7.38 (d, 2H, J = 8.1 Hz), 7.32 (s, 1H), 4.01 (s, 4H), 2.46 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.7 (C), 162.1 (C), 161.4 (C), 156.6 (C), 155.5 (C), 145.4 (C), 130.9 (CH), 130.2 (CH), 118.7 (C), 108.0 (CH), 53.2 (CH3), 34.4 (CH), 22.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H13N2O4+ 285.0870, found 285.0867.
Methyl 3-(3-(4-(tert-butyl)phenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate (3k). Compound 3k was prepared following the general procedure GP-A from 1-(3-(4-(tert-butyl)phenyl)-2H-azirin-2-yl)-2-diazoethan-1-one 1c (50 mg, 0.21 mmol), tert-butyl nitrite (99 μL, 1.05 mmol), and methyl propiolate 7a (135 μL, 1.05 mmol) in DCM (5 mL) to produce pure product in 41 mg (60% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, 2H, J = 8.4 Hz), 7.59 (d, 2H, J = 8.4 Hz), 7.31 (m, 1H), 4.01 (s, 4H), 1.36 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.7 (C), 162.1 (C), 161.3 (C), 158.4 (C), 156.6 (C), 155.5 (C), 130.8 (CH), 126.5 (CH), 118.6 (C), 108.8 (CH), 53.2 (CH), 35.4 (C), 34.4 (CH3), 31.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C18H19N2O4+ 327.1339, found 327.1343.
Methyl 3-(3-(4-methoxyphenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate (3l). Compound 3l was prepared following the general procedure GP-A from 2-diazo-1-(3-(4-methoxyphenyl)-2H-azirin-2-yl)ethan-1-one 1d (300 mg, 1.4 mmol), tert-butyl nitrite (910 μL, 7 mmol), and methyl propiolate 7a (665 μL, 7 mmol) in DCM (10 mL) to produce pure product in 366 mg (87% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1 + 5% CHCl3, (v/v)) as a light-brown solid: mp 127–129 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, 2H, J = 8.8 Hz), 7.31 (s, 1H), 7.06 (d, 2H, J = 8.8 Hz), 4.01 (s, 3H), 3.98 (s, 1H), 3.90 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.9 (C), 164.3 (C), 162.1 (C), 161.3 (C), 156.6 (C), 154.6 (C), 132.9 (CH), 115.0 (CH), 113.7 (C), 108.0 (CH), 55.7 (CH), 53.2 (CH3), 34.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H13N2O5+ 301.0819, found 301.0823.
(3-(4-Methoxyphenyl)-2H-azirin-2-yl)(5-(phenoxymethyl)isoxazol-3-yl)methanone (3m). Compound 3m was prepared following the general procedure GP-A from 2-diazo-1-(3-(4-methoxyphenyl)-2H-azirin-2-yl)ethan-1-one 1d (200 mg, 0.93 mmol), tert-butyl nitrite (610 μL, 4.65 mmol), and (prop-2-yn-1-yloxy)benzene 7e (580 μL, 4.65 mmol) in DCM (10 mL) to produce pure product in 220 mg (68% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1 + 5% CHCl3, (v/v)) as a light-brown solid: mp 125–127 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, 2H, J = 8.8 Hz), 7.34–7.30 (m, 2H), 7.05 (d, 2H, J = 8.8 Hz), 7.03–7.01 (m, 1H), 6.98–6.96 (m, 2H), 6.77 (s, 1H), 5.24 (s, 2H), 3.97 (s, 1H), 3.89 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.9 (C), 164.3 (C), 162.1 (C), 161.3 (C), 156.6 (C), 154.6 (C), 132.9 (CH), 129.7 (CH), 122.1 (CH), 115.0 (CH), 113.7 (CH), 108.0 (CH), 55.7 (CH), 53.2 (CH), 34.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C20H17N2O4+ 349.1183, found 349.1187.
(5-(Bromomethyl)isoxazol-3-yl)(3-(3-methoxyphenyl)-2H-azirin-2-yl)methanone (3n). Compound 3n was prepared following the general procedure GP-A from 2-diazo-1-(3-(3-methoxyphenyl)-2H-azirin-2-yl)ethan-1-one 1e (200 mg, 0.93 mmol), tert-butyl nitrite (610 μL, 4.65 mmol), and 3-bromoprop-1-yne 7h (413 μL, 4.65 mmol) in DCM (10 mL) to produce pure product in 177 mg (57% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.49–7.41 (m, 3H), 7.20–7.17 (m, 1H), 6.75 (s, 1H), 4.53 (s, 2H), 4.02 (s, 1H), 3.88 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.0 (C), 169.1 (C), 162.1 (C), 160.1 (C), 156.3 (C), 130.5 (CH), 123.4 (CH), 122.8 (C), 120.9 (CH), 114.5 (CH), 102.6 (CH), 55.6 (CH), 34.7 (CH3), 17.8 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C14H12BrN2O3+ 335.0019, found 335.0026.
(3-(3,4-Dimethoxyphenyl)-2H-azirin-2-yl)(5-(trimethylsilyl)isoxazol-3-yl)methanone (3o). Compound 3o was prepared following the general procedure GP-A from 2-diazo-1-(3-(3,4-dimethoxyphenyl)-2H-azirin-2-yl)ethan-1-one 1f (200 mg, 0.82 mmol), tert-butyl nitrite (520 μL, 4.1 mmol), and ethynyltrimethylsilane 7i (520 μL, 4.1 mmol) in DCM (10 mL) to produce pure product in 237 mg (84% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-yellow solid: mp 87–88 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, 1H, J = 1.9 Hz), 7.41 (dd, 1H, J = 8.2, 1.9 Hz), 6.98 (d, 1H, J = 8.2 Hz), 6.88 (s, 1H), 4.02 (s, 1H), 3.96 (s, 3H), 3.95 (s, 3H), 0.38 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz): δ 192.2 (C), 180.2 (C), 160.5 (C), 155.6 (C), 153.9 (C), 149.7 (C), 125.8 (CH), 114.2 (C), 111.8 (CH), 111.3 (CH), 111.1 (CH), 77.2 (CH), 56.2 (CH3), 35.1 (CH3), –1.97 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C17H20N2O4Si+ 345.1265, found 345.1262.
Methyl 3-(3-(4-fluorophenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate (3p). Compound 3p was prepared following the general procedure GP-A from 2-diazo-1-(3-(4-fluorophenyl)-2H-azirin-2-yl)ethan-1-one 1g (50 mg, 0.25 mmol), tert-butyl nitrite (160 μL, 1.25 mmol), and methyl propiolate 7a (120 μL, 1.25 mmol) in DCM (5 mL) to produce pure product in 48 mg (68% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-yellow solid: mp 112–114 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.94–7.90 (m, 2H), 7.32 (s, 1H), 7.31–7.27 (m, 2H), 4.04 (s, 1H), 4.01 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.4 (C), 166.2 (d, C, J = 257.5 Hz), 162.0 (C), 161.4 (C), 156.5 (C), 155.1 (C), 133.3 (d, CH, J = 9.8 Hz), 118.0 (d, C, J = 3.5 Hz), 117.0 (d, CH, J = 22.4 Hz), 107.9 (CH), 53.2 (CH3), 34.5 (CH); HRMS (ESI) m/z [M + H]+ calcd for C14H10FN2O4+ 289.0619, found 289.0621.
(3-(3-(4-Chlorophenyl)-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl benzenesulfonate (3q). Compound 3q was prepared following the general procedure GP-A from 1-(3-(4-chlorophenyl)-2H-azirin-2-yl)-2-diazoethan-1-one 1h (300 mg, 1.37 mmol), tert-butyl nitrite (860 μL, 6.83 mmol), and prop-2-yn-1-yl benzenesulfonate (1.1 mL, 6.83 mmol) in DCM (12 mL) to produce pure product in 389 mg (68% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 5:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.95–7.92 (m, 2H), 7.81 (d, 2H, J = 8.5 Hz), 7.71–7.68 (m, 1H), 7.60–7.55 (m, 4H), 6.69 (s, 1H), 5.24 (s, 2H), 3.96 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 185.8 (C), 161.3 (C), 157.0 (C), 150.8 (C), 136.0 (C), 130.7 (C), 129.7 (CH), 127.1 (CH), 125.2 (CH), 124.8 (CH), 123.3 (CH), 115.5 (C), 99.1 (CH), 55.8 (CH2), 29.8 (C); HRMS (ESI) m/z [M + H]+ calcd for C19H14ClN2O5S+ 417.0306, found 417.0304.
