

Antitumor Activity of Isalpinin from Paphiopedilum dianthum on Non-Small Cell Lung Cancer Cell Lines

 ,
,  , , and
, , and
Abstract

1. Introduction
2. Results
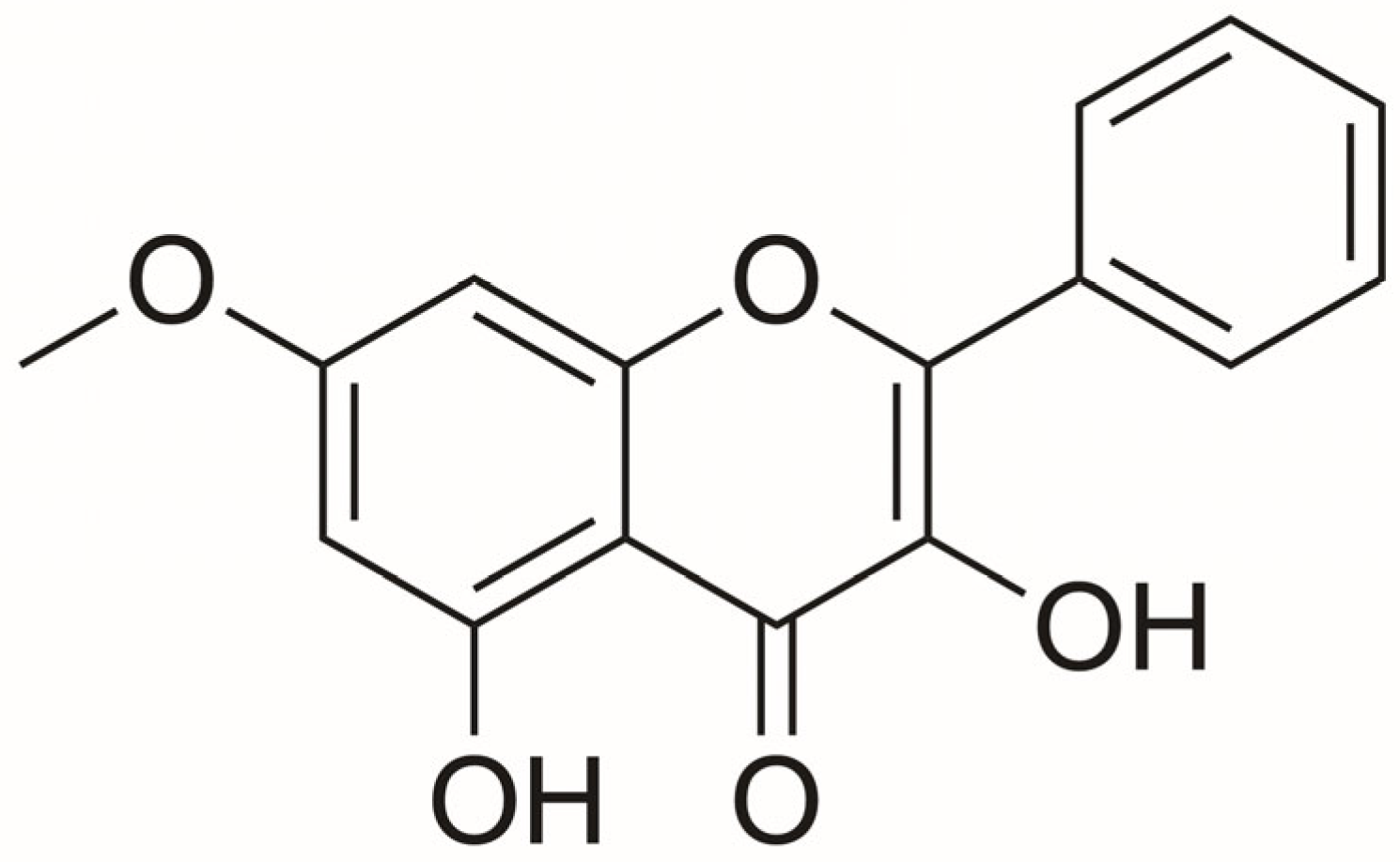
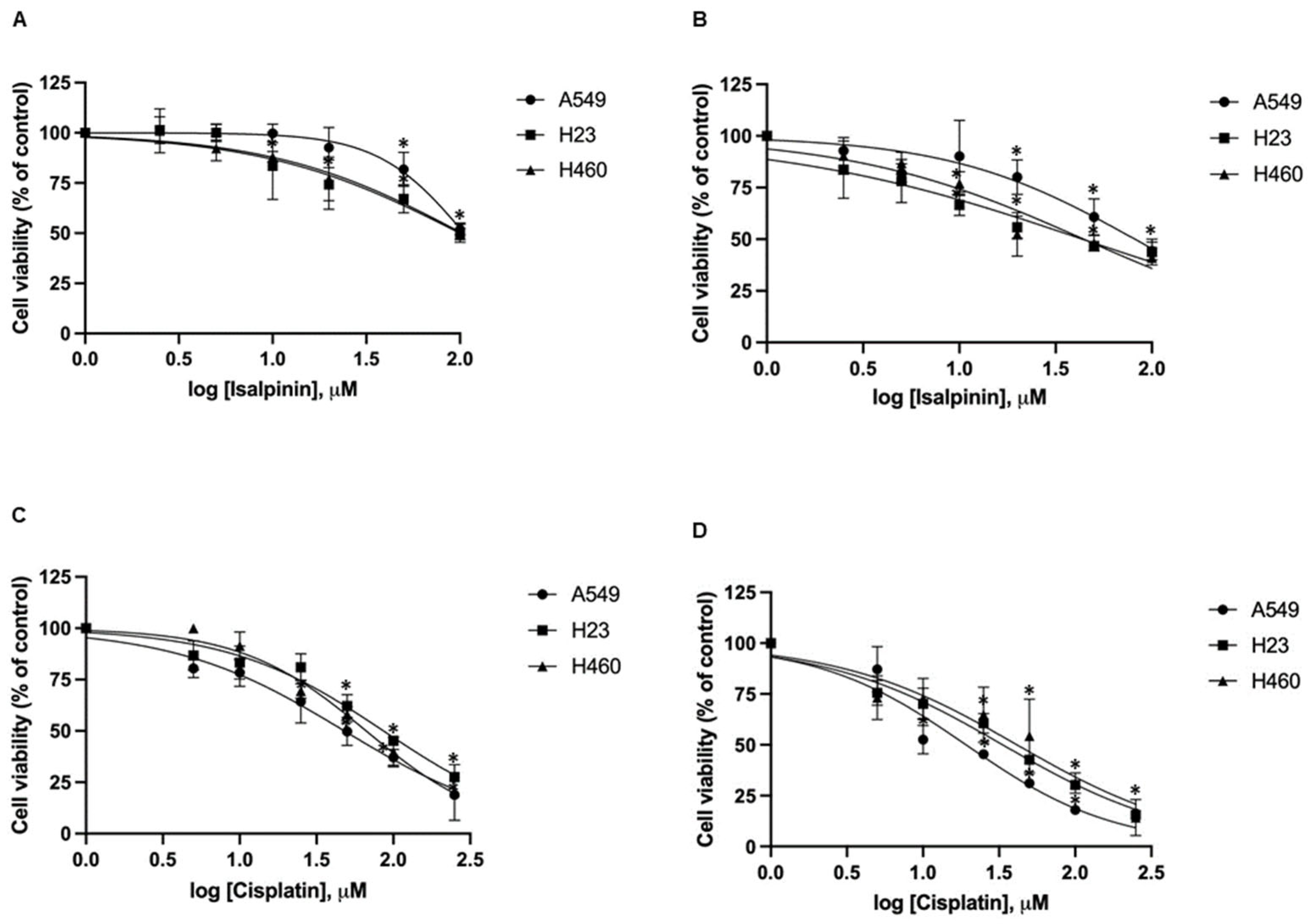
2.1. Cytotoxicity of Isalpinin and Cisplatin on NSCLC Cell Lines
2.2. Anti-Proliferative Effect of Isalpinin on A549, H23 and H460 NSCLC Cell Lines
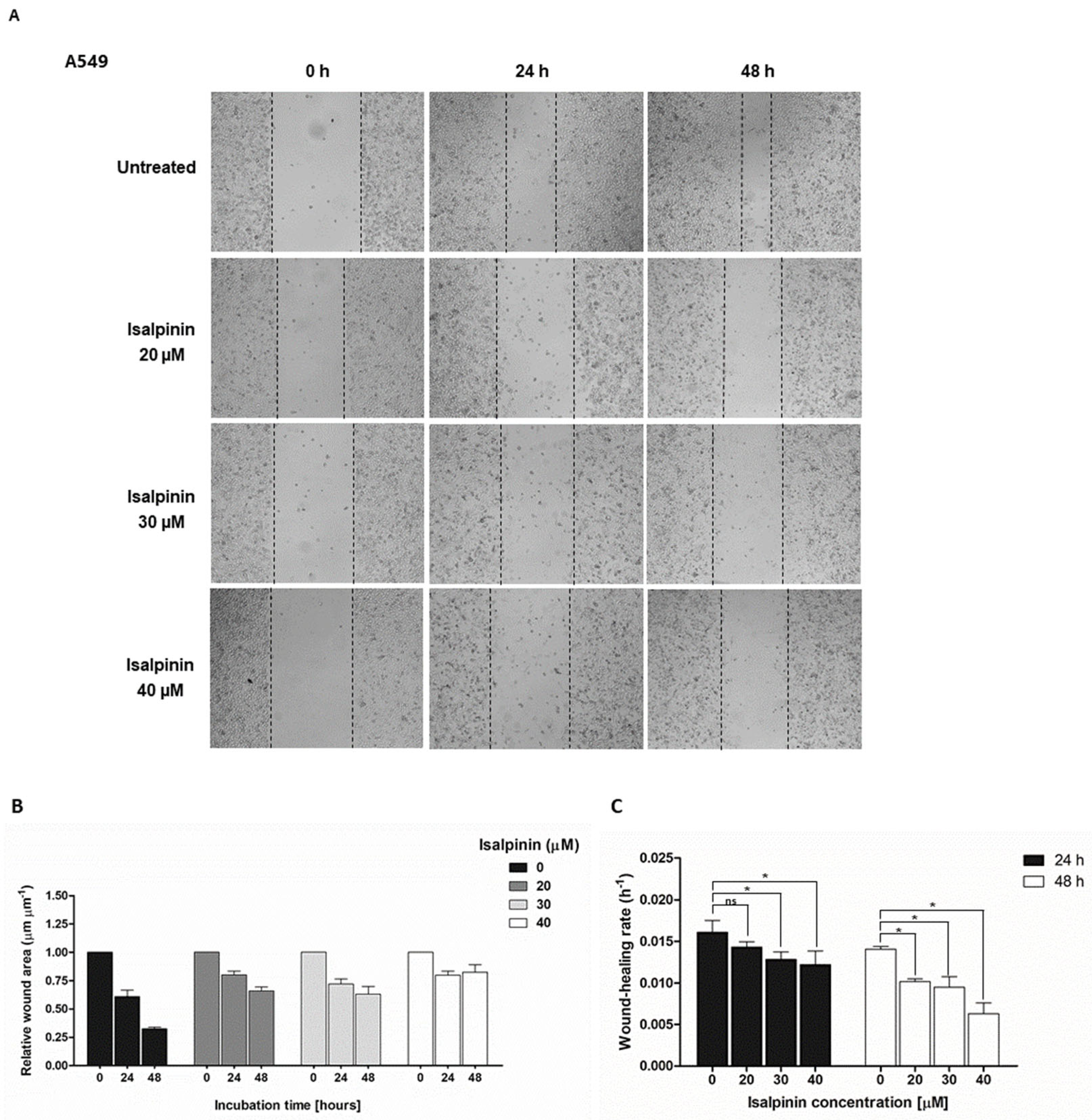
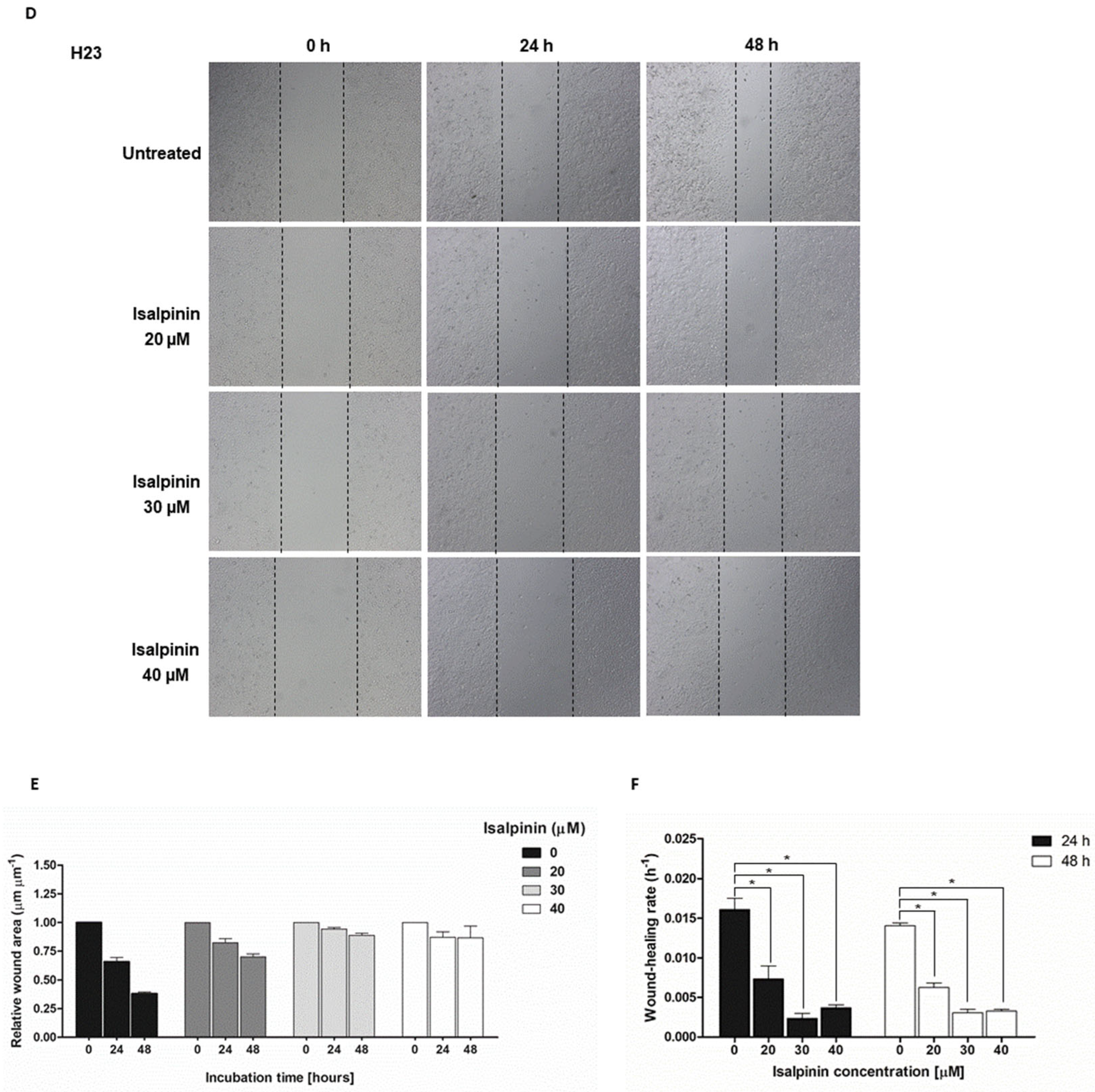
2.3. The Anti-Migratory Effect of Isalpinin on A549, H23, and H460 NSCLC Cell Lines
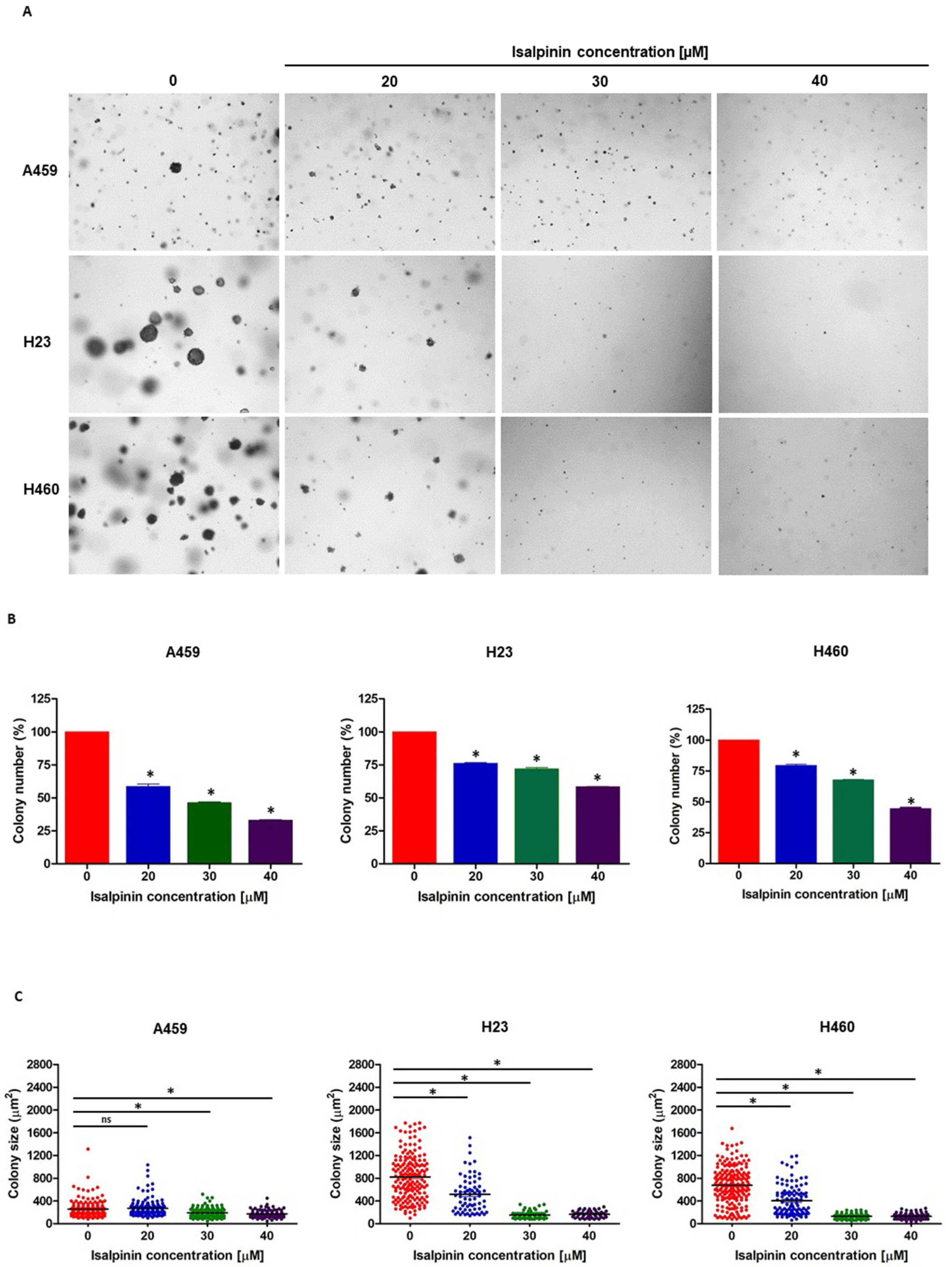
2.4. Isalpinin Inhibited Anchorage-Independent Growth in A549, H23, and H460 NSCLC Cell Lines
2.5. The Effect of Isalpinin on Intracellular Reactive Oxygen Species (ROS) Levels in A549, H23, and H460 NSCLC Cell Lines
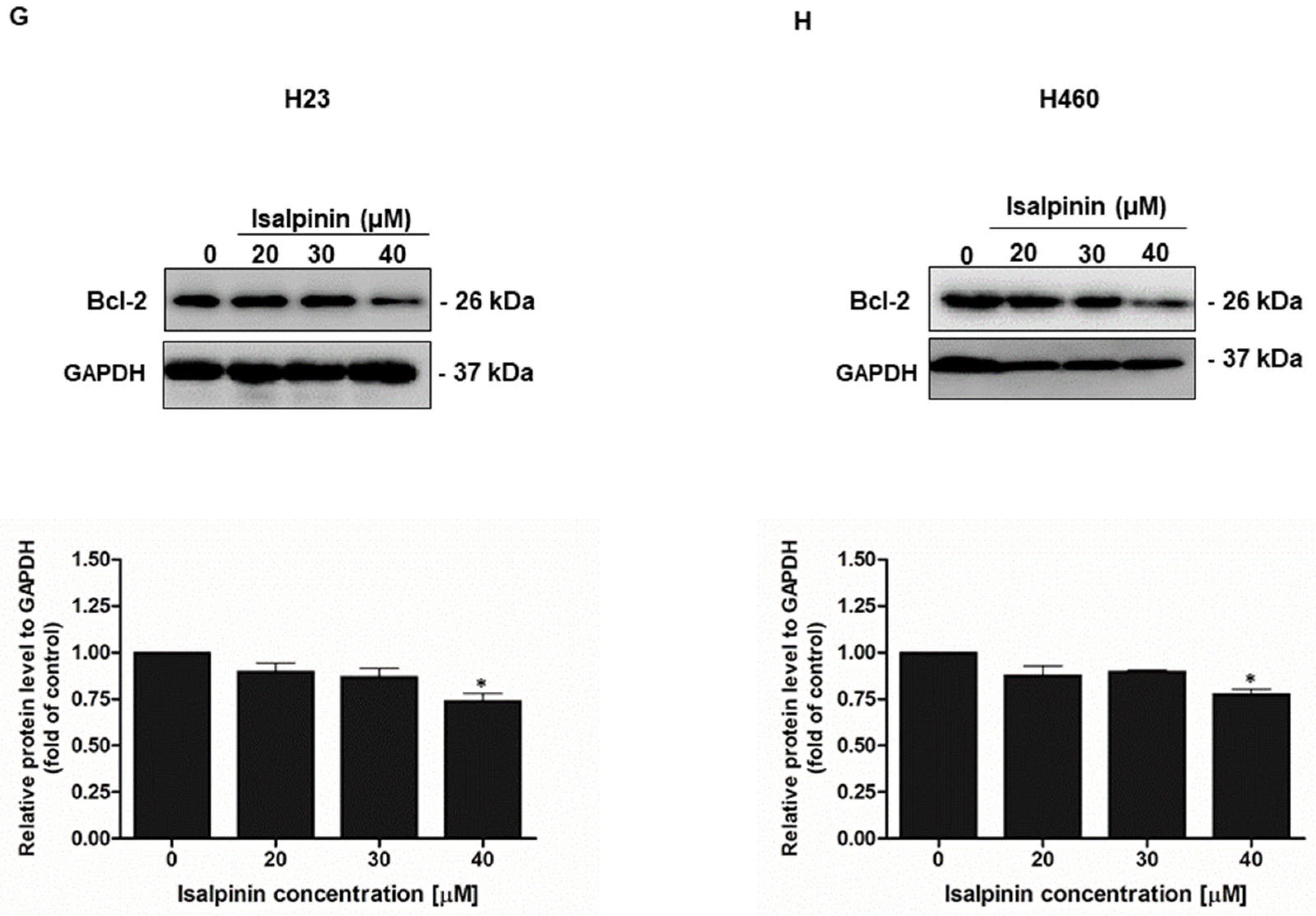
2.6. Effect of Isalpinin Induces Apoptosis and Modulates Anti-Apoptotic Bcl-2 Protein Expression in NSCLC Cell Lines
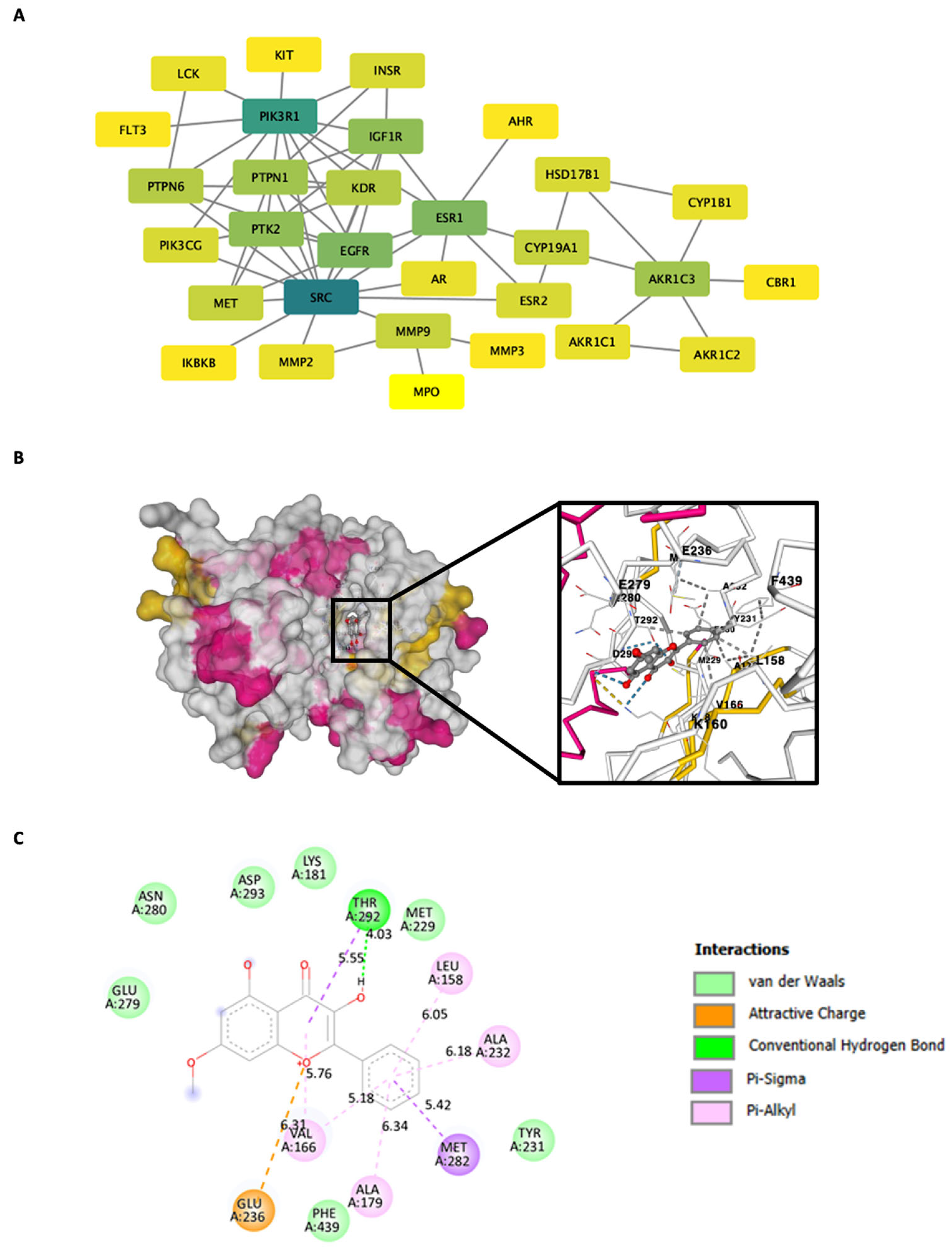
2.7. Gene Ontology (GO) and KEGG Pathway Enrichment Analysis of Isalpinin-Associated Targets
2.8. Construction of the Protein–Protein Interaction (PPI) Network and Computational Modeling of AKT with Molecular Docking
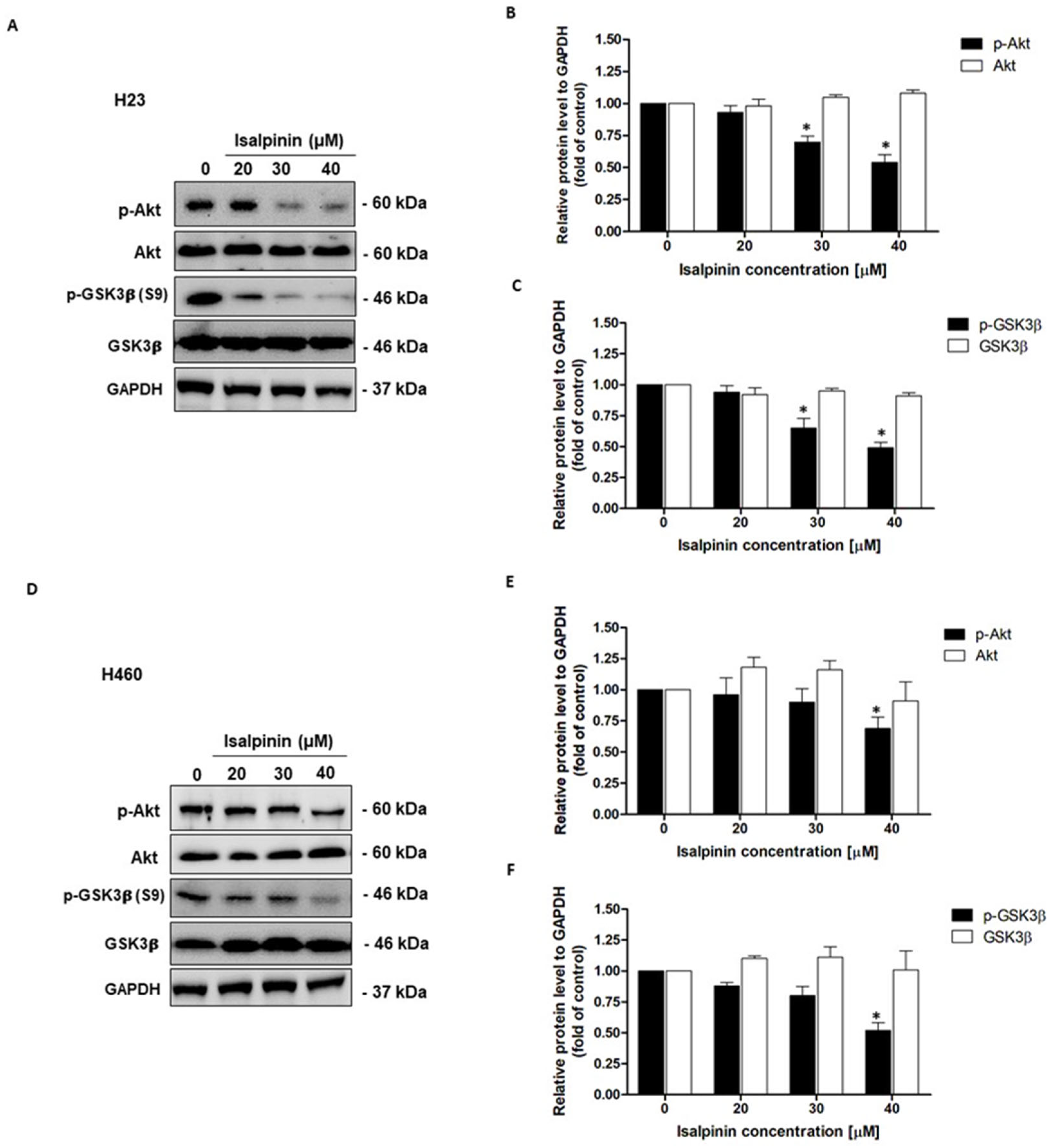
2.9. Effect of Isalpinin on Akt/GSK3β Pathway in NSCLC Cell Lines
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Isalpinin Preparation
4.2.2. Cell Culture
4.2.3. Cytotoxicity and Cell Proliferation Assays
4.2.4. Cell Migration Assays
4.2.5. Soft Agar Colony Formation Assay
4.2.6. Determination of Reactive Oxygen Species (ROS)
4.2.7. Cell Apoptosis
4.2.8. Western Blot Analysis
4.2.9. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
4.2.10. Construction of the Protein–Protein Interaction (PPI) Network and Computational Modeling of AKT with Molecular Docking
