
Identification and Characterization of Novel Inhibitors of Human Poly(ADP-Ribose) Polymerase-1

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. In Silico Pre-Selection of Potential PARPi
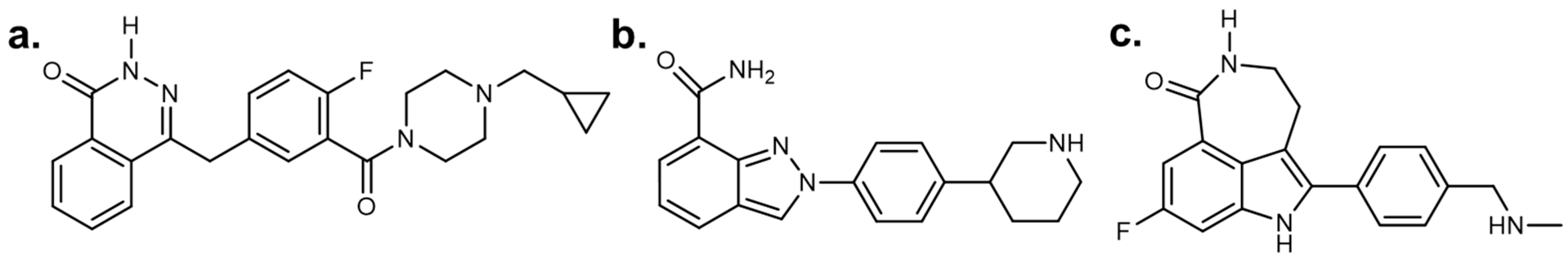
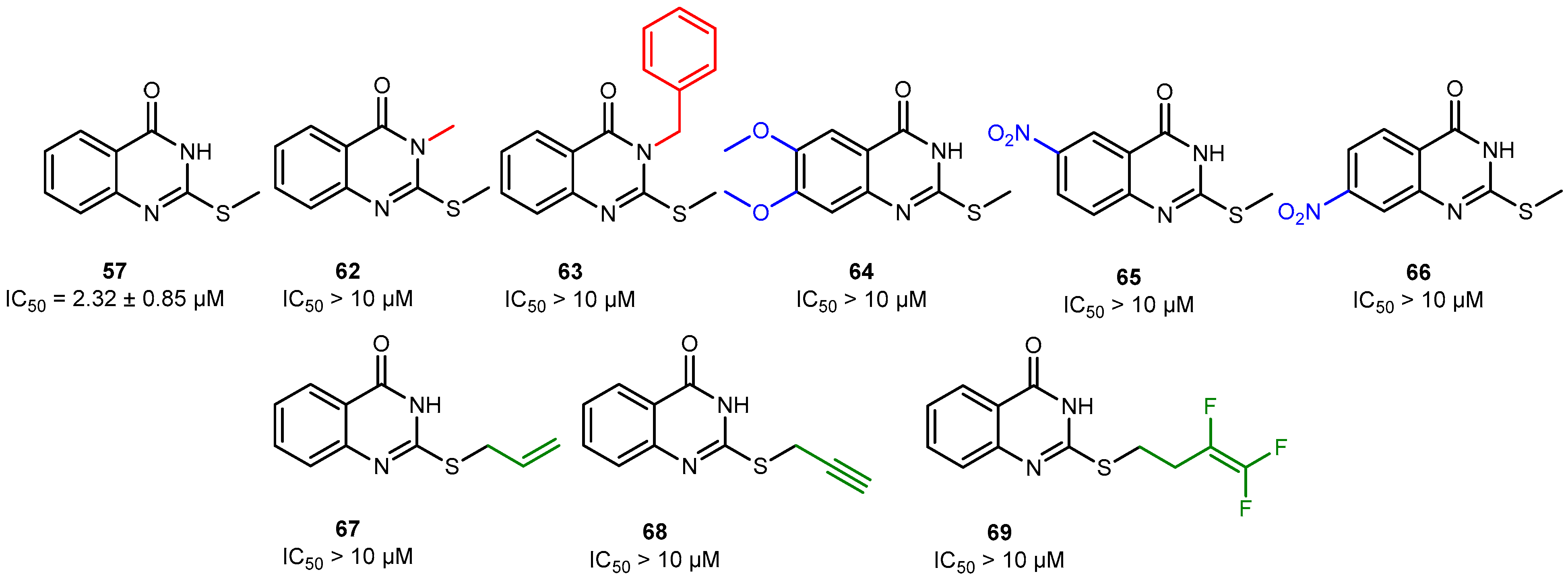
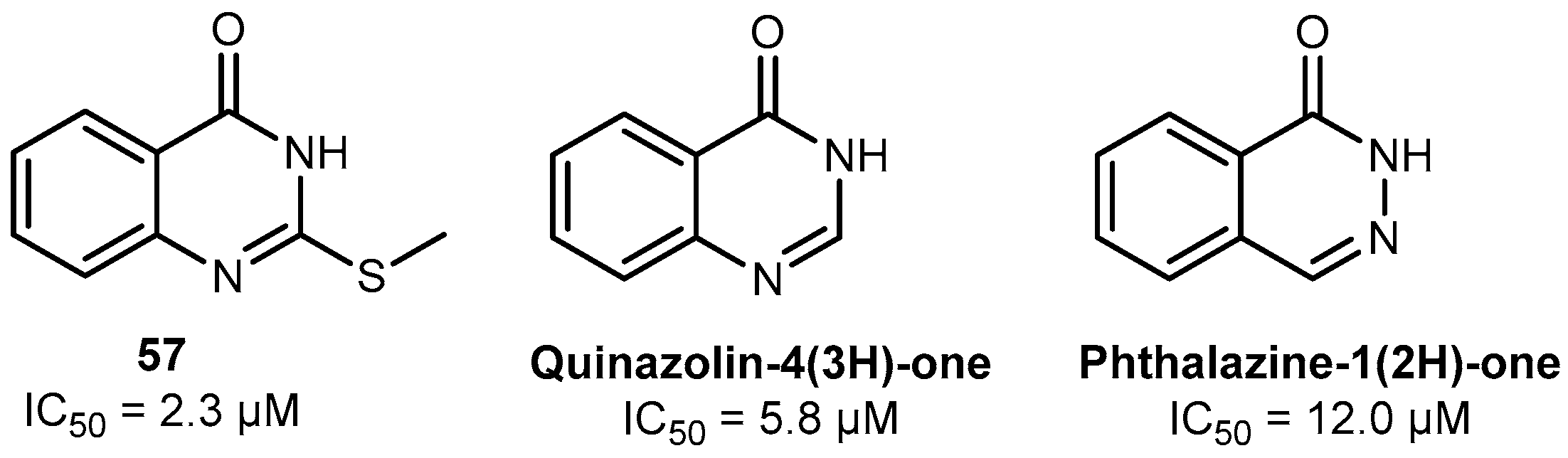
2.2. Discovery of Eight Novel PARP Inhibitors
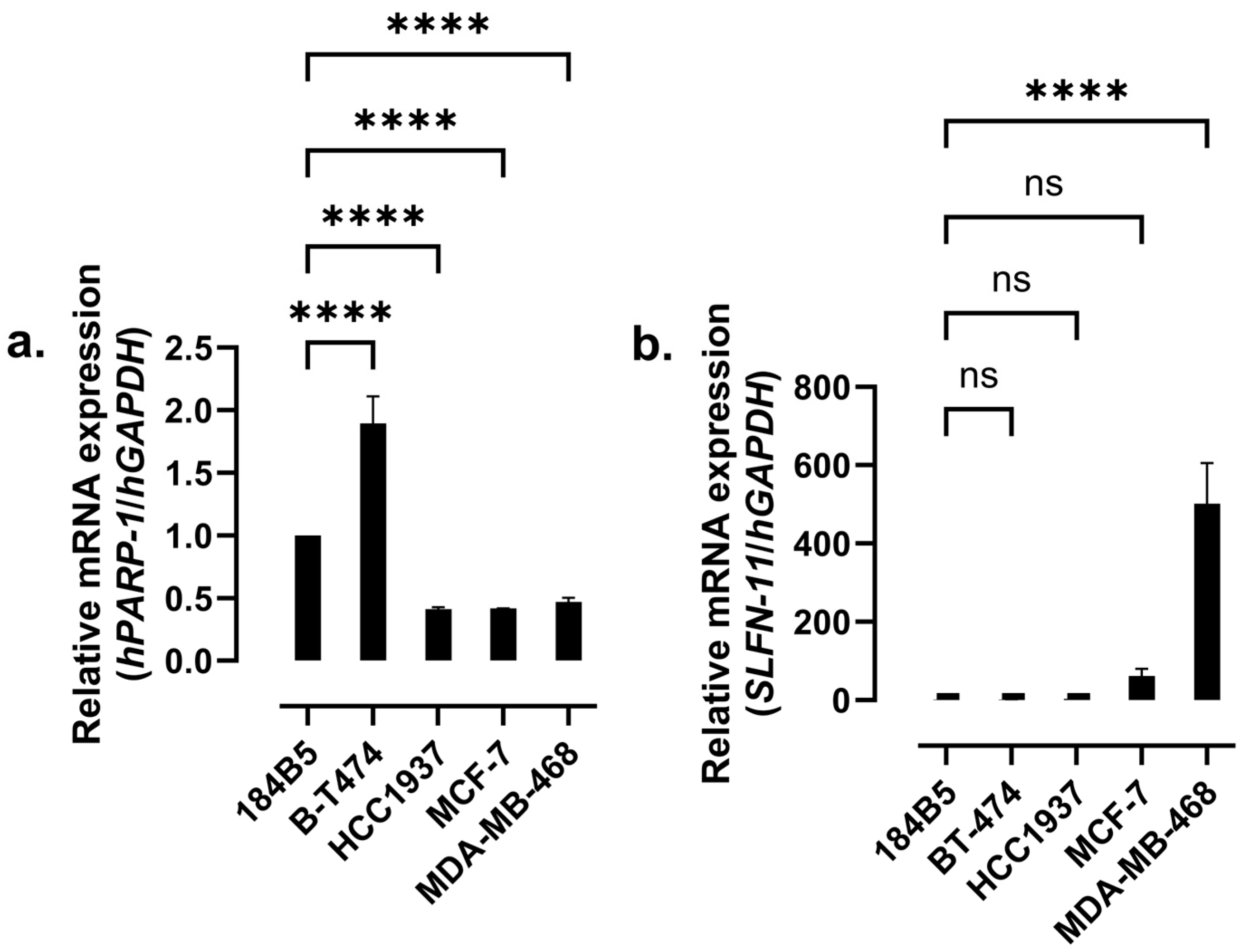
2.3. mRNA Expression Levels of hPARP-1 and SLFN-11
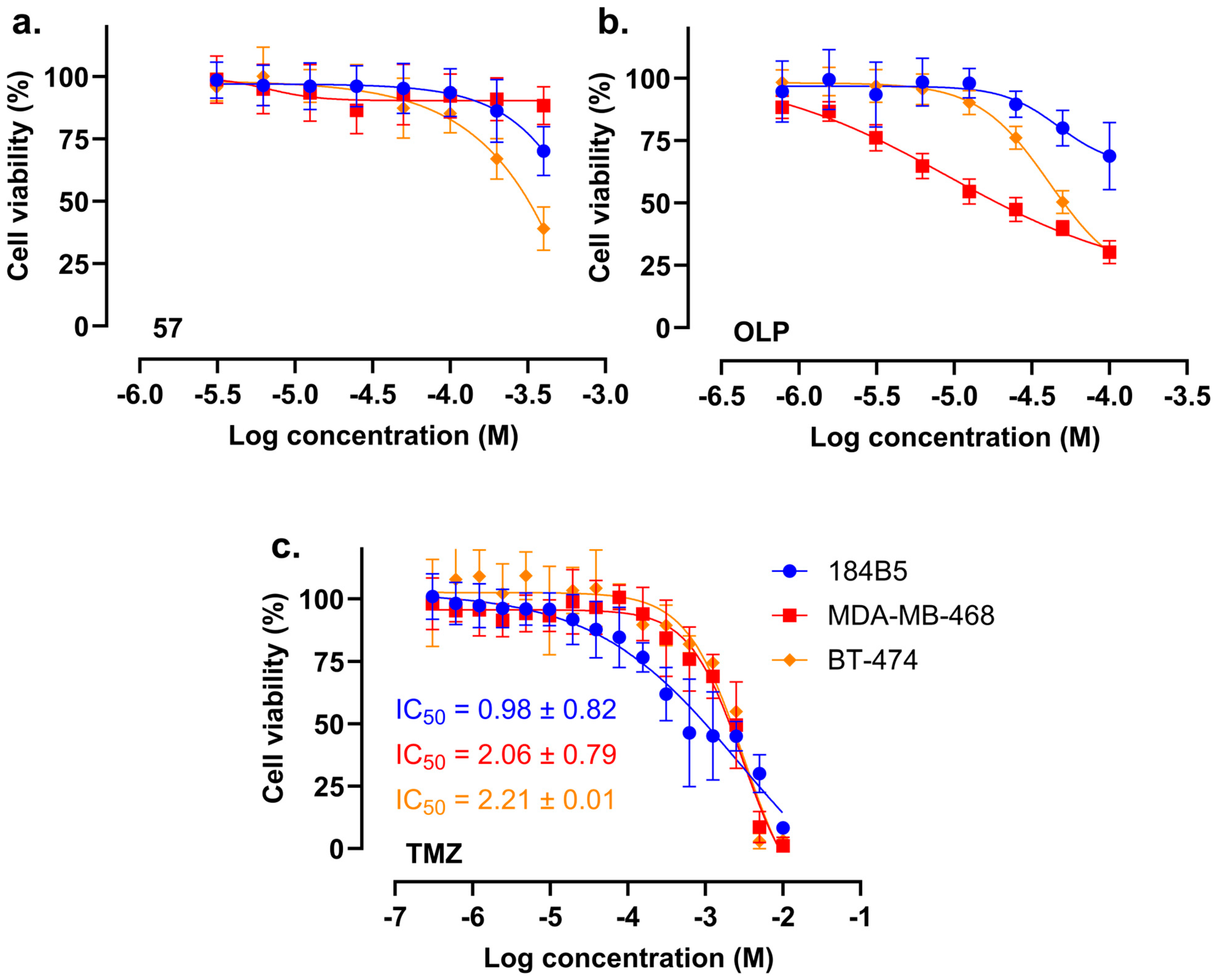
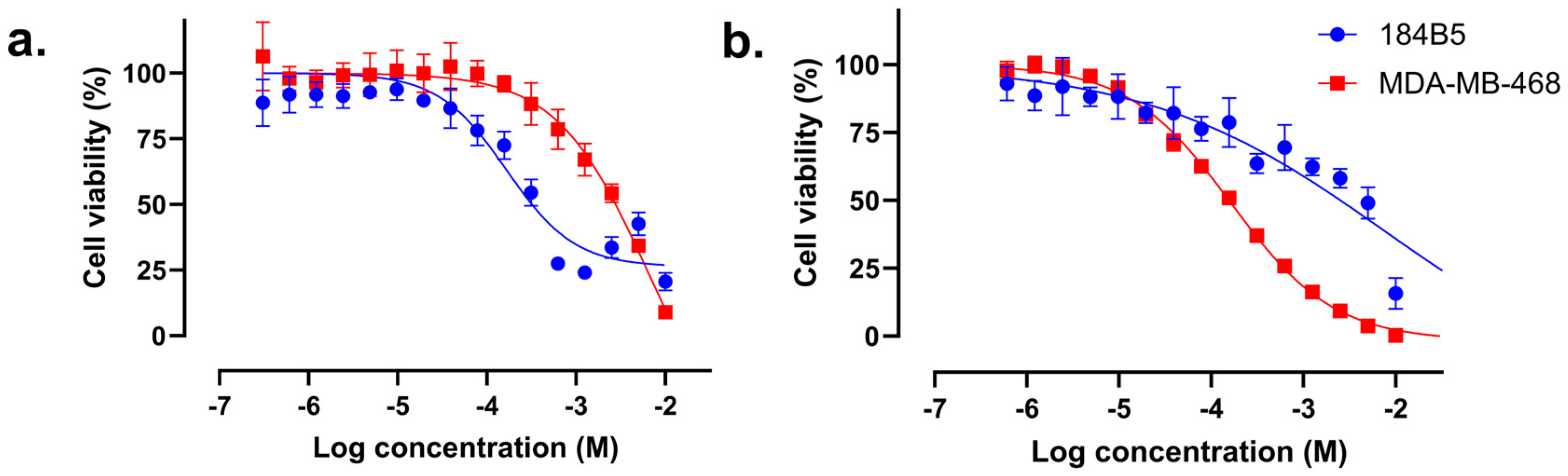
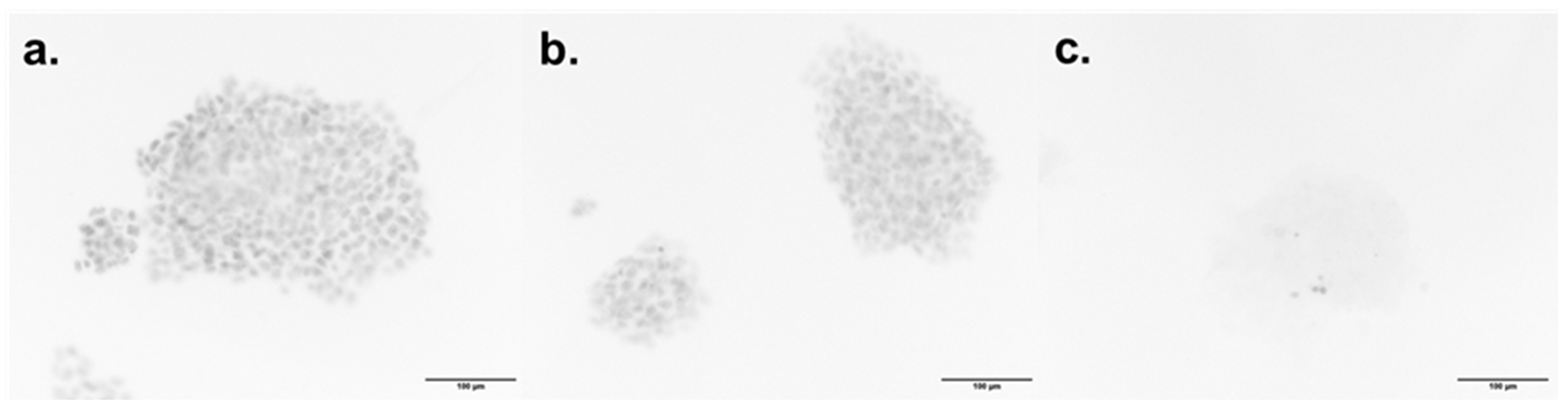
2.4. In Vitro Cytotoxicity of Compound 57
2.5. Compound 57 Did Not Exhibit Synergistic Effect in Combination with TMZ
2.6. Compound 57 Does Not Reach the Nucleus-Located hPARP-1 Enzyme
3. Discussion
4. Materials and Methods
4.1. Chemicals and Cell Lines
4.2. In Silico PARP-PARPi Molecular Docking
4.3. hPARP-1 Enzymatic Assay (New Variation)
4.4. Differential Gene Expression
4.5. In Vitro Cell Viability Assay
4.6. In Vitro Synergistic Cell Viability Assay