(3-(2-Bromophenyl)-2H-azirin-2-yl)(5-(chloromethyl)isoxazol-3-yl)methanone (3r). Compound 3r was prepared following the general procedure GP-A from 1-(3-(2-bromophenyl)-2H-azirin-2-yl)-2-diazoethan-1-one 1i (205 mg, 0.78 mmol), tert-butyl nitrite (500 μL, 3.88 mmol), and 3-chloroprop-1-yne 7h (400 μL, 3.88 mmol) in DCM (10 mL) to produce pure product in 197 mg (75% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.88–7.86 (m, 1H), 7.76–7.74 (m, 1H), 7.54–7.46 (m, 2H), 6.77 (s, 1H), 4.68 (s, 2H), 4.09 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.9 (C), 169.1 (C), 162.0 (C), 156.6 (C), 134.8 (CH), 134.2 (CH), 133.6 (CH), 128.0 (CH), 125.9 (C), 122.3 (C), 102.6 (CH), 35.5 (CH), 34.0 (CH2) HRMS (ESI) m/z [M+H]+ calcd for C13H9BrClN2O2+ 340.9509, found 340.9503.
(5-(Hydroxymethyl)isoxazol-3-yl)(3-(thiophen-2-yl)-2H-azirin-2-yl)methanone (3s). Compound 3s was prepared following the general procedure GP-A from 2-diazo-1-(3-(thiophen-2-yl)-2H-azirin-2-yl)ethan-1-one 1j (200 mg, 1.05 mmol), tert-butyl nitrite (680 μL, 5.25 mmol), and prop-2-yn-1-ol 7c (302 μL, 5.25 mmol) in DCM (10 mL) to produce pure product in 132 mg (51% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 2:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.88 (dd, 1H, J = 5.0, 1.2 Hz), 7.69 (dd, 1H, J = 3.7, 1.2 Hz), 7.26 (dd, 1H, J = 3.7, 1.2 Hz), 6.68 (s, 1H), 4.85 (s, 2H), 4.04 (s, 1H), 2.37 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 7.89–7.87 (m, 1H), 7.70–7.68 (m, 1H), 7.26–7.24 (m, 1H), 6.68 (s, 1H), 4.85 (s, 2H), 4.04 (s, 1H), 2.37 (br. s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.1 (C), 173.2 (C), 161.8 (C), 149.6 (C), 136.0 (CH), 135.9 (CH), 128.6 (CH), 123.8 (C), 100.7 (CH), 56.3 (CH2), 35.0 (CH); HRMS (ESI) m/z [M + H]+ calcd for C11H8N2O3S+ 249.0328, found 249.0329.
(5-(Chloromethyl)isoxazol-3-yl)(3-(thiophen-2-yl)-2H-azirin-2-yl)methanone (3t). Compound 3t was prepared following the general procedure GP-A from 2-diazo-1-(3-(thiophen-2-yl)-2H-azirin-2-yl)ethan-1-one 1j (200 mg, 1.05 mmol), tert-butyl nitrite (680 μL, 5.25 mmol), and 3-chloroprop-1-yne 7g (560 μL, 5.25 mmol) in DCM (10 mL) to produce pure product in 180 mg (65% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.89 (dd, 1H, J = 5.0, 1.2 Hz), 7.59 (dd, 1H, J = 3.7, 1.2 Hz), 7.27–7.25 (m, 1H), 6.76 (s, 1H), 4.68 (s, 2H), 4.04 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 190.7 (C), 169.1 (C), 162.0 (C), 149.5 (C), 136.0 (CH), 135.9 (CH), 128.6 (C), 123.9 (C), 102.6 (CH), 35.0 (CH), 34.0 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C11H7ClN2O2S+ 266.9990, found 266.9993.
Methyl 3-(3-(tert-butyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate (3u). Compound 3u was prepared following the general procedure GP-A from 1-(3-(tert-butyl)-2H-azirin-2-yl)-2-diazoethan-1-one 1k (200 mg, 1.21 mmol), tert-butyl nitrite (790 μL, 6.05 mmol), and methyl propiolate 7a (600 μL, 6.05 mmol) in DCM (10 mL) to produce pure product in 219 mg (72% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 106–108 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 7.60 (s, 1H), 3.94 (s, 3H), 3.71 (s, 1H), 1.26 (s, 9H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 191.0 (C), 164.7 (C), 162.3 (C), 161.7 (C), 156.7 (C), 108.4 (CH), 53.7 (CH), 35.0 (CH3), 33.8 (C), 25.9 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C12H15N2O4+ 251.1026, found 251.1030.
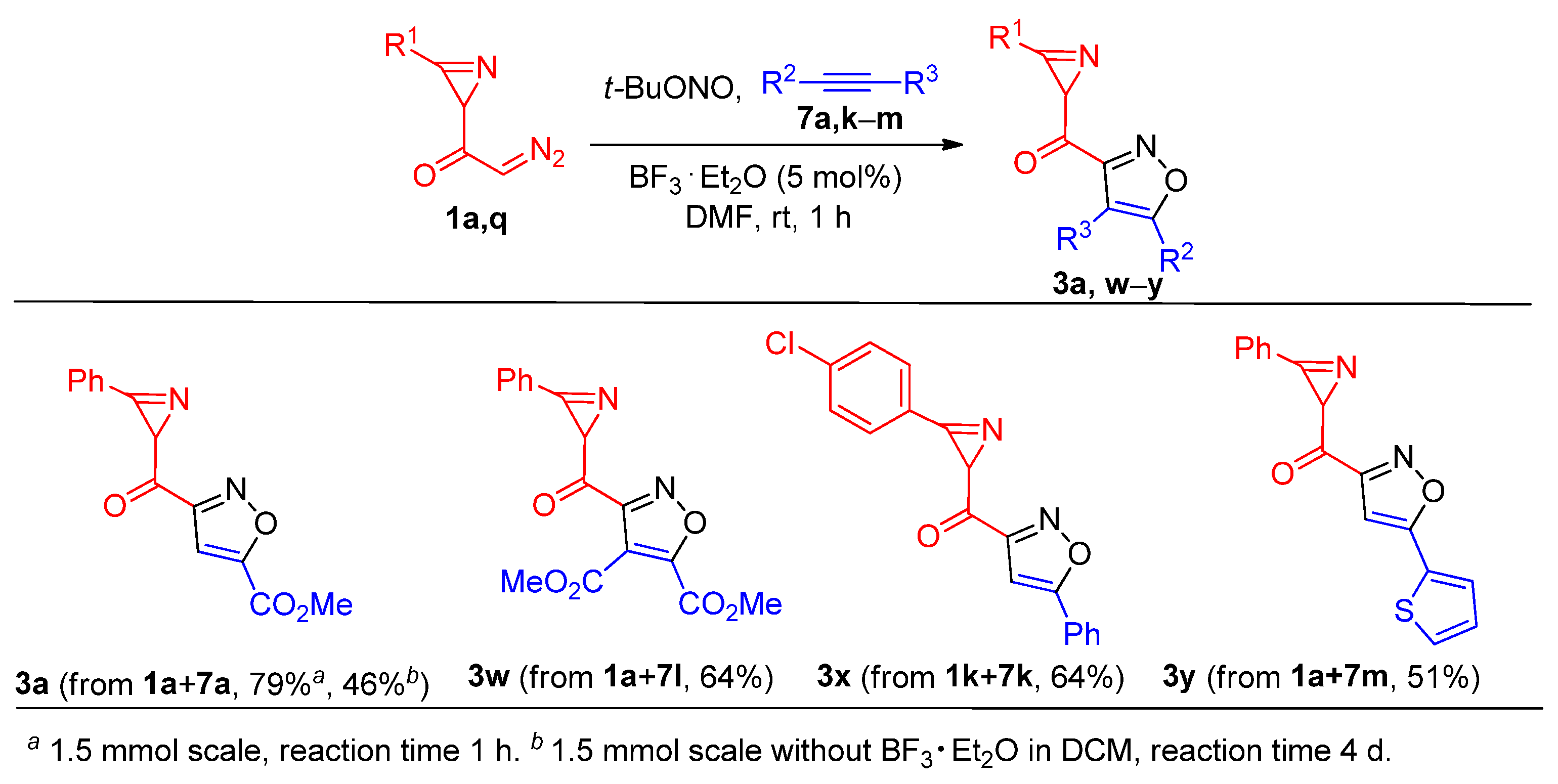
Dimethyl 3-(3-phenyl-2H-azirine-2-carbonyl)isoxazole-4,5-dicarboxylate (3w). Compound 3x was prepared following the general procedure GP-B from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (56 mg, 0.3 mmol), tert-butyl nitrite (193 μL, 1.5 mmol), dimethyl but-2-ynedioate 7l (185 μL, 1.5 mmol), and boron trifluoride etherate (2 μL, 0.02 mmol) in DMF (5 mL) to produce pure product in 64 mg (64% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-brown oil; 1H NMR (CDCl3, 400 MHz): δ 7.90–7.88 (m, 2H), 7.68–7.64 (m, 1H), 7.60–7.56 (m, 2H), 4.01 (s, 3H), 3.93 (m, 4H); 13C{1H} NMR (CDCl3, 100 MHz): δ 189.8 (C), 160.3 (C), 159.0 (C), 158.6 (C), 156.0 (C), 155.6 (C), 134.3 (CH), 130.9 (CH), 129.5 (CH), 121.4 (C), 116.8 (C), 53.6 (CH3), 53.5 (CH3), 34.9 (CH); IR (KBr, cm–1): 3048, 2958, 1757, 1734, 1686, 1452, 1341, 1305, 1272, 1179, 1126, 1099, 983, 841, 823, 768, 692, 568; HRMS (ESI) m/z [M + Na]+ calcd for C16H12N2O6Na+ 351.0588, found 351.0587.
