

Discrepancies in Mineral Oil Confirmation by Two-Dimensional Gas Chromatography–Mass Spectrometry: A Call for Harmonization


Abstract

1. Introduction
2. Results and Discussion
2.1. Laboratory Comparison of Quantitative Results Generated by LC-GC-FID
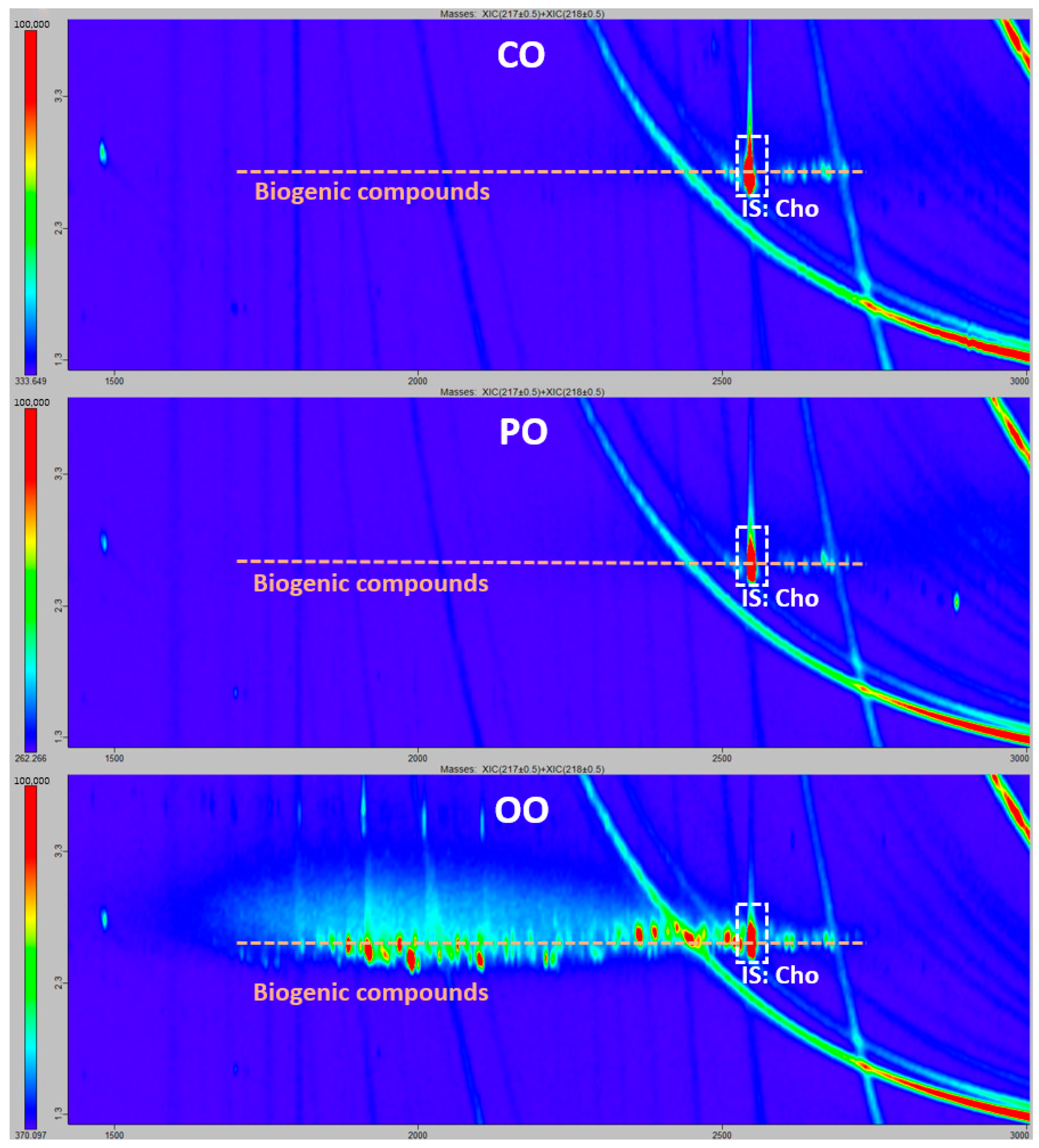
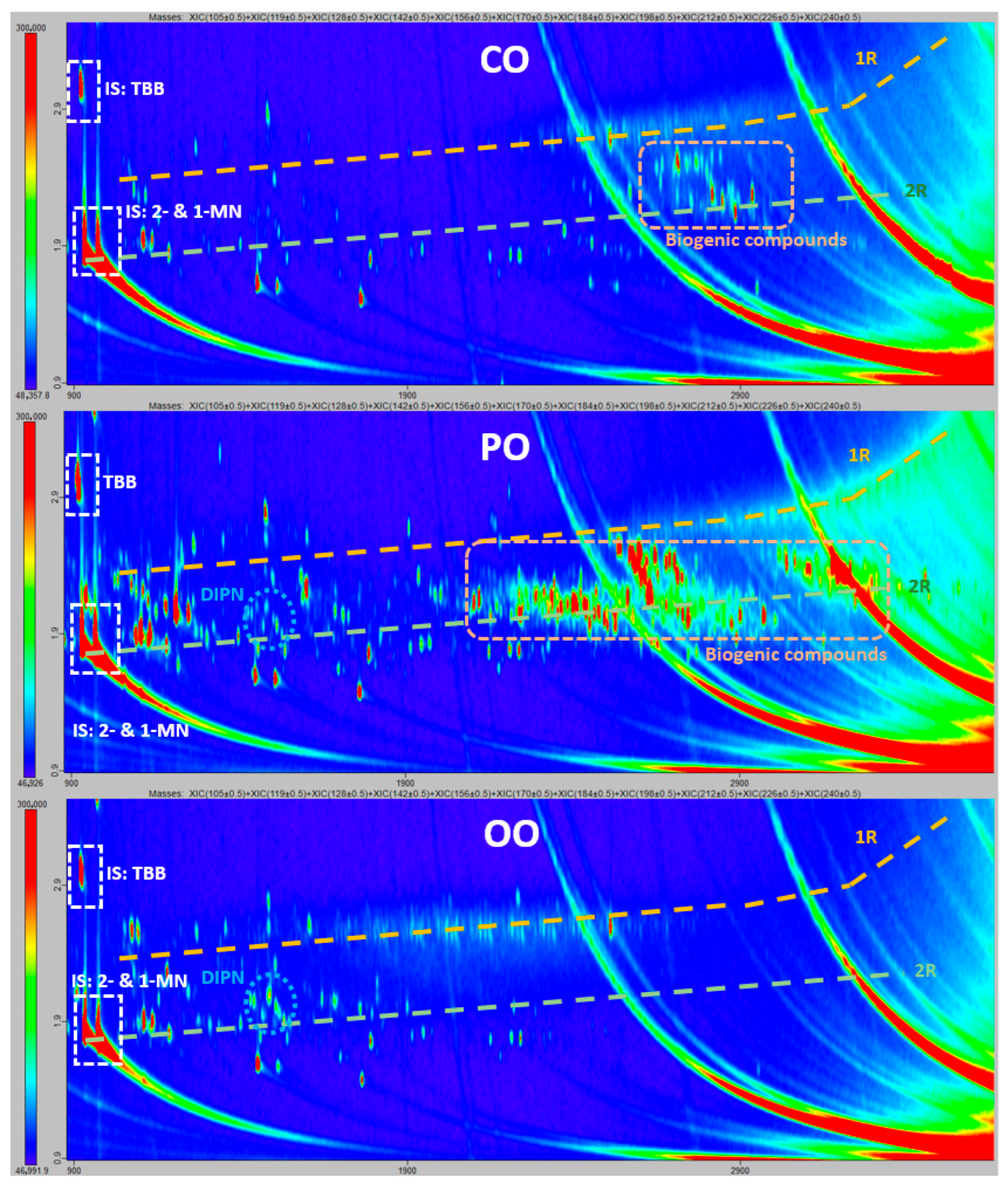
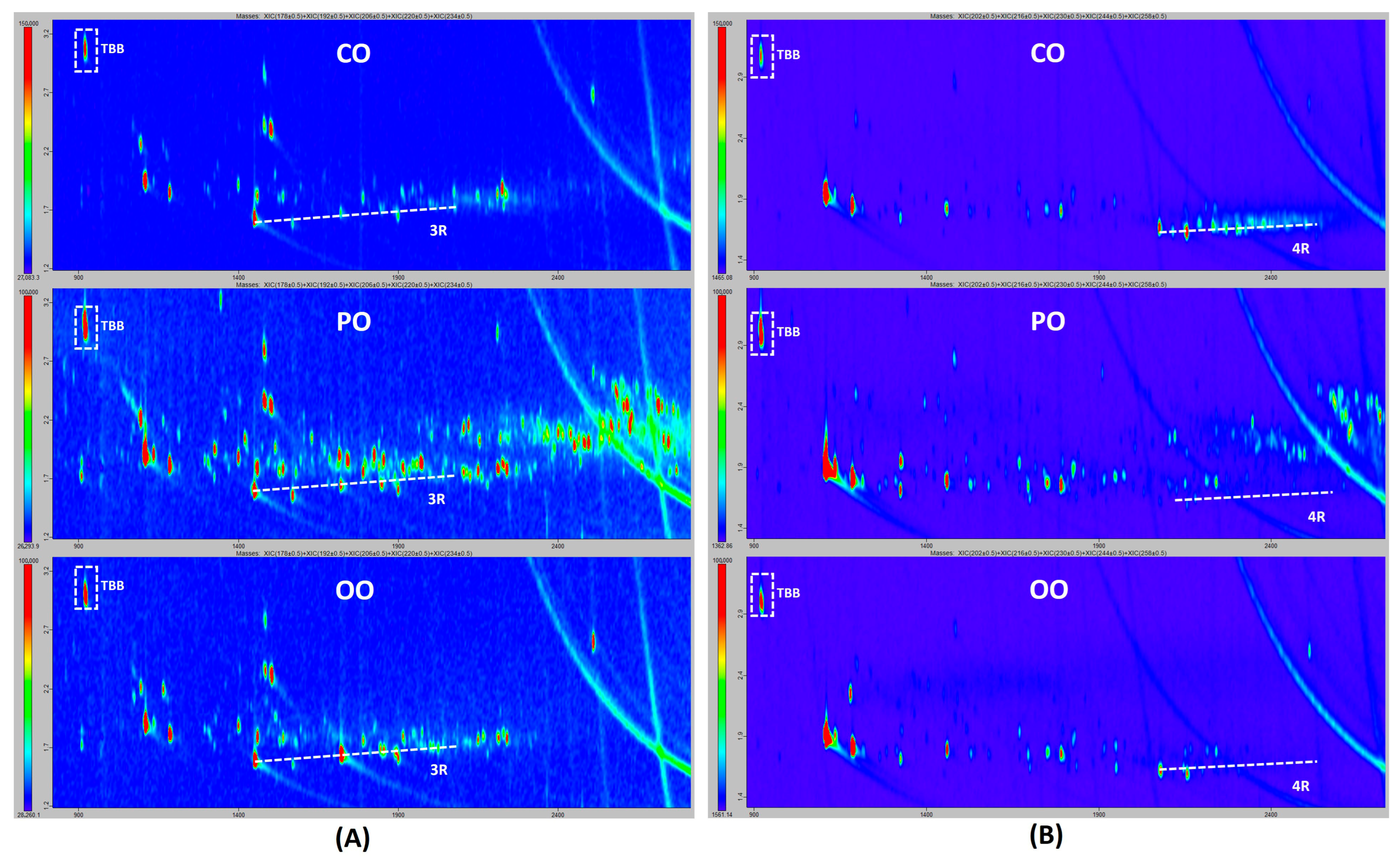
2.2. Laboratory Comparison for the Confirmation of Mineral Oil Components by GCxGC-ToF
3. Materials and Methods
3.1. Design of the Interlaboratory Trial
3.2. Quantification of MOSH and MOAH by LC-GC-FID
3.3. Confirmation of Mineral Oil Components by GCxGC-ToF
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1- and 2-MN | 1- and 2-Methylnaphthalene |
| 1R | mono-aromatics |
| 2R | di-aromatics |
| 3R | tri-aromatics |
| C10–C16 | Carbon range equivalent to an aliphatic chain with carbon numbers from 10 to 16 |
| C16–C20 | Carbon range equivalent to an aliphatic chain with carbon numbers from 16 to 20 |
| C16–C25 | Carbon range equivalent to an aliphatic chain with carbon numbers from 16 to 25 |
| C20–C25 | Carbon range equivalent to an aliphatic chain with carbon numbers from 20 to 25 |
| C25–C35 | Carbon range equivalent to an aliphatic chain with carbon numbers from 25 to 35 |
| C35–C50 | Carbon range equivalent to an aliphatic chain with carbon numbers from 35 to 50 |
| C40–C50 | Carbon range equivalent to an aliphatic chain with carbon numbers from 40 to 50 |