4.7. Fluorescence Microscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological types of breast cancer: How special are they? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Briest, S.; Stearns, V. Tamoxifen metabolism and its effect on endocrine treatment of breast cancer. Clin. Adv. Hematol. Oncol. HO 2009, 7, 185–192. [Google Scholar]
- Morgan, M.M.; Arendt, L.M.; Alarid, E.T.; Beebe, D.J.; Johnson, B.P. Mammary adipose stromal cells derived from obese women reduce sensitivity to the aromatase inhibitor anastrazole in an organotypic breast model. FASEB J. 2019, 33, 8623–8633. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.H.E.; Kyle, A.H.; Reinsberg, S.A.; Moosvi, F.; Patrick, H.M.; Cran, J.; Saatchi, K.; Häfeli, U.; Minchinton, A.I. Heterogeneous distribution of trastuzumab in HER2-positive xenografts and metastases: Role of the tumor microenvironment. Clin. Exp. Metastasis 2018, 35, 691–705. [Google Scholar] [CrossRef]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. New Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef]
- Tečić Vuger, A.; Šeparović, R.; Vazdar, L.; Pavlović, M.; Lepetić, P.; Šitić, S.; Bajić, Ž.; Šarčević, B.; Vrbanec, D. Characteristics and Prognosis of Triple-Negative Breast Cancer Patients: A Croatian Single Institution Retrospective Cohort Study. Acta Clin. Croat. 2020, 59, 97–108. [Google Scholar] [CrossRef]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef]
- Rissel, D.; Heym, P.P.; Thor, K.; Brandt, W.; Wessjohann, L.A.; Peiter, E. No silver bullet—Canonical Poly(ADP-Ribose) Polymerases (PARPs) are no universal factors of abiotic and biotic stress resistance of Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 59. [Google Scholar] [CrossRef]
- Lüscher, B.; Bütepage, M.; Eckei, L.; Krieg, S.; Verheugd, P.; Shilton, B.H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018, 118, 1092–1136. [Google Scholar] [CrossRef]
- Matta, E.; Kiribayeva, A.; Khassenov, B.; Matkarimov, B.T.; Ishchenko, A.A. Insight into DNA substrate specificity of PARP1-catalysed DNA poly(ADP-ribosyl)ation. Sci. Rep. 2020, 10, 3699. [Google Scholar] [CrossRef] [PubMed]
- van Beek, L.; McClay, É.; Patel, S.; Schimpl, M.; Spagnolo, L.; Maia de Oliveira, T. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22, 5112. [Google Scholar] [CrossRef]