(3-(4-Chlorophenyl)-2H-azirin-2-yl)(5-phenylisoxazol-3-yl)methanone (3x). Compound 3x was prepared following the general procedure GP-B from 1-(3-(4-chlorophenyl)-2H-azirin-2-yl)-2-diazoethan-1-one 1k (175 mg, 0.8 mmol), tert-butyl nitrite (507 μL, 3.98 mmol), phenylacetylene 7k (437 μL, 3.98 mmol), and boron trifluoride etherate (3 μL, 0.04 mmol) in DMF (5 mL) to produce pure product in 164 mg (64% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 140–142 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.87–7.82 (m, 4H), 7.58–7.52 (m, 2H), 7.51–7.50 (m, 3H), 6.94 (s, 1H), 4.08 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.3 (C), 171.6 (C), 162.4 (C), 155.8 (C), 140.6 (C), 131.9 (CH), 130.9 (CH), 129.9 (CH), 129.2 (CH), 126.6 (C), 126.0 (CH), 120.4 (C), 98.0 (CH), 34.6 (CH); HRMS (ESI) m/z [M + Na]+ calcd for C18H11ClN2O2Na+ 345.0401, found 345.0400.
(3-Phenyl-2H-azirin-2-yl)(5-(thiophen-2-yl)isoxazol-3-yl)methanone (3y). Compound 3y was prepared following the general procedure GP-B from 2-diazo-1-(3-phenyl-2H-azirin-2-yl)ethan-1-one 1a (100 mg, 0.54 mmol), tert-butyl nitrite (350 μL, 2.70 mmol), 2-ethynylthiophene 7m (270 μL, 2.70 mmol), and boron trifluoride etherate (3 μL, 0.03 mmol) in DMF (5 mL) to produce pure product in 81 mg (51% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 2:1, (v/v)) as a yellow solid: mp 85–87 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.92–7.90 (m, 2H), 7.67–7.63 (m, 1H), 7.59–7.56 (m, 3H), 7.52–7.51 (dd, 1H, J = 4.9, 1.2 Hz), 7.18–7.16 (dd, 1H, J = 5.0, 3.7 Hz), 6.80 (s, 1H), 4.06 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 191.3 (C), 166.5 (C), 162.4 (C), 156.3 (C), 134.1 (CH), 130.8 (CH), 129.4 (CH), 129.0 (CH), 128.3 (C), 128.3 (CH), 127.9 (CH), 121.8 (C), 97.6 (CH), 34.5 (CH); HRMS (ESI) m/z [M + Na]+ calcd for C16H10N2O2SNa+ 317.0355, found 317.0357.
Methyl 3’-phenyl-[3,5’-biisoxazole]-5-carboxylate (4a). Compound 4a was prepared following the general procedure GP-C from methyl 3-(3-phenyl-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3a (42 mg, 0.16 mmol) in acetonitrile (7 mL) to produce pure product in 21 mg (50% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 121–122 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.88–7.86 (m, 2H), 7.51–7.49 (m, 3H), 7.40 (s, 1H), 7.22 (s, 1H), 4.03 (m, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 163.3 (C), 161.4 (C), 159.6 (C), 156.5 (C), 153.3 (C), 130.6 (CH), 129.1 (CH), 128.1 (CH), 127.0 (CH), 107.6 (CH), 102.0 (CH), 53.2 (CH3); IR (KBr, cm–1): 3122, 2949, 1735, 1581, 1482, 1455, 1433, 1394, 1302, 1215, 1141, 997, 930, 834, 771, 694; HRMS (ESI) m/z [M + H]+ calcd for C14H11N2O4+ 271.0713, found 271.0709.
2-((3’-Phenyl-[3,5’-biisoxazol]-5-yl)methyl)isoindoline-1,3-dione (4b). Compound 4b was prepared following the general procedure GP-C from 2-((3-(3-phenyl-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl)isoindoline-1,3-dione 3b (54 mg, 0.15 mmol) in acetonitrile (9 mL) to produce pure product in 9 mg (17% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1 + 10% CHCl3, (v/v)).
Compound 4b was also prepared following the general procedure GP-D from 2-((3-(3-phenyl-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl)isoindoline-1,3-dione 3b (52 mg, 0.15 mmol) and hydroquinone (18 mg, 0.17 mmol) in acetonitrile (9 mL) to produce pure product in 25 mg (48% yield), after column chromatography on silica (light petroleum/ethyl acetate, 4:1 + 10% CHCl3, (v/v)) as a colorless solid: mp 236–238 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 7.96–7.88 (m, 6H), 7.78 (s, 1H), 7.55–7.54 (m, 3H), 7.17 (s, 1H), 5.08 (s, 2H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 174.3 (C), 172.2 (C), 167.9 (C), 165.3 (C), 157.5 (C), 140.0 (CH), 136.8 (C), 135.8 (CH), 134.4 (CH), 133.1 (C), 132.0 (CH), 128.6 (CH), 108.6 (CH), 107.0 (C), 38.4 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H14N3O4+ 372.0979, found 372.0982.
(3’-Phenyl-[3,5’-biisoxazol]-5-yl)methanol (4c). Compound 4c was prepared following the general procedure GP-C from (5-(hydroxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3c (50 mg, 0.21 mmol) in acetonitrile (8 mL) to produce pure product in 9 mg (18% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)).
Compound 4c was also prepared following the general procedure GP-D from (5-(hydroxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3c (50 mg, 0.21 mmol) and hydroquinone (25 mg, 0.23 mmol) in acetonitrile (8 mL) to produce pure product in 23 mg (46% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 140–141 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6 400 MHz): δ 7.97–7.94 (m, 2H), 7.80 (s, 1H), 7.57–7.55 (m, 3H), 6.97 (s, 1H), 5.85–5.82 (t, 1H, J = 6.1 Hz), 4.69–4.68 (d, 2H, J = 6.1 Hz); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 175.5 (C), 163.1 (C), 160.9 (C), 152.3 (C), 131.2 (CH), 129.7 (CH), 128.2 (C), 127.3 (CH), 103.6 (CH), 100.9 (CH), 55.2 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C13H11N2O3+ 243.0764, found 243.0761.
5-(Methoxymethyl)-3’-phenyl-3,5’-biisoxazole (4d). Compound 4d was prepared following the general procedure GP-C from (5-(methoxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3d (53 mg, 0.21 mmol) in acetonitrile (8 mL) to produce pure product in 33 mg (62% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 3:1, (v/v)) as a colorless solid: mp 125–126 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.88–7.85 (m, 2H), 7.50–7.49 (m, 3H), 7.15 (s, 1H), 6.76 (s, 1H), 4.64 (s, 2H), 3.49 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 170.9 (C), 163.1 (C), 160.7 (C), 152.7 (C), 130.4 (CH), 129.1 (CH), 128.3 (C), 126.9 (CH), 101.4 (CH), 101.3 (CH), 65.2 (CH2), 59.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H13N2O4+ 285.0870, found 285.0874.