4.2.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, C.; Lei, S.; Ding, L.; Xu, Y.; Wu, X.; Wang, H.; Zhang, Z.; Gao, T.; Zhang, Y.; Li, L. Global Burden and Trends of Lung Cancer Incidence and Mortality. Chin. Med. J. 2023, 136, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global Cancer Statistics. CA. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Hecht, S.S. Lung Carcinogenesis by Tobacco Smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Lung Cancer Guide|What You Need to Know|American Cancer Society. Available online: https://www.cancer.org/cancer/types/lung-cancer.html (accessed on 5 April 2025).
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Mir, S.A.; Dar, A.; Hamid, L.; Nisar, N.; Malik, J.A.; Ali, T.; Bader, G.N. Flavonoids as Promising Molecules in the Cancer Therapy: An Insight. Curr. Res. Pharmacol. Drug Discov. 2024, 6, 100167. [Google Scholar] [CrossRef]
- Wang, X.; Huang, M.; Xie, W.; Ding, Q.; Wang, T. Eupafolin Regulates Non-Small-Cell Lung Cancer Cell Proliferation, Migration, and Invasion by Suppressing MMP9 and RhoA via FAK/PI3K/AKT Signaling Pathway. J. Biosci. 2023, 48, 1. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, F.; Che, X.; Li, Y.; Li, N.; Jiang, Z.; Li, X. Angelica Acutiloba Kitagawa Flower Induces A549 Cell Pyroptosis via the NF-κB/NLRP3 Pathway for Anti-Lung Cancer Effects. Cell Div. 2023, 18, 19. [Google Scholar] [CrossRef]
- Śliwiński, T.; Kowalczyk, T.; Sitarek, P.; Kolanowska, M. Orchidaceae-Derived Anticancer Agents: A Review. Cancers 2022, 14, 754. [Google Scholar] [CrossRef] [PubMed]
- Lertnitikul, N.; Liangsakul, J.; Jianmongkol, S.; Suttisri, R. Three New Cytotoxic Stilbene Dimers from Paphiopedilum Dianthum. Nat. Prod. Res. 2023, 37, 3685–3693. [Google Scholar] [CrossRef]
- Farhan, M. Insights on the Role of Polyphenols in Combating Cancer Drug Resistance. Biomedicines 2023, 11, 1709. [Google Scholar] [CrossRef]