| CO | Coconut oil |
| DIPN | di-isopropyl-naphthalene |
| FID | Flame ionization detector |
| GCxGC | Comprehensive two-dimensional gas chromatography |
| IS | Internal standard |
| LC-GC | Liquid chromatography hyphenated to gas chromatography |
| LOQ | Limit of quantification |
| MOAH | Mineral oil aromatic hydrocarbons |
| MOH | Mineral oil hydrocarbons |
| MOSH | Mineral oil saturated hydrocarbons |
| OO | Olive oil |
| PAO | Polyalphaolefins |
| PO | Palm olein |
| POSH | Polyolefin saturated hydrocarbons |
| ToF | Time-of-flight mass spectrometry analyzer |
References
- Lehotay, S.J.; Chen, Y. Hits and misses in research trends to monitor contaminants in foods. Anal. Bioanal. Chem. 2018, 410, 5331–5351. [Google Scholar] [CrossRef] [PubMed]
- Brühl, L. Occurrence, determination, and assessment of mineral oils in oilseeds and vegetable oils. Eur. J. Lipid Sci. Technol. 2016, 118, 361–372. [Google Scholar] [CrossRef]
- Grob, K. Mineral oil hydrocarbons in food: A review. Food Addit. Contam.-Part A Chem. Anal. Control. Expo. Risk Assess. 2018, 35, 1845–1860. [Google Scholar] [CrossRef] [PubMed]
- El Bassoussi, A.A.; El-Sabagh, S.M.; Harb, F.M.; El Nady, M.M. Characterization and correlation of crude oils from some wells in the North Western Desert, Egypt. Pet. Sci. Technol. 2018, 36, 384–391. [Google Scholar] [CrossRef]