- Ali, A.A.E.; Timinszky, G.; Arribas-Bosacoma, R.; Kozlowski, M.; Hassa, P.O.; Hassler, M.; Ladurner, A.G.; Pearl, L.H.; Oliver, A.W. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012, 19, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef]
- Herceg, Z.; Wang, Z.-Q. Functions of poly (ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2001, 477, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, P.A.; Cuneo, M.J.; Mueller, G.A.; DeRose, E.F.; Gabel, S.A.; London, R.E. Structural studies of the PARP-1 BRCT domain. BMC Struct. Biol. 2011, 11, 37. [Google Scholar] [CrossRef]
- Cuneo, M.J.; Gabel, S.A.; Krahn, J.M.; Ricker, M.A.; London, R.E. The structural basis for partitioning of the XRCC1/DNA ligase III-α BRCT-mediated dimer complexes. Nucleic Acids Res. 2011, 39, 7816–7827. [Google Scholar] [CrossRef]
- Beniey, M.; Haque, T.; Hassan, S. Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience 2019, 6, 287. [Google Scholar] [CrossRef]
- Spigel, D.R. PARP Inhibitors in Lung Cancer. J. Thorac. Oncol. 2012, 7, S392–S393. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Farago, A.F.; Yeap, B.Y.; Stanzione, M.; Hung, Y.P.; Heist, R.S.; Marcoux, J.P.; Zhong, J.; Rangachari, D.; Barbie, D.A.; Phat, S.; et al. Combination Olaparib and Temozolomide in Relapsed Small-Cell Lung Cancer. Cancer Discov. 2019, 9, 1372–1387. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, M.; Mountzios, G.; Papadimitriou, C.A. The role of PARP inhibition in triple-negative breast cancer: Unraveling the wide spectrum of synthetic lethality. Cancer Treat. Rev. 2018, 67, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Y.; Zhang, C.; Chu, J.; Wu, Y.; Li, Y.; Liu, J.; Li, Q.; Li, S.; Shi, Q.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9, 1595. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Zoppoli, G.; Regairaz, M.; Leo, E.; Reinhold, W.C.; Varma, S.; Ballestrero, A.; Doroshow, J.H.; Pommier, Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. USA 2012, 109, 15030–15035. [Google Scholar] [CrossRef]
- Coleman, N.; Zhang, B.; Byers, L.A.; Yap, T.A. The role of Schlafen 11 (SLFN11) as a predictive biomarker for targeting the DNA damage response. Br. J. Cancer 2021, 124, 857–859. [Google Scholar] [CrossRef]