5-(Phenoxymethyl)-3’-phenyl-3,5’-biisoxazole (4e). Compound 4e was prepared following the general procedure GP-C from (5-(phenoxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3e (50 mg, 0.16 mmol) in acetonitrile (8 mL) to produce pure product in 20 mg (40% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1 + 10% CHCl3, (v/v)) as a colorless solid: mp 147–148 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 7.88–7.85 (m, 2H), 7.50–7.49 (m, 3H), 7.36–7.32 (m, 2H), 7.16 (s, 1H), 7.06–7.02 (m, 1H), 7.01–6.99 (m, 2H), 6.84 (s, 1H), 5.26 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz): δ 169.7 (C), 163.1 (C), 160.6 (C), 157.6 (C), 152.8 (C), 130.5 (CH), 129.8 (CH), 129.1 (CH), 128.3 (C), 126.9 (CH), 122.2 (CH), 114.8 (CH), 101.8 (CH), 101.5 (CH), 61.2 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O3+ 319.1077, found 319.1079.
(3’-Phenyl-[3,5’-biisoxazol]-5-yl)methyl benzenesulfonate (4f). Compound 4f was prepared following the general procedure GP-C from (3-(3-phenyl-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl benzenesulfonate 3f (50 mg, 0.13 mmol) in acetonitrile (8 mL) to produce pure product in 26 mg (52% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 150–151 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 7.96–7.94 (m, 2H), 7.87–7.84 (m, 2H), 7.71–7.67 (m, 1H), 7.60–7.57 (m, 2H), 7.50–7.49 (m, 3H), 7.13 (s, 1H), 6.75 (s, 1H), 5.27 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz): δ 165.9 (C), 163.2 (C), 160.0 (C), 152.8 (C), 135.5 (C), 134.4 (CH), 130.5 (CH), 129.5 (CH), 129.1 (CH), 128.1 (C), 128.0 (CH), 126.9 (CH), 103.5 (CH), 101.6 (CH), 60.7 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O5S+ 383.0696, found 383.0697.
5-(Chloromethyl)-3’-phenyl-3,5’-biisoxazole (4g). Compound 4g was prepared following the general procedure GP-C from (5-(chloromethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3g (50 mg, 0.19 mmol) in acetonitrile (8 mL) to produce pure product in 31 mg (62% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 175–176 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 7.88–7.86 (m, 2H), 7.51–7.49 (m, 3H), 7.17 (s, 1H), 6.83 (s, 1H), 4.71 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz): δ 169.1 (C), 163.2 (C), 160.3 (C), 153.0 (C), 130.5 (CH), 129.1 (CH), 128.2 (C), 126.9 (CH), 102.2 (CH), 101.5 (CH), 34.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C13H10ClN2O2+ 261.0425, found 261.0421.
5-(Bromomethyl)-3’-phenyl-3,5’-biisoxazole (4h). Compound 4h was prepared following the general procedure GP-C from (5-(bromomethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3h (50 mg, 0.16 mmol) in acetonitrile (8 mL) to produce pure product in 8 mg (15% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)).
Compound 4h was also prepared following the general procedure GP-D from (5-(bromomethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3h (52 mg, 0.17 mmol) and hydroquinone (21 mg, 0.19 mmol) in acetonitrile (8 mL) to produce pure product in 28 mg (52% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 190–192 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 7.88–7.85 (m, 2H), 7.51–7.49 (m, 3H), 7.17 (s, 1H), 6.82 (s, 1H), 4.55 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz): δ 169.1 (C), 163.2 (C), 160.3 (C), 153.0 (C), 130.5 (CH), 129.1 (CH), 128.2 (C), 127.0 (CH), 102.2 (CH), 101.5 (CH), 17.9 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C13H10N2O2+ 304.9920, found 304.9922.
3’-Phenyl-5-(trimethylsilyl)-3,5’-biisoxazole (4i). Compound 4i was prepared following the general procedure GP-C from (3-phenyl-2H-azirin-2-yl)(5-(trimethylsilyl)isoxazol-3-yl)methanone 3i (50 mg, 0.18 mmol) in acetonitrile (8 mL) to produce pure product in 33 mg (67% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 77–79 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.88–7.86 (m, 2H), 7.50–7.48 (m, 3H), 7.15 (s, 1H), 6.92 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 180.2 (C), 163.1 (C), 161.3 (C), 151.1 (C), 130.4 (CH), 129.0 (CH), 128.5 (C), 126.9 (CH), 110.8 (CH), 101.2 (CH), –1.96 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H17N2O3Si+ 285.1054, found 285.1049.
Methyl 3’-(p-tolyl)-[3,5’-biisoxazole]-5-carboxylate (4j). Compound 4j was prepared following the general procedure GP-C from methyl 3-(3-(p-tolyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3j (45 mg, 0.16 mmol) in acetonitrile (8 mL) to produce pure product in 18 mg (40% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 177–179 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 7.76 (d, 2H, J = 8.1 Hz), 7.39 (s, 1H), 7.31 (d, 2H, J = 8.1 Hz), 7.20 (s, 1H), 4.03 (s, 3H), 2.42 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 163.2 (C), 161.4 (C), 159.4 (C), 156.6 (C), 153.3 (C), 140.9 (C), 129.8 (CH), 126.8 (CH), 125.2 (C), 107.6 (CH), 102.0 (CH), 53.2 (CH3), 21.5 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H13N2O4+ 285.0870, found 285.0874.
Methyl 3’-(4-(tert-butyl)phenyl)-[3,5’-biisoxazole]-5-carboxylate (4k). Compound 4k was prepared following the general procedure GP-C from methyl 3-(3-(4-(tert-butyl)phenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3k (34 mg, 0.1 mmol) in acetonitrile (6 mL) to produce pure product in 14 mg (41% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 137–138 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.80 (d, 2H, J = 8.4 Hz), 7.52 (d, 2H, J = 8.4 Hz), 7.40 (s, 1H), 7.21 (s, 1H), 4.03 (s, 3H), 1.37 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz): δ 163.2 (C), 161.4 (C), 159.4 (C), 156.5 (C), 154.0 (C), 153.3 (C), 126.7 (CH), 126.1 (CH), 125.2 (C), 107.6 (CH), 102.0 (CH), 53.2 (CH), 34.9 (CH3), 31.2 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C18H19N2O4+ 327.1345, found 327.1338.
Methyl 3’-(4-methoxyphenyl)-[3,5’-biisoxazole]-5-carboxylate (4l). Compound 4l was prepared following the general procedure GP-C from methyl 3-(3-(4-methoxyphenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3l (42 mg, 0.14 mmol) in acetonitrile (8 mL) to produce pure product in 27 mg (67% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1 + 10% CHCl3, (v/v)) as a colorless solid: mp 120–122 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 7.89–7.87 (m, 3H), 7.84 (s, 1H), 7.12 (d, 2H, J = 8.8 Hz), 3.96 (s, 3H), 3.84 (s, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 162.8 (C), 161.6 (C), 161.5 (C), 159.3 (C), 156.7 (C), 153.4 (C), 128.8 (CH), 120.4 (C), 115.2 (CH), 108.8 (CH), 104.6 (CH), 55.9 (CH3), 53.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H13N2O5+ 301.0819, found 301.0812.
3’-(4-Methoxyphenyl)-5-(phenoxymethyl)-3,5’-biisoxazole (4m). Compound 4m was prepared following the general procedure GP-C from ((3-(4-methoxyphenyl)-2H-azirin-2-yl)(5-(phenoxymethyl)isoxazol-3-yl)methanone 3m (43 mg, 0.12 mmol) in acetonitrile (8 mL) to produce pure product in 22 mg (50% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1 + 10% CHCl3, (v/v)) as a light-brown solid: mp 179–181 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 7.89 (d, 2H, J = 8.9 Hz), 7.77 (s, 1H), 7.36 (m, 2H), 7.20 (s, 1H), 7.12–7.08 (m, 4H), 7.03–6.99 (m, 1H), 5.42 (s, 2H), 3.83 (s, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 170.3 (C), 162.7 (C), 161.6 (C), 160.2 (C), 157.9 (C), 152.6 (C), 130.2 (CH), 128.8 (CH), 122.1 (CH), 120.5 (C), 115.3 (CH), 115.1 (CH), 103.7 (CH), 103.2 (CH), 60.6 (CH2), 55.8 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C20H17N2O4+ 349.1183, found 349.1187.