- Moura, A.K.S.; Ribeiro, D.O.; Carmo, I.S.D.; Araujo, B.Q.; Pereira, V.B.; Azevedo, D.A.; Citó, A.M.G.L. An Assay on Alkyl Aromatic Hydrocarbons, Unexpected Group-Type Separation of Diaromatic Hydrocarbons in Cretaceous Crude Oils from Brazilian Marginal Basin. Energy Fuels 2019, 33, 691–699. [Google Scholar] [CrossRef]
- Burazer, N.; Šajnović, A.; Vasić, N.; Kašanin-Grubin, M.; Životić, D.; Filho, J.G.M.; Vulić, P.; Jovančićević, B. Influence of paleoenvironmental conditions on distribution and relative abundance of saturated and aromatic hydrocarbons in sediments from the NW part of the Toplica basin, Serbia. Mar. Pet. Geol. 2020, 115, 104252. [Google Scholar] [CrossRef]
- Bratinova, S.; Robouch, P.; Hoekstra, E. Guidance on Sampling, Analysis and Data Reporting for the Monitoring of Mineral Oil Hydrocarbons in Food and Food Contact Materials, 2nd ed.; EUR 31473 EN; Publications Office of the European Union: Luxembourg, 2023; ISBN 978-92-68-01789-0. JRC 133174. [Google Scholar] [CrossRef]
- Biedermann, M.; Grob, K. On-line coupled high performance liquid chromatography-gas chromatography for the analysis of contamination by mineral oil. Part 1, Method of analysis. J. Chromatogr. A 2012, 1255, 56–75. [Google Scholar] [CrossRef]
- Biedermann, M.; Grob, K. On-line coupled high performance liquid chromatography-gas chromatography for the analysis of contamination by mineral oil. Part 2, Migration from paperboard into dry foods, Interpretation of chromatograms. J. Chromatogr. A 2012, 1255, 76–99. [Google Scholar] [CrossRef]