- Stewart, C.A.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef]
- Wang, R.; Cong, Y.; Li, M.; Bao, J.; Qi, Y.; Zhang, J.Z.H. Molecular Mechanism of Selective Binding of NMS-P118 to PARP-1 and PARP-2: A Computational Perspective. Front. Mol. Biosci. 2020, 7, 50. [Google Scholar] [CrossRef]
- Heym, P.-P.; Brandt, W.; Wessjohann, L.A.; Niclas, H.-J. Virtual screening for plant PARP inhibitors—What can be learned from human PARP inhibitors? J. Cheminformatics 2012, 4, O24. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Differential trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Ryu, H.; Ahn, J.; Choi, H.K. Novel Benzamide Derivatives: Synthesis and Bioactivity as Potent PARP-1 Inhibitors. Bull. Korean Chem. Soc. 2017, 38, 935–943. [Google Scholar] [CrossRef]
- Berger, R.; Wessjohann, L.; Gonzalez Ceballos, L.; Heym, P.P. Quinazolinones as Phytoeffectors. Patent EP 24,190,327.7, 2024. [Google Scholar]
- Berger, R. Design, Synthesis and Evaluation of Drought Stress Tolerance-Inducing Compounds. Ph.D. Thesis, Martin-Luther-Universität Halle-Wittenberg, Halle (Saale), Germany, 2023.