- Sein, K.L.; Lertnitikul, N.; Suttisri, R.; Jianmongkol, S. Anticancer and Chemosensitizing Activities of Stilbenoids from Three Orchid Species. Naunyn. Schmiedebergs Arch. Pharmacol. 2023, 396, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the Hallmarks of Cancer. Am. J. Cancer Res. 2017, 7, 1016. [Google Scholar]
- Huang, Q.; Li, Y.; Huang, Y.; Wu, J.; Bao, W.; Xue, C.; Li, X.; Dong, S.; Dong, Z.; Hu, S. Advances in Molecular Pathology and Therapy of Non-Small Cell Lung Cancer. Signal Transduct. Target. Ther. 2025, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.S.; Jeong, J.C.; Jeong, Y.S.; Kim, E.J.; Um, S.J. Quercetin Potentiates Apoptosis by Inhibiting Nuclear Factor-kappaB Signaling in H460 Lung Cancer Cells. Biol. Pharm. Bull. 2013, 36, 944–951. [Google Scholar] [CrossRef]
- Tsui, K.C.; Chiang, T.H.; Wang, J.S.; Lin, L.J.; Chao, W.C.; Chen, B.H.; Lu, J.F. Flavonoids from Gynostemma Pentaphyllum Exhibit Differential Induction of Cell Cycle Arrest in H460 and A549 Cancer Cells. Molecules 2014, 19, 17663–17681. [Google Scholar] [CrossRef]
- Li, H.L.; Li, S.M.; Luo, Y.H.; Xu, W.T.; Zhang, Y.; Zhang, T.; Zhang, D.J.; Jin, C.H. Kaempferide Induces G0/G1 Phase Arrest and Apoptosis via ROS-Mediated Signaling Pathways in A549 Human Lung Cancer Cells. Nat. Prod. Commun. 2020, 15, 1934578X2093522. [Google Scholar] [CrossRef]
- Sak, K.; Lust, H.; Kase, M.; Jaal, J. Cytotoxic Action of Methylquercetins in Human Lung Adenocarcinoma Cells. Oncol. Lett. 2017, 15, 1973. [Google Scholar] [CrossRef]
- Chen, L.; Teng, H.; Xie, Z.; Cao, H.; Cheang, W.S.; Skalicka-Woniak, K.; Georgiev, M.I.; Xiao, J. Modifications of Dietary Flavonoids towards Improved Bioactivity: An Update on Structure–Activity Relationship. Crit. Rev. Food Sci. Nutr. 2018, 58, 513–527. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, Y.; Xiang, W.; Wang, Q.; Cao, Y. Molecular Mechanisms of the Action of Myricetin in Cancer. Mini-Rev. Med. Chem. 2019, 20, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Park, S.J.; Choi, Y.S.; Jeon, W.K.; Kim, B.C. Kaempferol Suppresses Transforming Growth Factor-Β1–Induced Epithelial-to-Mesenchymal Transition and Migration of A549 Lung Cancer Cells by Inhibiting Akt1-Mediated Phosphorylation of Smad3 at Threonine-179. Neoplasia 2015, 17, 525–537. [Google Scholar] [CrossRef]