5-(Bromomethyl)-3’-(3-methoxyphenyl)-3,5’-biisoxazole (4n). Compound 4n was prepared following the general procedure GP-C from (5-(bromomethyl)isoxazol-3-yl)(3-(3-methoxyphenyl)-2H-azirin-2-yl)methanone 3n (42 mg, 0.13 mmol) in acetonitrile (6 mL) to produce pure product in 8 mg (18% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)).
Compound 4n was also prepared following the general procedure GP-D from (5-(bromomethyl)isoxazol-3-yl)(3-(3-methoxyphenyl)-2H-azirin-2-yl)methanone 3n (50 mg, 0.15 mmol) and hydroquinone (18 mg, 0.17 mmol) in acetonitrile (6 mL) to produce pure product in 26 mg (51% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 161–163 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.43–7.40 (m, 3H), 7.15 (s, 1H), 7.05–7.02 (m, 1H), 6.81 (s, 1H), 4.54 (s, 2H), 3.88 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 161.1 (C), 163.1 (C), 160.3 (C), 160.1 (C), 153.0 (C), 130.2 (CH), 129.4 (C), 119.4 (CH), 116.6 (CH), 111.9 (CH), 102.2 (CH), 101.6 (CH), 55.4 (CH3), 17.8 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C14H12BrN2O3+ 335.0026, found 335.0019.
3’-(3,4-Dimethoxyphenyl)-5-(trimethylsilyl)-3,5’-biisoxazole (4o). Compound 4o was prepared following the general procedure GP-C from (3-(3,4-dimethoxyphenyl)-2H-azirin-2-yl)(5-(trimethylsilyl)isoxazol-3-yl)methanone 3o (47 mg, 0.14 mmol) in acetonitrile (8 mL) to produce pure product in 26 mg (55% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 112–114 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.47 (d, 1H, J = 2.0 Hz), 7.37 (dd, 1H, J = 8.3, 2.0 Hz), 7.10 (s, 1H), 6.96 (d, 1H, J = 8.3 Hz), 6.91 (s, 1H), 3.97 (s, 3H), 3.95 (s, 3H), 0.41 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz): δ 180.1 (C), 162.8 (C), 161.1 (C), 151.1 (C), 150.9 (C), 149.4 (C), 121.2 (C), 120.1 (CH), 111.2 (CH), 110.8 (CH), 109.4 (CH), 101.1 (CH), 56.1 (CH3), 56.0 (CH3), –1.97 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C21H17N2O4Si+ 345.1271, found 345.1274.
Methyl 3’-(4-fluorophenyl)-[3,5’-biisoxazole]-5-carboxylate (4p). Compound 4p was prepared following the general procedure GP-C from methyl 3-(3-(4-fluorophenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3p (42 mg, 0.15 mmol) in acetonitrile (7 mL) to produce pure product in 26 mg (62% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 202–204°C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 8.02–7.98 (m, 2H), 7.90 (s, 1H), 7.89 (s, 1H), 7.44–7.40 (m, 2H), 3.96 (s, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 162.3 (C), 161.5 (C), 159.8 (C), 156.7 (C), 146.9 (d, C, J = 242.8 Hz), 129.7 (d, CH, J = 8.7 Hz), 124.6 (d, C, J = 3.3 Hz), 116.9 (d, CH, J = 22.0 Hz), 108.7 (CH), 104.7 (CH), 51.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C14H10FN2O4+ 289.0619, found 289.0618.
(3’-(4-Chlorophenyl)-[3,5’-biisoxazol]-5-yl)methyl benzenesulfonate (4q). Compound 4q was prepared following the general procedure GP-C from (3-(3-(4-chlorophenyl)-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl benzenesulfonate 3q (50 mg, 0.12 mmol) in acetonitrile (7 mL) to produce pure product in 33 mg (66% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-yellow solid: mp 158–159 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.96–7.94 (m, 2H), 7.80–7.78 (m, 2H), 7.71–7.67 (m, 1H), 7.61–7.57 (m, 2H), 7.49–7.46 (m, 2H), 7.10 (s, 1H), 6.76 (s, 1H), 5.27 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz): δ 166.0 (C), 162.2 (C), 160.3 (C), 152.7 (C), 136.7 (C), 135.5 (C), 134.5 (CH), 129.5 (CH), 129.4 (CH), 128.2 (CH), 128.0 (CH), 126.6 (C), 103.4 (CH), 101.5 (CH), 60.6 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C19H14ClN2O5S+ 417.0306, found 417.0294.
3’-(2-Bromophenyl)-5-(chloromethyl)-3,5’-biisoxazole (4r). Compound 4r was prepared following the general procedure GP-C from 3-(3-(2-bromophenyl)-2H-azirin-2-yl)-5-(chloromethyl)isoxazole 3r (50 mg, 0.15 mmol) in acetonitrile (7 mL) to produce pure product in 26 mg (52% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-brown solid: mp 110–111 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.74–7.68 (m, 2H), 7.46–7.42 (m, 1H), 7.37–7.33 (m, 1H), 7.30 (s, 1H), 6.83 (s, 1H), 4.71 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz): δ 169.1 (C), 163.1 (C), 159.5 (C), 152.9 (C), 133.8 (CH), 131.4 (CH), 131.4 (CH), 129.6 (C), 127.8 (CH), 122.3 (C), 104.9 (CH), 102.2 (CH), 34.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C13H9BrClN2O2+ 338.9530, found 338.9526.
(3’-(Thiophen-2-yl)-[3,5’-biisoxazol]-5-yl)methanol (4s). Compound 4s was prepared following the general procedure GP-C from (5-(hydroxymethyl)isoxazol-3-yl)(3-(thiophen-2-yl)-2H-azirin-2-yl)methanone 3s (38 mg, 0.15 mmol) in acetonitrile (8 mL) to produce pure product in 27 mg (71% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 1:1, (v/v)) as a colorless solid: mp 139–141 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.53 (dd, 1H, J = 3.6, 1.2 Hz), 7.46 (dd, 1H, J = 5.1, 1.2 Hz), 7.14 (dd, 1H, J = 5.1, 3.6 Hz), 7.06 (s, 1H), 6.73 (s, 1H), 4.84 (s, 2H), 3.48 (br. s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 175.5 (C), 160.8 (C), 158.6 (C), 152.2 (C), 130.2 (CH), 129.8 (CH), 129.4 (C), 128.7 (CH), 103.4 (CH), 100.9 (CH), 55.2 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C11H9N2O3S+ 249.0328, found 249.0321.
5-(Chloromethyl)-3’-(thiophen-2-yl)-3,5’-biisoxazole (4t). Compound 4t was prepared following the general procedure GP-C from (5-(chloromethyl)isoxazol-3-yl)(3-(thiophen-2-yl)-2H-azirin-2-yl)methanone 3t (38 mg, 0.14 mmol) in acetonitrile (8 mL) to produce pure product in 18 mg (47% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 158–160 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.54 (dd, 1H, J = 3.7, 1.2 Hz), 7.47 (dd, 1H, J = 5.1, 1.2 Hz), 7.16 (dd, 1H, J = 5.1, 3.7 Hz), 7.09 (s, 1H), 6.82 (s, 1H), 4.70 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz): δ 169.2 (C), 160.2 (C), 158.4 (C), 152.8 (C), 129.8 (C), 128.3 (CH), 128.1 (CH), 127.8 (CH), 102.2 (CH), 101.5 (CH), 34.0 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C11H8ClN2O2S+ 266.9900, found 266.9988.
Methyl 3’-(tert-butyl)-[3,5’-biisoxazole]-5-carboxylate (4u). Compound 4u was prepared following the general procedure GP-C from methyl 3-(3-(tert-butyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3u (50 mg, 0.2 mmol) in acetonitrile (8 mL) to produce pure product in 22 mg (44% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 95–97 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.32 (s, 1H), 6.82 (s, 1H), 4.02 (s, 3H), 1.39 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz): δ 172.9 (C), 161.2 (C), 158.5 (C), 156.6 (C), 153.5 (C), 107.5 (CH), 102.1 (CH), 53.1 (CH3), 32.2 (C), 29.5 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C12H15N2O4+ 251.1026, found 251.1022.