- Weber, S.; Schrag, K.; Mildau, G.; Kuballa, T.; Walch, S.G.; Lachenmeier, D.W. Analytical Methods for the Determination of Mineral Oil Saturated Hydrocarbons (MOSH) and Mineral Oil Aromatic Hydrocarbons (MOAH)—A Short Review. Anal. Chem. Insights 2018, 13, 1177390118777757. [Google Scholar] [CrossRef]
- Spack, L.W.; Leszczyk, G.; Varela, J.; Simian, H.; Gude, T.; Stadler, R.H. Understanding the contamination of food with mineral oil: The need for a confirmatory analytical and procedural approach. Food Addit. Contam.-Part A 2017, 34, 1052–1071. [Google Scholar] [CrossRef]
- Koster, S.; Varela, J.; Stadler, R.H.; Moulin, J.; Cruz-Hernandez, C.; Hielscher, J.; Lesueur, C.; Roïz, J.; Simian, H. Mineral oil hydrocarbons in foods, Is the data reliable? Food Addit. Contam.-Part A Chem. Anal. Control. Expo. Risk Assess. 2020, 37, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Bratinova, S.; Hoekstra, E. Guidance on Sampling, Analysis and Data Reporting for the Monitoring of Mineral Oil Hydrocarbons in Food and Food Contact Materials; EUR 29666 EN; Publications Office of the European Union: Luxembourg, 2019; ISBN 978-92-76-00172-0. JRC115694. [Google Scholar] [CrossRef]
- European Commission Commission decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of the results (2002/657/EC). Off. J. Eur. Communities 2002, L221, 8–36.