- Zhu, X.; Ma, P.; Peng, D.; Wang, Y.; Wang, D.; Chen, X.; Zhang, X.; Song, Y. Quercetin Suppresses Lung Cancer Growth by Targeting Aurora B Kinase. Cancer Med. 2016, 5, 3156–3165. [Google Scholar] [CrossRef]
- Luo, W.; Liu, Q.; Jiang, N.; Li, M.; Shi, L. Isorhamnetin Inhibited Migration and Invasion via Suppression of Akt/ERK-Mediated Epithelial-to-Mesenchymal Transition (EMT) in A549 Human Non-Small-Cell Lung Cancer Cells. Biosci. Rep. 2019, 39, BSR20190159. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Cui, W.; Yang, X.; Tong, B. Kaempferol Inhibits the Growth and Metastasis of Cholangiocarcinoma in Vitro and in Vivo. Acta Biochim. Biophys. Sin. 2016, 48, 238–245. [Google Scholar] [CrossRef]
- Gong, G.; Guan, Y.-Y.; Zhang, Z.-L.; Rahman, K.; Wang, S.-J.; Zhou, S.; Luan, X.; Zhang, H. Isorhamnetin: A Review of Pharmacological Effects. Biomed. Pharmacother. 2020, 128, 110301. [Google Scholar] [CrossRef]
- Guo, H.; Ding, H.; Tang, X.; Liang, M.; Li, S.; Zhang, J.; Cao, J. Quercetin Induces Pro-Apoptotic Autophagy via SIRT1/AMPK Signaling Pathway in Human Lung Cancer Cell Lines A549 and H1299 in Vitro. Thorac. Cancer 2021, 12, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Amjad, E.; Sokouti, B.; Asnaashari, S. A Systematic Review of Anti-Cancer Roles and Mechanisms of Kaempferol as a Natural Compound. Cancer Cell Int. 2022, 22, 8. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, S.; Kim, E.J.; Park, J.H.; Yang, S.H.; Jeong, E.T.; Park, C.; Youn, M.J.; So, H.S.; Park, R. Suppression of Nrf2-Driven Heme Oxygenase-1 Enhances the Chemosensitivity of Lung Cancer A549 Cells toward Cisplatin. Lung Cancer 2008, 60, 47–56. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Fu, Q.; Ni, D.; Zhang, J.; Li, X.; Lu, S. The Chemical Diversity and Structure-Based Discovery of Allosteric Modulators for the PIF-Pocket of Protein Kinase PDK1. J. Enzyme Inhib. Med. Chem. 2019, 34, 361–374. [Google Scholar] [CrossRef]
- Agullo, G.; Gamet-Payrastre, L.; Manenti, S.; Viala, C.; Rémésy, C.; Chap, H.; Payrastre, B. Relationship between Flavonoid Structure and Inhibition of Phosphatidylinositol 3-Kinase: A Comparison with Tyrosine Kinase and Protein Kinase C Inhibition. Biochem. Pharmacol. 1997, 53, 1649–1657. [Google Scholar] [CrossRef]
- Aparicio, S. A Systematic Computational Study on Flavonoids. Int. J. Mol. Sci. 2010, 11, 2017–2038. [Google Scholar] [CrossRef]
- Suhail, M.; AlZahrani, W.M.; Shakil, S.; Tarique, M.; Tabrez, S.; Zughaibi, T.A.; Rehan, M. Analysis of Some Flavonoids for Inhibitory Mechanism against Cancer Target Phosphatidylinositol 3-Kinase (PI3K) Using Computational Tools. Front. Pharmacol. 2023, 14, 1236173. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Marfell, B.J.; Waterhouse, N.J. Analyzing Cell Death by Nuclear Staining with Hoechst 33342. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087205. [Google Scholar] [CrossRef]
- Chaisit, S.; Jianmongkol, S. Apoptosis Inducing Activity of Rhinacanthin-C in Doxorubicin-Resistant Breast Cancer MCF-7 Cells. Biol. Pharm. Bull. 2021, 44, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Jimoh, T.O.; Nuamnaichati, N.; Sungthong, R.; Chansriniyom, C.; Chanvorachote, P.; Likhitwitayawuid, K.; Chaotham, C.; Sritularak, B. Phytochemicals from Vanda Bensonii and Their Bioactivities to Inhibit Growth and Metastasis of Non-Small Cell Lung Cancer Cells. Molecules 2022, 27, 7902. [Google Scholar] [CrossRef]