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A Novel Bioavailable Inhibitor of Poly(ADP-ribose) Polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Maksimainen, M.M.; Murthy, S.; Sowa, S.T.; Galera-Prat, A.; Rolina, E.; Heiskanen, J.P.; Lehtiö, L. Analogs of TIQ-A as inhibitors of human mono-ADP-ribosylating PARPs. Bioorganic Med. Chem. 2021, 52, 116511. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.J.; Srinivasan, S.; Bowman, K.; Calvert, A.H.; Curtin, N.J.; Newell, D.R.; Pemperton, L.C.; Golding, B.T. Resistance-modifying agents. 5. Synthesis and biological properties of quinazolinone inhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP). J. Med. Chem. 1998, 41, 5247–5256. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Singh, S.; Shah, J.R.; Low, W.-K.; Talele, T.T. Synthesis and SAR optimization of quinazolin-4(3H)-ones as poly(ADP-ribose)polymerase-1 inhibitors. Eur. J. Med. Chem. 2012, 50, 264–273. [Google Scholar] [CrossRef]
- Rudolph, J.; Jung, K.; Luger, K. Inhibitors of PARP: Number crunching and structure gazing. Proc. Natl. Acad. Sci. USA 2022, 119, e2121979119. [Google Scholar] [CrossRef]
- Tutt, A.N.J.; Lord, C.J.; McCabe, N.; Farmer, H.; Turner, N.; Martin, N.M.; Jackson, S.P.; Smith, G.C.M.; Ashworth, A. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 139–148. [Google Scholar] [CrossRef]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.-W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP Side of the Nucleus: Molecular Actions, Physiological Outcomes, and Clinical Targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Banasik, M.; Komura, H.; Shimoyama, M.; Ueda, K. Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. J. Biol. Chem. 1992, 267, 1569–1575. [Google Scholar] [CrossRef]
- Cockcroft, X.; Dillon, K.J.; Dixon, L.; Drzewiecki, J.; Kerrigan, F.; Loh, V.M.; Martin, N.M.B.; Menear, K.A.; Smith, G.C.M. Phthalazinones 2: Optimisation and synthesis of novel potent inhibitors of poly(ADP-ribose)polymerase. Bioorganic Med. Chem. Lett. 2006, 16, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Johannes, J.W.; Almeida, L.; Daly, K.; Ferguson, A.D.; Grosskurth, S.E.; Guan, H.; Howard, T.; Ioannidis, S.; Kazmirski, S.; Lamb, M.L.; et al. Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorganic Med. Chem. Lett. 2015, 25, 5743–5747. [Google Scholar] [CrossRef] [PubMed]
- Coussens, N.P.; Braisted, J.C.; Peryea, T.; Sittampalam, G.S.; Simeonov, A.; Hall, M.D. Small-Molecule Screens: A Gateway to Cancer Therapeutic Agents with Case Studies of Food and Drug Administration-Approved Drugs. Pharmacol. Rev. 2017, 69, 479–496. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Ruberte, A.C.; Plano, D.; Encío, I.; Aydillo, C.; Sharma, A.K.; Sanmartín, C. Novel selenadiazole derivatives as selective antitumor and radical scavenging agents. Eur. J. Med. Chem. 2018, 157, 14–27. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Nottbohm, A.C.; Dothager, R.S.; Putt, K.S.; Hoyt, M.T.; Hergenrother, P.J. A Colorimetric Substrate for Poly(ADP-Ribose) Polymerase-1, VPARP, and Tankyrase-1. Angew. Chem. 2007, 119, 2112–2115. [Google Scholar] [CrossRef]