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on Mineral Oil Hydrocarbons in Food. EFSA J. 2012, 10, 2704–2889. [Google Scholar]
- Vendeuvre, C.; Bertoncini, F.; Duval, L.; Duplan, J.-L.; Thiébaut, D.; Hennion, M.-C. Comparison of conventional gas chromatography and comprehensive two-dimensional gas chromatography for the detailed analysis of petrochemical samples. J. Chromatogr. A 2004, 1056, 155–162. [Google Scholar] [CrossRef]
- Ventura, G.T.; Kenig, F.; Reddy, C.M.; Frysinger, G.S.; Nelson, R.K.; Van Mooy, B.; Gaines, R.B. Analysis of unresolved complex mixtures of hydrocarbons extracted from Late Archean sediments by comprehensive two-dimensional gas chromatography (GC×GC). Org. Geochem. 2008, 39, 846–867. [Google Scholar] [CrossRef]
- Kumar, S.; Dutta, S. Utility of comprehensive GC×GC-TOFMS in elucidation of aromatic hydrocarbon biomarkers. Fuel 2021, 283, 118890. [Google Scholar] [CrossRef]
- Biedermann, M.; Grob, K. Comprehensive two-dimensional gas chromatography for characterizing mineral oils in foods and distinguishing them from synthetic hydrocarbons. J. Chromatogr. A 2015, 1375, 146–153. [Google Scholar] [CrossRef]
- Funk, M.; Hillmann, H.; Derra, R.; Leist, U. Comparability of mineral oil testing for dry food and cardboard samples–Perspectives from different PT rounds. Food Addit. Contam.-Part A Chem. Anal. Control. Expo. Risk Assess. 2018, 35, 305–315. [Google Scholar] [CrossRef]
- Albert, C.; Humpf, H.U.; Brühl, L. Determining MOSH and MOAH with High Sensitivity in Vegetable Oil—A New, Reliable, and Comparable Approach Using Online LC-GC-FID─Evaluation of Method Precision Data. J. Agric. Food Chem. 2022, 70, 10337–10348. [Google Scholar] [CrossRef]
- Biedermann, M.; Munoz, C.; Grob, K. Epoxidation for the analysis of the mineral oil aromatic hydrocarbons in food. An update. J. Chromatogr. A 2020, 1624, 461236. [Google Scholar] [CrossRef]
- EN 16995:2017; Foodstuffs-Vegetable Oils and Foodstuff on Basis of Vegetable Oils-Determination of Mineral Oil Saturated Hydrocarbons (MOSH) and Mineral Oil Aromatic Hydrocarbons (MOAH) with Online HPLC-GC-FID Analysis. European Committee for Standardization: Brussels, Belgium, 2017.
- Bauwens, G.; Pantó, S.; Purcaro, G. Mineral oil saturated and aromatic hydrocarbons quantification, Mono- and two-dimensional approaches. J. Chromatogr. A 2021, 1643, 462044. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion-Update of the risk assessment of mineral oil hydrocarbons (MOH) in food. EFSA J. 2023, 21, 8215–8360. [Google Scholar]
- Tranchida, P.Q.; Zoccali, M.; Mondello, L. Views on the uses of comprehensive two-dimensional gas chromatography-mass spectrometry in food analysis over the literature period 2018–2023. TrAC Trends Anal. Chem. 2024, 174, 117671. [Google Scholar] [CrossRef]
- Method Ring Test MOSH/MOAH in Chocolate by GCxGC-TOF P2101-MRT. Summary. 2019. Available online: https://www.proof-acs.de/competenceschemes/36/summary (accessed on 25 May 2025).