Dimethyl 3’-phenyl-[3,5’-biisoxazole]-4,5-dicarboxylate (4w). Compound 4w was prepared following the general procedure GP-C from dimethyl 3-(3-phenyl-2H-azirine-2-carbonyl)isoxazole-4,5-dicarboxylate 3w (58 mg, 0.18 mmol) in acetonitrile (10 mL) to produce pure product in 24 mg (41% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 150–152 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.88–7.86 (m, 2H), 7.51–7.49 (m, 3H), 7.32 (s, 1H), 4.05 (s, 3H), 4.01 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 163.0 (C), 160.3 (C), 160.2 (C), 158.0 (C), 155.9 (C), 151.0 (C), 130.6 (CH), 129.1 (CH), 128.0 (C), 127.0 (CH), 115.1 (C), 104.4 (CH), 53.7 (CH3), 53.5 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C16H13N2O6+ 329.0768, found 329.0775.
3’-(4-Chlorophenyl)-5-phenyl-3,5’-biisoxazole (4x). Compound 4x was prepared following the general procedure GP-C from (3-(4-chlorophenyl)-2H-azirin-2-yl)(5-phenylisoxazol-3-yl)methanone 3x (51 mg, 0.16 mmol) in acetonitrile (10 mL) to produce pure product in 30 mg (59% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 203–205 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 8.03–8.01 (d, 2H, J = 8.6 Hz), 8.00–7.98 (m, 2H), 7.85 (s, 1H), 7.67 (s, 1H), 7.66–7.64 (d, 2H, J = 8.7 Hz), 7.62–7.60 (m, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 174.4 (C), 171.3 (C), 162.3 (C), 161.0 (C), 153.2 (C), 135.9 (C), 131.7 (CH), 129.9 (CH), 129.9 (CH), 129.1 (CH), 127.1 (C), 126.5 (C), 126.4 (CH), 103.5 (CH), 99.4 (CH); HRMS (ESI) m/z [M + H]+ calcd for C18H12ClN2O2+ 323.0582, found 323.0583.
3’-Phenyl-5-(thiophen-2-yl)-3,5’-biisoxazole (4y). Compound 4y was prepared following the general procedure GP-C from (3-phenyl-2H-azirin-2-yl)(5-(thiophen-2-yl)isoxazol-3-yl)methanone 3y (38 mg, 0.13 mmol) in acetonitrile (10 mL) to produce pure product in 20 mg (53% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a colorless solid: mp 194–196 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 7.90–7.87 (m, 2H), 7.63–7.62 (m, 1H), 7.54–7.49 (m, 4H), 7.19–7.17 (m, 2H), 6.87 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 174.4 (C), 166.5 (C), 163.2 (C), 160.7 (C), 153.2 (C), 130.5 (CH), 129.1 (CH), 128.9 (CH), 128.3 (C), 128.3 (CH), 127.9 (CH), 127.0 (CH), 101.4 (CH), 97.4 (CH); HRMS (ESI) m/z [M + H]+ calcd for C16H11N2O2S+ 295.0536, found 295.0542.
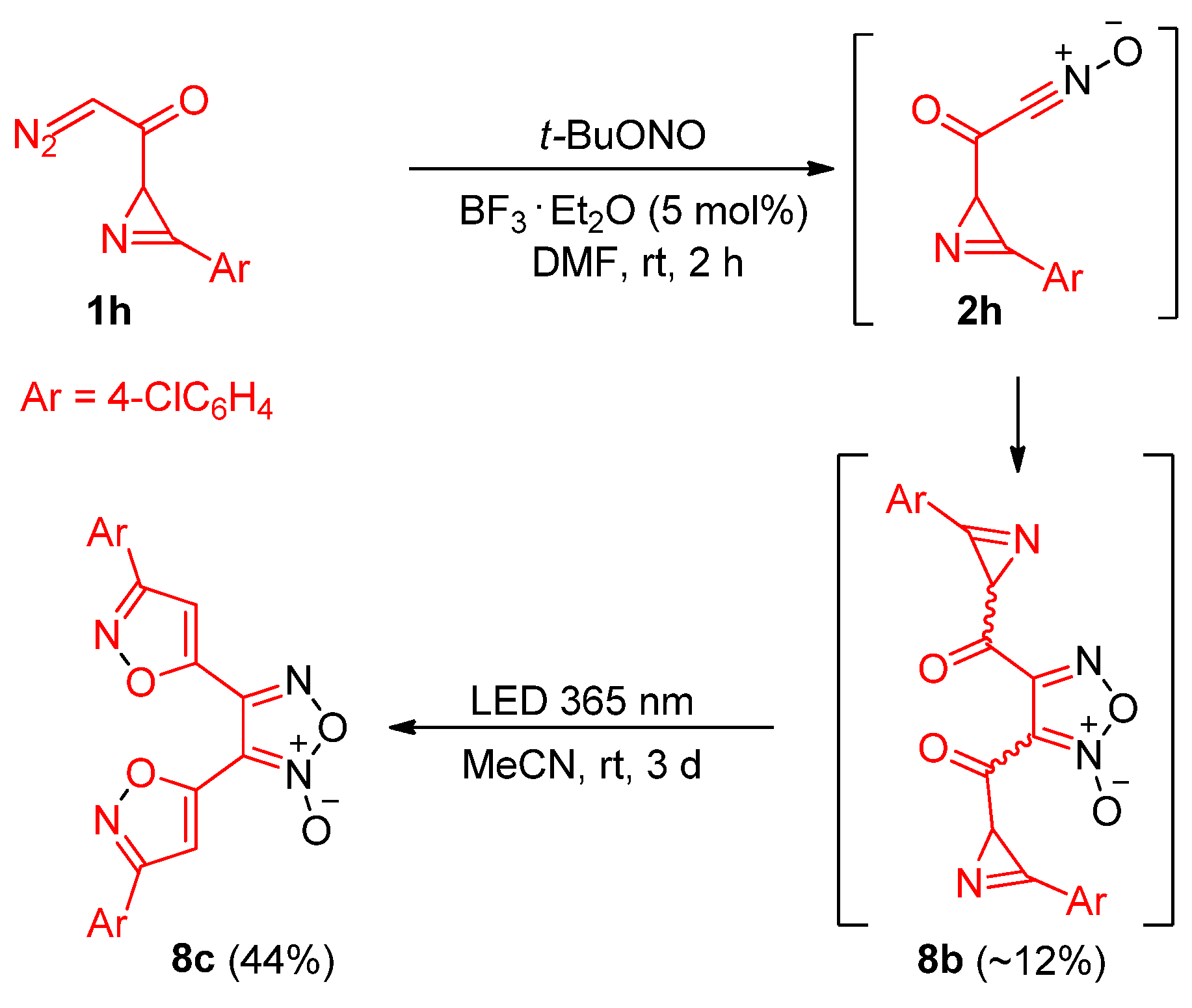
3,4-bis(3-(4-Chlorophenyl)isoxazol-5-yl)-1,2,5-oxadiazole 2-oxide (8c). A mixture of 1-(3-(4-chlorophenyl)-2H-azirin-2-yl)-2-diazoethan-1-one 1h (300 mg, 1.37 mmol), tert-butyl nitrite (533 μL, 4.11 mmol), and boron trifluoride etherate (9 μL, 70 μmol) in dimethylformamide (4 mL) was stirred at rt for 1 h (monitored by TLC). The reaction mixture was diluted with water and extracted with ethyl acetate. The organic phase was washed with water, dried over Na2SO4, and the solvent was evaporated. The residue was dissolved in acetonitrile, and the solution was flushed with argon and irradiated using LED 365 at rt for 2 days (monitored by TLC). The solvent was evaporated, and the residue was purified by column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) or washed with diethyl ether or acetonitrile to produce pure compound 8c in 24 mg (44% yield) as a yellow solid: mp 224–225 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 8.08–8.06 (m, 4H), 8.03 (s, 1H), 7.95 (s, 1H), 7.68–7.63 (m, 4H); 13C{1H} NMR (CDCl3, 100 MHz): δ 162.4 (C), 162.1 (C), 157.3 (C), 155.2 (C), 145.3 (C), 136.2 (C), 130.0 (CH), 129.9 (C), 129.9 (CH), 129.3 (CH), 129.3 (CH), 126.6 (C), 126.5 (C), 107.8 (C), 107.0 (CH), 105.2 (CH); HRMS (ESI) m/z [M + H]+ calcd for C20H11Cl2N4O4+ 441.0152, found 441.0150.