- Kanamori, K.S.; de Oliveira, G.C.; Auxiliadora-Martins, M.; Schoon, R.A.; Reid, J.M.; Chini, E.N. Two Different Methods of Quantification of Oxidized Nicotinamide Adenine Dinucleotide (NAD+) and Reduced Nicotinamide Adenine Dinucleotide (NADH) Intracellular Levels: Enzymatic Coupled Cycling Assay and Ultra-performance LiquidChromatography (UPLC)-Mass Spectrometry. Bio-Protoc. 2018, 8, e2937. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(–delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath 2013, 3, 71–85. [Google Scholar]
- Rahn, J.; Lennicke, C.; Kipp, A.P.; Müller, A.S.; Wessjohann, L.A.; Lichtenfels, R.; Seliger, B. Altered protein expression pattern in colon tissue of mice upon supplementation with distinct selenium compounds. Proteomics 2017, 17, 1600486. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087379. [Google Scholar] [CrossRef] [PubMed]
- Seixas, N.; Ravanello, B.B.; Morgan, I.; Kaluđerović, G.N.; Wessjohann, L.A. Chlorambucil Conjugated Ugi Dendrimers with PAMAM-NH2 Core and Evaluation of Their Anticancer Activity. Pharmaceutics 2019, 11, 59. [Google Scholar] [CrossRef]
- Sultani, H.N.; Morgan, I.; Hussain, H.; Roos, A.H.; Haeri, H.H.; Kaluđerović, G.N.; Hinderberger, D.; Westermann, B. Access to New Cytotoxic Triterpene and Steroidal Acid-TEMPO Conjugates by Ugi Multicomponent-Reactions. Int. J. Mol. Sci. 2021, 22, 7125. [Google Scholar] [CrossRef] [PubMed]
- Morgan, I.; Wessjohann, L.A.; Kaluđerović, G.N. In Vitro Anticancer Screening and Preliminary Mechanistic Study of A-Ring Substituted Anthraquinone Derivatives. Cells 2022, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.; Lee, J.-H.; Kim, S.H.; Go, K.-O.; Ji, S.Y.; Han, J.H.; Kim, C.-Y. The Combination PARP Inhibitor Olaparib With Temozolomide in an Experimental Glioblastoma Model. In Vivo 2021, 35, 2015–2023. [Google Scholar] [CrossRef]
- Irwin, C.P.; Portorreal, Y.; Brand, C.; Zhang, Y.; Desai, P.; Salinas, B.; Weber, W.A.; Reiner, T. PARPi-FL—A Fluorescent PARP1 Inhibitor for Glioblastoma Imaging. Neoplasia 2014, 16, 432–440. [Google Scholar] [CrossRef]
- Salinas, B.; Irwin, C.P.; Kossatz, S.; Bolaender, A.; Chiosis, G.; Pillarsetty, N.; Weber, W.A.; Reiner, T. Radioiodinated PARP1 tracers for glioblastoma imaging. EJNMMI Res. 2015, 5, 46. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment\IC50 [mM] | 184B5 | MDA-MB-468 |
|---|---|---|
| TMZ | 0.98 ± 0.82 | 2.06 ± 0.79 |
| TMZ + compound 57 | 0.32 ± 0.07 | 2.77 ± 0.25 |
| TMZ + OLP | 4.95 ± 3.90 | 0.13 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, I.; Rennert, R.; Berger, R.; Hassanin, A.; Davari, M.D.; Eisenschmidt-Bönn, D.; Wessjohann, L.A. Identification and Characterization of Novel Inhibitors of Human Poly(ADP-Ribose) Polymerase-1. Molecules 2025, 30, 2728. https://doi.org/10.3390/molecules30132728
Morgan I, Rennert R, Berger R, Hassanin A, Davari MD, Eisenschmidt-Bönn D, Wessjohann LA. Identification and Characterization of Novel Inhibitors of Human Poly(ADP-Ribose) Polymerase-1. Molecules. 2025; 30(13):2728. https://doi.org/10.3390/molecules30132728
Chicago/Turabian StyleMorgan, Ibrahim, Robert Rennert, Robert Berger, Ahmed Hassanin, Mehdi D. Davari, Daniela Eisenschmidt-Bönn, and Ludger A. Wessjohann. 2025. "Identification and Characterization of Novel Inhibitors of Human Poly(ADP-Ribose) Polymerase-1" Molecules 30, no. 13: 2728. https://doi.org/10.3390/molecules30132728
APA StyleMorgan, I., Rennert, R., Berger, R., Hassanin, A., Davari, M. D., Eisenschmidt-Bönn, D., & Wessjohann, L. A. (2025). Identification and Characterization of Novel Inhibitors of Human Poly(ADP-Ribose) Polymerase-1. Molecules, 30(13), 2728. https://doi.org/10.3390/molecules30132728