- Reference Material MOSH/MOAH GCxGC-TOF Cheese (P2206-RMCh1). Summary. 2022. Available online: https://www.proof-acs.de/upload/media/default/0001/01/bbc15cd3f114ea1efa900bedece0047907753ecc.pdf (accessed on 25 May 2025).
- ISO 20122:2024; Vegetable Oils—Determination of Mineral Oil Saturated Hydrocarbons (MOSH) and Mineral Oil Aromatic Hydrocarbons (MOAH) with Online-Coupled High Performance Liquid Chromatography-Gas Chromatography-Flame Ionization Detection (HPLC-GC-FID) Analysis—Method for Low Limit of Quantification. International Organization for Standarization: Geneva, Switzerland, 2024.
- Nestola, M. Automated workflow utilizing saponification and improved epoxidation for the sensitive determination of mineral oil saturated and aromatic hydrocarbons in edible oils and fats. J. Chromatogr. A 2022, 1682, 463523. [Google Scholar] [CrossRef] [PubMed]
- Van der Westhuizen, R.; Ajam, M.; Coning, P.D.; Beens, J.; de Villiers, A.; Sandra, P. Comprehensive two-dimensional gas chromatography for the analysis of synthetic and crude-derived jet fuels. J. Chromatogr. A 2011, 1218, 4478–4486. [Google Scholar] [CrossRef]
- Hu, S.Z.; Li, S.F.; Cao, J.; Zhang, D.M.; Ma, J.; He, S.; Wang, X.L.; Wu, M. A Comparison of Normal and Reversed Phase Columns in Oil Analysis by Comprehensive Two-dimensional Gas Chromatography With Time-of-flight Mass Spectrometry. Pet. Sci. Technol. 2014, 32, 565–574. [Google Scholar] [CrossRef]
- Biedermann, M.; Grob, K. Advantages of comprehensive two-dimensional gas chromatography for comprehensive analysis of potential migrants from food contact materials. Anal. Chim. Acta 2019, 1057, 11–17. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vegetable Oil | Laboratory | MOSH [mg/kg] | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C10–C16 | U | C16–C20 | U | C20–C25 | U | C25–C35 | U | C35–C40 | U | C40–C50 | U | Total | U | ||
| Coconut oil | A | <LOQ (1.0) | - | <LOQ (1.0) | - | <LOQ (1.0) | - | 11.0 | ±4.4 | 5.0 | ±2.0 | 4.7 | ±1.9 | 20.7 | ±8.3 |
| B | <LOQ (0.4) | - | <LOQ (0.4) | - | 0.8 | ±0.2 | 18.0 | ±5.2 | 7.1 | ±2.1 | 8.1 | ±2.3 | 34.0 | ±9.9 | |
| C | <LOQ (1.0) | - | <LOQ (1.0) | - | <LOQ (1.0) | - | 14.0 | ±4.2 | 5.0 | ±1.5 | 5.1 | ±1.5 | 24.1 | ±7.2 | |
| D | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | 9.2 | ±2.1 | 4.0 | ±1.0 | 3.2 | ±0.9 | 17.0 | ±4.0 | |
| E | <LOQ (0.5) | - | <LOQ (0.5) | - | 0.7 | ±0.2 | 8.8 | ±2.6 | 4.0 | ±1.2 | 3.9 | ±1.2 | 17.4 | ±5.2 | |
| F | <LOQ (0.5) | - | <LOQ (0.5) | - | 0.7 | ±0.2 | 11.8 | ±4.1 | 4.5 | ±1.6 | 3.9 | ±1.4 | 20.9 | ±7.3 | |
| Median | - | - | 0.7 | 11.4 | 4.8 | 4.3 | 20.8 | ||||||||
| SD | 0.1 | 3.4 | 1.2 | 1.7 | 6.3 | ||||||||||
| Palm olein | A | <LOQ (1.0) | - | <LOQ (1.0) | - | <LOQ (1.0) | - | 11.0 | ±4.4 | 9.5 | ±3.8 | 13.0 | ±5.2 | 33.5 | ±13.4 |
| B | <LOQ (0.4) | - | <LOQ (0.4) | - | 0.5 | ±0.1 | 13.4 | ±3.9 | 12.1 | ±3.5 | 18.5 | ±5.4 | 44.5 | ±12.9 | |