- Wen, X.; Walle, T. Methylated Flavonoids Have Greatly Improved Intestinal Absorption and Metabolic Stability. Drug Metab. Dispos. 2006, 34, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Iida, M.; Keinath, S.; Iyi, F.F.; Mueck, K.; Fehrenbacher, B.; Mansour, W.Y.; Schaller, M.; Wheeler, D.L.; Rodemann, H.P. Dual Targeting of PI3K and MEK Enhances the Radiation Response of K-RAS Mutated Non-Small Cell Lung Cancer. Oncotarget 2016, 7, 43746–43761. [Google Scholar] [CrossRef]
- Shukuya, T.; Yamada, T.; Koenig, M.J.; Xu, J.; Okimoto, T.; Li, F.; Amann, J.M.; Carbone, D.P. The Effect of LKB1 Activity on the Sensitivity to PI3K/mTOR Inhibition in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 1061–1076. [Google Scholar] [CrossRef]
- Yamamoto, H.; Shigematsu, H.; Nomura, M.; Lockwood, W.W.; Sato, M.; Okumura, N.; Soh, J.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; et al. PIK3CA Mutations and Copy Number Gains in Human Lung Cancers. Cancer Res. 2008, 68, 6913–6921. [Google Scholar] [CrossRef]
- Zughaibi, T.A.; Suhail, M.; Tarique, M.; Tabrez, S. Targeting PI3K/Akt/mTOR Pathway by Different Flavonoids: A Cancer Chemopreventive Approach. Int. J. Mol. Sci. 2021, 22, 12455. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.-J.; Razi, S.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTOR Pathway in Lung Cancer; Oncogenic Alterations, Therapeutic Opportunities, Challenges, and a Glance at the Application of Nanoparticles. Transl. Oncol. 2022, 18, 101364. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-M.; Kim, H.-K.; Shim, W.; Anwar, M.A.; Kwon, J.-W.; Kwon, H.-K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef]
- Mirzaei, S.; Hushmandi, K.; Zabolian, A.; Saleki, H.; Torabi, S.M.R.; Ranjbar, A.; SeyedSaleh, S.; Sharifzadeh, S.O.; Khan, H.; Ashrafizadeh, M.; et al. Elucidating Role of Reactive Oxygen Species (ROS) in Cisplatin Chemotherapy: A Focus on Molecular Pathways and Possible Therapeutic Strategies. Molecules 2021, 26, 2382. [Google Scholar] [CrossRef]