Methyl 3-(4-acetyl-5-methyl-3-phenyl-1H-pyrrole-2-carbonyl)isoxazole-5-carboxylate (5a). Compound 5a was prepared following the general procedure GP-E from methyl 3-(3-phenyl-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3a (50 mg, 0.19 mmol), acetylacetone (60 μL, 0.57 mmol), and Ni(acac)2 (10 mg, 0.04 mmol) in acetonitrile (5 mL) at 40 °C to produce pure product in 37 mg (57% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-yellow solid: mp 125–127 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 10.67 (s, 1H), 7.44–7.40 (m, 3H), 7.33–7.31 (m, 2H), 7.19 (s, 1H), 3.99 (s, 3H), 2.66 (s, 3H), 3.09 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 196.6 (C), 170.3 (C), 163.3 (C), 160.5 (C), 156.4 (C), 141.5 (C), 137.4 (C), 134.7 (C), 129.5 (CH), 128.3 (CH), 128.1 (CH), 125.0 (C), 124.8 (C), 109.5 (CH), 53.2 (CH3), 30.9 (CH3), 15.0 (CH3); IR (KBr, cm–1): 3229, 3140, 2924, 2854, 1739, 1643, 1546, 1514, 1424, 1326, 1286, 1259, 1213, 1072, 994, 890, 755, 701, 558; HRMS (ESI) m/z [M + H]+ calcd for C19H16N2O5Na+ 375.0951, found 375.0948.
1-(5-(5-(Chloromethyl)isoxazole-3-carbonyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethan-1-one (5b). Compound 5b was prepared following the general procedure GP-E from (5-(chloromethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3g (55 mg, 0.21 mmol), acetylacetone (65 μL, 0.63 mmol) and Ni(acac)2 (11 mg, 0.04 mmol) in acetonitrile (5 mL) at 40 °C to produce pure product in 42 mg (55% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a light-yellow solid: mp 153–155 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 10.78 (s, 1H), 7.46–7.40 (m, 3H), 7.34–7.31 (m, 2H), 4.63 (s, 2H), 2.65 (s, 3H), 1.83 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 196.7 (C), 170.8 (C), 168.3 (C), 163.3 (C), 141.1 (C), 137.0 (C), 134.9 (C), 129.5 (CH), 128.2 (CH), 128.0 (CH), 125.0 (C), 124.7 (C), 104.2 (CH), 33.8 (CH2), 30.9 (CH3), 15.0 (CH3); HRMS (ESI) m/z [M + Na]+ calcd for C18H15ClN2O3Na+ 365.0663, found 365.0662.
Methyl 3-(4-acetyl-3-(4-methoxyphenyl)-5-methyl-1H-pyrrole-2-carbonyl)isoxazole-5-carboxylate (5c). Compound 5c was prepared following the general procedure GP-E from methyl 3-(3-(4-methoxyphenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3l (60 mg, 0.2 mmol), acetylacetone (60 μL, 0.6 mmol), and Ni(acac)2 (10 mg, 0.04 mmol) in acetonitrile (5 mL) at 40 °C to produce pure product in 53 mg (69% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 159–160 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3 400 MHz): δ 10.63 (s, 1H), 7.23 (d, 2H, J = 8.6 Hz), 7.15 (s, 1H), 6.95 (d, 2H, J = 8.6 Hz), 3.99 (s, 3H), 3.85 (s, 3H), 2.64 (s, 3H), 1.87 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 196.8 (C), 170.1 (C), 163.4 (C), 160.4 (C), 159.5 (C), 158.5 (C), 141.3 (C), 137.3 (C), 130.8 (CH), 126.4 (C), 124.98 (C), 124.96 (C), 113.7 (CH), 109.5 (CH), 55.2 (CH3), 53.2 (CH3), 30.9 (CH3), 15.0 (CH3); HRMS (ESI) m/z [M + Na]+ calcd for C20H19N2O6+ 383.1238, found 383.1237.
(3-(4-Acetyl-3-(4-chlorophenyl)-5-methyl-1H-pyrrole-2-carbonyl)isoxazol-5-yl)methyl benzenesulfonate (5d). Compound 5d was prepared following the general procedure GP-E from (5(3-(3-(4-chlorophenyl)-2H-azirine-2-carbonyl)isoxazol-5-yl)methyl benzenesulfonate 3q (50 mg, 0.12 mmol), acetylacetone (37 μL, 0.36 mmol), and Ni(acac)2 (5 mg, 0.02 mmol) in acetonitrile (5 mL) at 40 °C to produce pure product in 14 mg (24% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow oil; 1H NMR (CDCl3 400 MHz): δ 10.75 (s, 1H), 7.94–7.92 (m, 2H), 7.71–7.67 (m, 1H), 7.60–7.56 (m, 2H), 7.42–7.40 (m, 2H), 7.26 (m, 2H), 6.66 (s, 1H), 5.21 (s, 2H), 2.64 (s, 3H), 1.86 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 196.2 (C), 170.2 (C), 165.3 (C), 163.1 (C), 141.1 (C), l35.6 (C), 135.3 (C), 134.5 (CH), 134.1 (C), 133.3 (C), 130.9 (CH), 129.5 (CH), 128.6 (CH), 128.0 (CH), 124.68 (C), 125.67 (C), 105.5 (CH), 60.3 (CH2), 31.0 (CH3), 15.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C24H20ClN2O6S+ 499.0731, found 499.0723.
Methyl 3-(3-phenyl-5-(thiophen-2-yl)-4-(thiophene-2-carbonyl)-1H-pyrrole-2-carbonyl)isoxazole-5-carboxylate (5e). Compound 5e was prepared following the general procedure GP-E from methyl 3-(3-phenyl-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3a (70 mg, 0.26 mmol), 1,3-di(thiophen-2-yl)propane-1,3-dione (69 mg, 0.29 mmol), and Co(acac)3 (4 mg, 0.01 mmol) in acetonitrile (5 mL) at 70 °C to produce pure product in 102 mg (80% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 2:1, (v/v)) as a yellow solid: mp 214–215 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 13.00 (s, 1H), 7.87 (dd, 1H, J = 5.0, 1.1 Hz), 7.64 (dd, 1H, J = 3.6, 1.3 Hz), 7.61 (dd, 1H, J = 5.0, 1.1 Hz), 7.35 (dd, 1H, J = 3.8, 1.3 Hz), 7.15 (s, 1H), 7.12 (dd, 1H, J = 5.0, 3.8 Hz), 7.05 (s, 5H), 6.96 (dd, 1H, J = 5.0, 3.8 Hz), 3.85 (s, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 185.4 (C), 175.0 (C), 162.6 (C), 159.4 (C), 156.6 (C), 145.1 (C), 136.6 (CH), 136.2 (CH), 135.1 (C), 132.8 (C), 132.4 (C), 131.2 (C), 130.6 (CH), 129.2 (CH), 129.0 (CH), 128.9 (CH), 128.2 (CH), 127.8 (CH), 127.8 (CH), 124.0 (C), 110.1 (C), 53.5 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C25H17N2O5S2+ 489.0573, found 489.0581.
(5-(Chloromethyl)isoxazol-3-yl)(4-(4-methoxybenzoyl)-5-(4-methoxyphenyl)-3-phenyl-1H-pyrrol-2-yl)methanone (5f). Compound 5f was prepared following the general procedure GP-E from (5-(chloromethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3g (70 mg, 0.27 mmol), 1,3-bis(4-methoxyphenyl)propane-1,3-dione (85 mg, 0.30 mmol), and Co(acac)3 (4 mg, 0.01 mmol) in acetonitrile (5 mL) at 70 °C to produce pure product in 131 mg (92% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 2:1, (v/v)) as a light-yellow solid: mp 86–88 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 12.61 (s, 1H), 7.57 (d, 2H, J = 8.9 Hz) 7.46 (d, 2H, J = 8.9 Hz), 7.04 (s, 5H), 6.92 (d, 2H, J = 8.9 Hz), 6.80 (d, 2H, J = 8.9 Hz), 6.61 (s, 1H), 4.79 (s, 2H), 3.73 (s, 3H), 3.71 (s, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 192.8 (C), 175.7 (C), 168.1 (C), 163.6 (C), 162.4 (C), 160.1 (C), 138.2 (C), 135.2 (C), 133.4 (C), 132.2 (CH), 131.0 (C), 130.5 (CH), 130.0 (CH), 127.9 (C), 127.7 (CH), 127.6 (CH), 123.7 (C), 122.8 (C), 114.5 (CH), 114.2 (CH), 104.6 (CH), 55.9 (CH3), 55.7 (CH3), 34.3 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C30H24ClN2O5+ 527.1368, found 527.1378.