| C | <LOQ (1.0) | - | <LOQ (1.0) | - | <LOQ (1.0) | - | 13.0 | ±3.9 | 9.9 | ±3.0 | 14.0 | ±4.2 | 36.9 | ±11.1 | |
| D | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | 8.6 | ±2.0 | 7.6 | ±1.8 | 9.7 | ±2.2 | 26.0 | ±5.0 | |
| E | <LOQ (0.5) | - | <LOQ (0.5) | - | <LOQ (0.5) | - | 8.9 | ±2.7 | 6.8 | ±2.0 | 8.8 | ±2.6 | 24.5 | ±7.4 | |
| F | <LOQ (0.5) | - | <LOQ (0.5) | - | <LOQ (0.5) | - | 11.2 | ±3.9 | 8.5 | ±3.0 | 10.8 | ±3.8 | 30.7 | ±10.7 | |
| Median | - | - | - | 11.1 | 9.0 | 11.9 | 32.1 | ||||||||
| SD | 2.0 | 1.9 | 3.5 | 7.4 | |||||||||||
| Olive oil | A | <LOQ (1.0) | - | <LOQ (1.0) | - | 2.9 | ±1.2 | 9.4 | ±3.8 | 2.3 | ±0.9 | 1.5 | ±0.6 | 16.1 | ±6.4 |
| B | <LOQ (0.4) | - | 0.8 | ±0.2 | 3.5 | ±1.0 | 13.3 | ±3.9 | 4.4 | ±1.3 | 3.4 | ±1.0 | 25.4 | ±7.4 | |
| C | <LOQ (1.0) | - | <LOQ (1.0) | - | 3.6 | ±1.1 | 12.0 | ±3.6 | 2.8 | ±0.8 | 1.9 | ±0.6 | 20.3 | ±6.1 | |
| D | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | 6.7 | ±1.6 | 2.2 | ±0.6 | <LOQ (2.0) | - | 12.0 | ±3.0 | |
| E | <LOQ (0.5) | - | <LOQ (0.5) | - | 0.8 | ±0.2 | 5.1 | ±1.5 | 1.1 | ±0.3 | 0.7 | ±0.2 | 7.6 | ±2.3 | |
| F | <LOQ (0.5) | - | 1.2 | ±0.4 | 3.4 | ±1.2 | 15.5 | ±5.4 | 2.8 | ±1.0 | 1.4 | ±0.5 | 24.3 | ±8.5 | |
| Median | - | - | - | - | 3.4 | 10.7 | 2.6 | 1.5 | 18.2 | ||||||
| SD | 1.2 | 4.0 | 1.1 | 1.0 | 7.0 | ||||||||||
| Vegetable Oil | Laboratory | MOAH [mg/kg] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C10–C16 | U | C16–C25 | U | C25–C35 | U | C35–C50 | U | Total | U | ||
| Coconut oil | A | <LOQ (1.0) | - | <LOQ (1.0) | - | 1.7 | ±0.7 | 2.1 | ±0.8 | 3.8 | ±1.5 |
| B | <LOQ (0.4) | - | <LOQ (0.4) | - | 2.4 | ±0.7 | 3.2 | ±0.9 | 5.6 | ±1.6 | |
| C | <LOQ (1.0) | - | <LOQ (1.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (4.0) | - | |
| D | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | 2.4 | ±0.7 | |
| E | <LOQ (0.5) | - | <LOQ (0.5) | - | 2.2 | ±0.7 | 1.8 | ±0.5 | 4.0 | ±1.2 | |
| F | <LOQ (0.5) | - | <LOQ (0.5) | - | 2.8 | ±1.0 | <LOQ (0.5) | - | 3.5 | ±1.2 | |
| Median | - | - | 1.95 | 2.1 | 3.8 | ||||||
| SD | 0.7 | 0.7 | 1.2 | ||||||||
| Palm olein | A | <LOQ (1.0) | - | <LOQ (1.0) | - | 1.3 | ±0.5 | 5.2 | ±2.1 | 6.5 | ±2.6 |
| B | <LOQ (0.4) | - | 1.0 | ±0.3 | 4.0 | ±1.2 | 6.7 | ±1.9 | 11.7 | ±3.4 | |
| C | <LOQ (1.0) | - | <LOQ (1.0) | - | 2.3 | ±0.7 | 6.0 | ±1.8 | 8.3 | ±2.5 | |
| D | <LOQ (2.0) | - | <LOQ (2.0) | - | 2.0 | ±0.6 | 4.3 | ±1.1 | 6.5 | ±1.6 | |
| E | <LOQ (0.5) | - | <LOQ (0.5) | - | 2.2 | ±0.7 | 3.9 | ±1.2 | 6.1 | ±1.8 | |
| F | <LOQ (0.5) | - | <LOQ (0.5) | - | 1.7 | ±0.6 | 5.5 | ±1.9 | 7.2 | ±2.5 | |
| Median | - | - | 2.1 | 5.4 | 6.9 | ||||||
| SD | 0.9 | 1.0 | 2.1 | ||||||||
| Olive oil | A | <LOQ (1.0) | - | <LOQ (1.0) | - | 1.4 | ±0.6 | <LOQ (1.0) | - | 1.4 | ±0.6 |
| B | <LOQ (0.4) | - | 0.7 | ±0.2 | 1.5 | ±0.4 | 1.0 | ±0.3 | 3.2 | ±0.9 | |
| C | <LOQ (1.0) | - | <LOQ (1.0) | - | 1.2 | ±0.4 | <LOQ (1.0) | - | 1.2 | ±0.4 | |