- Liskova, A.; Samec, M.; Koklesova, L.; Brockmueller, A.; Zhai, K.; Abdellatif, B.; Siddiqui, M.; Biringer, K.; Kudela, E.; Pec, M.; et al. Flavonoids as an Effective Sensitizer for Anti-Cancer Therapy: Insights into Multi-Faceted Mechanisms and Applicability towards Individualized Patient Profiles. EPMA J. 2021, 12, 155–176. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, M.; Huang, H.; Jin, W.-L. Drug Repurposing for Cancer Therapy. Signal Transduct. Target. Ther. 2024, 9, 92. [Google Scholar] [CrossRef]
- Chaisit, T.; Siripong, P.; Jianmongkol, S. Rhinacanthin-C Enhances Doxorubicin Cytotoxicity via Inhibiting the Functions of P-Glycoprotein and MRP2 in Breast Cancer Cells. Eur. J. Pharmacol. 2017, 795, 50–57. [Google Scholar] [CrossRef]
- Witayateeraporn, W.; Arunrungvichian, K.; Pothongsrisit, S.; Doungchawee, J.; Vajragupta, O.; Pongrakhananon, V. A7-Nicotinic Acetylcholine Receptor Antagonist QND7 Suppresses Non-Small Cell Lung Cancer Cell Proliferation and Migration via Inhibition of Akt/mTOR Signaling. Biochem. Biophys. Res. Commun. 2020, 521, 977–983. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The Soft Agar Colony Formation Assay. J. Vis. Exp. JoVE 2014, 92, e51998. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Murphy, D. Tutorial: Application of Ggplot2 to Pharmacometric Graphics. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e79. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, L.; Wang, J.; Zhang, M.; Song, Z.; Ni, B.; You, Y. Identification of Key Biomarkers and Immune Infiltration in Systemic Lupus Erythematosus by Integrated Bioinformatics Analysis. J. Transl. Med. 2021, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Gao, Z.; Zhai, X.; Yan, G.; Tian, Y.; Huang, X.; Wu, Q.; Yuan, L.; Su, L. Bioinformatics Analyses of Gene Expression Profile to Identify Pathogenic Mechanisms for COVID-19 Infection and Cutaneous Lupus Erythematosus. Front. Immunol. 