Methyl 3-(4-benzoyl-3-(4-fluorophenyl)-5-phenyl-1H-pyrrole-2-carbonyl)isoxazole-5-carboxylatebenzenesulfonate (5g). Compound 5g was prepared following the general procedure GP-E from 3-(3-(4-fluorophenyl)-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3p (70 mg, 0.24 mmol), 1,3-diphenylpropane-1,3-dione (58 mg, 0.26 mmol), and Co(acac)3 (4 mg, 0.01 mmol) in acetonitrile (5 mL) at 70 °C to produce pure product in 101 mg (85% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 2:1, (v/v)) as a yellow solid mp 185–187 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6 400 MHz): δ 12.97 (s, 1H), 7.58–7.56 (m, 2H), 7.49–7.47 (m, 2H), 7.42–7.38 (m, 1H), 7.34–7.32 (m, 3H), 7.26–7.23 (m, 2H), 7.21 (s, 1H), 7.10–7.07 (m, 2H), 6.89–6.84 (m, 2H), 3.87 (s, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 193.7 (C), 175.1 (C), 162.7 (C), 161.9 (d, C, J = 245.0 Hz), 159.6 (C), 156.6 (C), 139.9 (C), 138.0 (C), 134.6 (C), 133.6 (CH), 132.9 (d, CH, J = 8.3 Hz), 130.1 (C), 129.7 (CH), 129.43 (CH), 129.40 (C), 129.0 (CH), 128.9 (CH), 128.8 (CH), 128.5 (C), 124.1 (C), 114.7 (d, CH, J = 21.5 Hz), 110.0 (CH), 53.5 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C29H20FN2O5+ 495.1351, found 495.1361.
3-(2-phenyloxazol-5-yl)isoxazole-5-carboxylic acid (6a). Compound 6a was prepared following the general procedure GP-F from methyl 3-(3-phenyl-2H-azirine-2-carbonyl)isoxazole-5-carboxylate 3a (207 mg, 0.77 mmol) and K2CO3 (425 mg, 3.08 mmol) in methanol (6 mL) to produce pure product in 124 mg (63% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 1:1 + 20% CHCl3, (v/v)) as a brown solid: mp 208–210 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 8.12–8.10 (m, 3H), 7.78 (s, 1H), 7.61–7.59 (m, 3H), 3.34 (br, 1H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 162.7 (C), 162.6 (C), 157.8 (C), 153.3 (C), 141.1 (C), 132.0 (CH), 130.9 (CH), 129.8 (CH), 127.0 (CH), 126.5 (C), 107.8 (CH); HRMS (ESI) m/z [M + H]+ calcd for C13H9N2O4+ 257.0557, found 257.0559.
5-(Methoxymethyl)-3-(2-phenyloxazol-5-yl)isoxazole (6b). Compound 6b was prepared following the general procedure GP-F from (5-(methoxymethyl)isoxazol-3-yl)(3-phenyl-2H-azirin-2-yl)methanone 3d (97 mg, 0.38 mmol) and K2CO3 (210 mg, 1.52 mmol) in methanol (4 mL) to produce pure product in 33 mg (34% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 71–72 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 8.15–8.12 (m, 2H), 7.66 (s, 1H), 7.51–7.49 (m, 3H), 6.60 (s, 1H), 4.62 (s, 2H), 3.49 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 170.2 (C), 162.8 (C), 152.5 (C), 141.6 (C), 131.1 (CH), 128.9 (CH), 128.4 (CH), 126.8 (CH), 126.7 (C), 100.6 (CH), 65.3 (CH2), 59.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C14H13N2O3+ 257.0921, found 257.0926.
3-(2-Phenyloxazol-5-yl)isoxazole (6c). Compound 6c was prepared following the general procedure GP-F from (3-phenyl-2H-azirin-2-yl)(5-(trimethylsilyl)isoxazol-3-yl)methanone 3i (165 mg, 0.58 mmol) and K2CO3 (321 mg, 2.32 mmol) in methanol (4 mL) to produce pure product in 71 mg (58% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 115–117 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 8.52–8.51 (d, 1H, J = 1.7 Hz), 8.15–8.13 (m, 2H), 7.68 (s, 1H), 7.51–7.49 (s, 3H), 6.70 (d, 1H, J = 1.8 Hz); 13C{1H} NMR (CDCl3, 100 MHz): δ 162.8 (C), 159.1 (CH), 151.7 (C), 141.6 (C), 131.1 (CH), 128.9 (CH), 128.4 (CH), 126.8 (CH), 126.7 (C), 10.3 (CH); HRMS (ESI) m/z [M + H]+ calcd for C12H9N2O2+ 213.0659, found 213.0662.
3-(2-(3,4-Dimethoxyphenyl)oxazol-5-yl)isoxazole (6d). Compound 6d was prepared following the general procedure GP-F from (3-(3,4-dimethoxyphenyl)-2H-azirin-2-yl)(5-(trimethylsilyl)isoxazol-3-yl)methanone 3o (176 mg, 0.51 mmol) and K2CO3 (282 mg, 2.04 mmol) in methanol (4 mL) to produce pure product in 67 mg (48% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 150–152 °C (light petroleum/ethyl acetate); 1H NMR (CDCl3, 400 MHz): δ 8.51–8.50 (d, 1H, J = 1.7 Hz), 7.75–7.73 (dd, 1H, J = 8.4, 2.0 Hz), 7.63 (s, 2H), 6.97–6.95 (d, 1H, J = 8.3 Hz), 6.68 (d, 1H, J = 1.7 Hz), 3.99 (s, 3H), 3.96 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 163.0 (C), 159.0 (CH), 151.7 (C), 151.6 (C), 149.3 (C), 141.1 (C), 128.5 (CH), 120.3 (CH), 119.5 (C), 111.1 (CH), 109.4 (CH), 102.2 (CH), 56.1 (CH3), 56.0 (CH3); IR (KBr, cm–1): 3110, 2917, 1609, 1496, 1438, 1281, 1248, 1139, 1024, 877, 815, 768, 737; HRMS (ESI) m/z [M + H]+ calcd for C14H13N2O4+ 273.0870, found 273.0875.
3-(2-(4-Chlorophenyl)oxazol-5-yl)-5-phenylisoxazole (6e). Compound 6e was prepared following the general procedure GP-F from (3-(4-chlorophenyl)-2H-azirin-2-yl)(5-phenylisoxazol-3-yl)methanone 3x (99 mg, 0.31 mmol) and K2CO3 (171 mg, 1.24 mmol) in methanol (4 mL) to produce pure product in 34 mg (34% yield), after column chromatography on silica gel (eluent: light petroleum/ethyl acetate, 4:1, (v/v)) as a yellow solid: mp 190–192 °C (light petroleum/ethyl acetate); 1H NMR (DMSO-d6, 400 MHz): δ 8.12–8.10 (d, 2H, J = 8.6 Hz), 8.04 (s, 1H), 7.95–7.93 (m, 2H), 7.70–7.68 (d, 2H, J = 8.6 Hz), 7.61 (s, 1H), 7.60–7.56 (m, 3H); 13C{1H} NMR (DMSO-d6, 100 MHz): δ 170.5 (C), 161.5 (C), 153.2 (C), 142.1 (C), 136.6 (C), 131.4 (CH), 130.0 (CH), 129.9 (CH), 129.9 (CH), 128.6 (CH), 126.7 (C), 126.2 (CH), 125.5 (C), 98.9 (CH); HRMS (ESI) m/z [M + H]+ calcd for C18H12ClN2O2+ 323.0582, found 323.0586.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