| D | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | <LOQ (2.0) | - | |
| E | <LOQ (0.5) | - | <LOQ (0.5) | - | 1.3 | ±0.4 | <LOQ (0.5) | - | <LOQ (2.8) | - | |
| F | <LOQ (0.5) | - | <LOQ (0.5) | - | 0.8 | ±0.3 | <LOQ (0.5) | - | 1.2 | ±0.4 | |
| Median | - | - | 1.3 | - | 1.3 | ||||||
| SD | 0.3 | 1.0 | |||||||||
| Fraction | Sub-Group | Coconut Oil | Palm Olein | Olive Oil | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Laboratory | Laboratory | Laboratory | |||||||||||||||||
| A | B | C | D | E | F | A | B | C | D | E | F | A | B | C | D | E | F | ||
| MOSH | n-Alkanes | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | |||||
| iso-Alkanes | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | |||||||
| Cycloalkanes | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ||||||||
| Hopanes | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ||||
| Biogenic compounds | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | |||||||||||
| POSH | ☑ | ☑ | ☑ | ☑ | ☑ | ||||||||||||||
| MOAH | Alkyl-monoaromatics | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ||||
| Alkyl-diaromatics | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ||||||||
| 3–4 PACs | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | |||||||||||
| 5–7 PACs | ☑ | ☑ | ☑ | ☑ | |||||||||||||||
| Thiophenes | ☑ | ☑ | ☑ | ||||||||||||||||
| Biogenic compounds | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | |||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huertas-Pérez, J.F.; Cruz-Hernández, C.; Núñez-Galindo, A.; Dubois, M.; Perring, L.; Tarres, A.; Nicolay, J.; Vocat, C.; Delatour, T. Discrepancies in Mineral Oil Confirmation by Two-Dimensional Gas Chromatography–Mass Spectrometry: A Call for Harmonization. Molecules 2025, 30, 2830. https://doi.org/10.3390/molecules30132830
Huertas-Pérez JF, Cruz-Hernández C, Núñez-Galindo A, Dubois M, Perring L, Tarres A, Nicolay J, Vocat C, Delatour T. Discrepancies in Mineral Oil Confirmation by Two-Dimensional Gas Chromatography–Mass Spectrometry: A Call for Harmonization. Molecules. 2025; 30(13):2830. https://doi.org/10.3390/molecules30132830
Chicago/Turabian StyleHuertas-Pérez, José Fernando, Cristina Cruz-Hernández, Antonio Núñez-Galindo, Mathieu Dubois, Loïc Perring, Adrienne Tarres, Julie Nicolay, Céline Vocat, and Thierry Delatour. 2025. "Discrepancies in Mineral Oil Confirmation by Two-Dimensional Gas Chromatography–Mass Spectrometry: A Call for Harmonization" Molecules 30, no. 13: 2830. https://doi.org/10.3390/molecules30132830
APA StyleHuertas-Pérez, J. F., Cruz-Hernández, C., Núñez-Galindo, A., Dubois, M., Perring, L., Tarres, A., Nicolay, J., Vocat, C., & Delatour, T. (2025). Discrepancies in Mineral Oil Confirmation by Two-Dimensional Gas Chromatography–Mass Spectrometry: A Call for Harmonization. Molecules, 30(13), 2830. https://doi.org/10.3390/molecules30132830