2023, 14, 1268912. [Google Scholar] [CrossRef]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved Services and an Expanding Collection of Metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved Protein–Ligand Blind Docking by Integrating Cavity Detection, Docking and Homologous Template Fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NSCLC Cell Lines/Normal Mouse Fibroblast Embryonic Cell Lines | a IC50 of Isalpinin (μM ± SEM) | a IC50 of Cisplatin (μM ± SEM) | ||
|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |
| A549 | 105.2 ± 14.46 | 81.88 ± 23.36 | 48.03 ± 4.71 * | 19.17 ± 3.34 * |
| H23 | 100.3 ± 24.39 | 44.34 ± 16.34 * | 86.34 ± 8.91 | 33.39 ± 2.95 * |
| H460 | 103.1 ± 29.42 | 44.46 ± 13.40 * | 66.55 ± 7.59 | 41.43 ± 8.58 * |
| NIH/3T3 | >100 | >100 | 36.25 ± 5.64 | 14.65 ± 2.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pouyfung, P.; Lertnitikul, N.; Bai, H.; Chookaew, A.; Pongrakhananon, V.; Chonsut, P.; Chaisit, S. Antitumor Activity of Isalpinin from Paphiopedilum dianthum on Non-Small Cell Lung Cancer Cell Lines. Molecules 2025, 30, 2762. https://doi.org/10.3390/molecules30132762
Pouyfung P, Lertnitikul N, Bai H, Chookaew A, Pongrakhananon V, Chonsut P, Chaisit S. Antitumor Activity of Isalpinin from Paphiopedilum dianthum on Non-Small Cell Lung Cancer Cell Lines. Molecules. 2025; 30(13):2762. https://doi.org/10.3390/molecules30132762
Chicago/Turabian StylePouyfung, Phisit, Nonthalert Lertnitikul, Hua Bai, Achitphol Chookaew, Varisa Pongrakhananon, Piriya Chonsut, and Suwichak Chaisit. 2025. "Antitumor Activity of Isalpinin from Paphiopedilum dianthum on Non-Small Cell Lung Cancer Cell Lines" Molecules 30, no. 13: 2762. https://doi.org/10.3390/molecules30132762
APA StylePouyfung, P., Lertnitikul, N., Bai, H., Chookaew, A., Pongrakhananon, V., Chonsut, P., & Chaisit, S. (2025). Antitumor Activity of Isalpinin from Paphiopedilum dianthum on Non-Small Cell Lung Cancer Cell Lines. Molecules, 30(13), 2762. https://doi.org/10.3390/molecules30132762