
On the Question of the Course of the Hetero Diels–Alder Reactions Between N-(2,2,2-Trichloroethylidene)Carboxamides and Dicyclohexylcarbodiimide: A New Case of the Stepwise Zwitterionic Cycloaddition Process



Abstract
1. Introduction
2. Results and Discussion
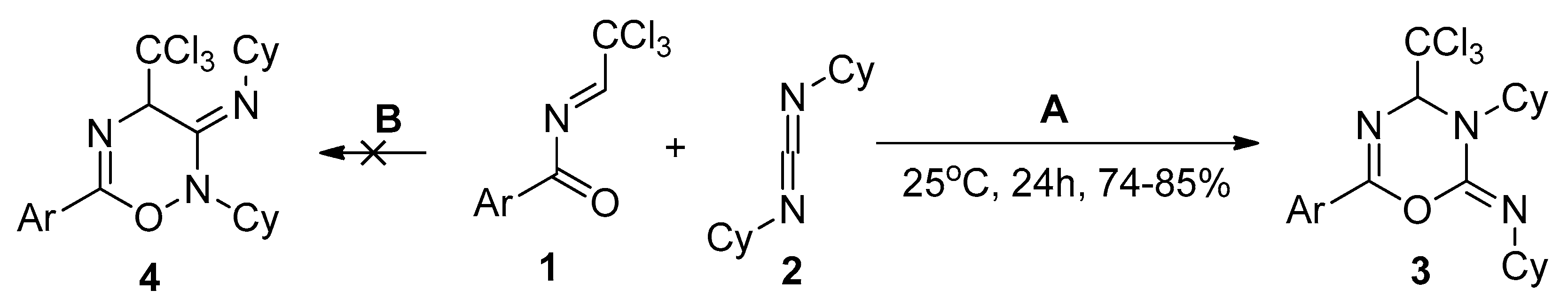
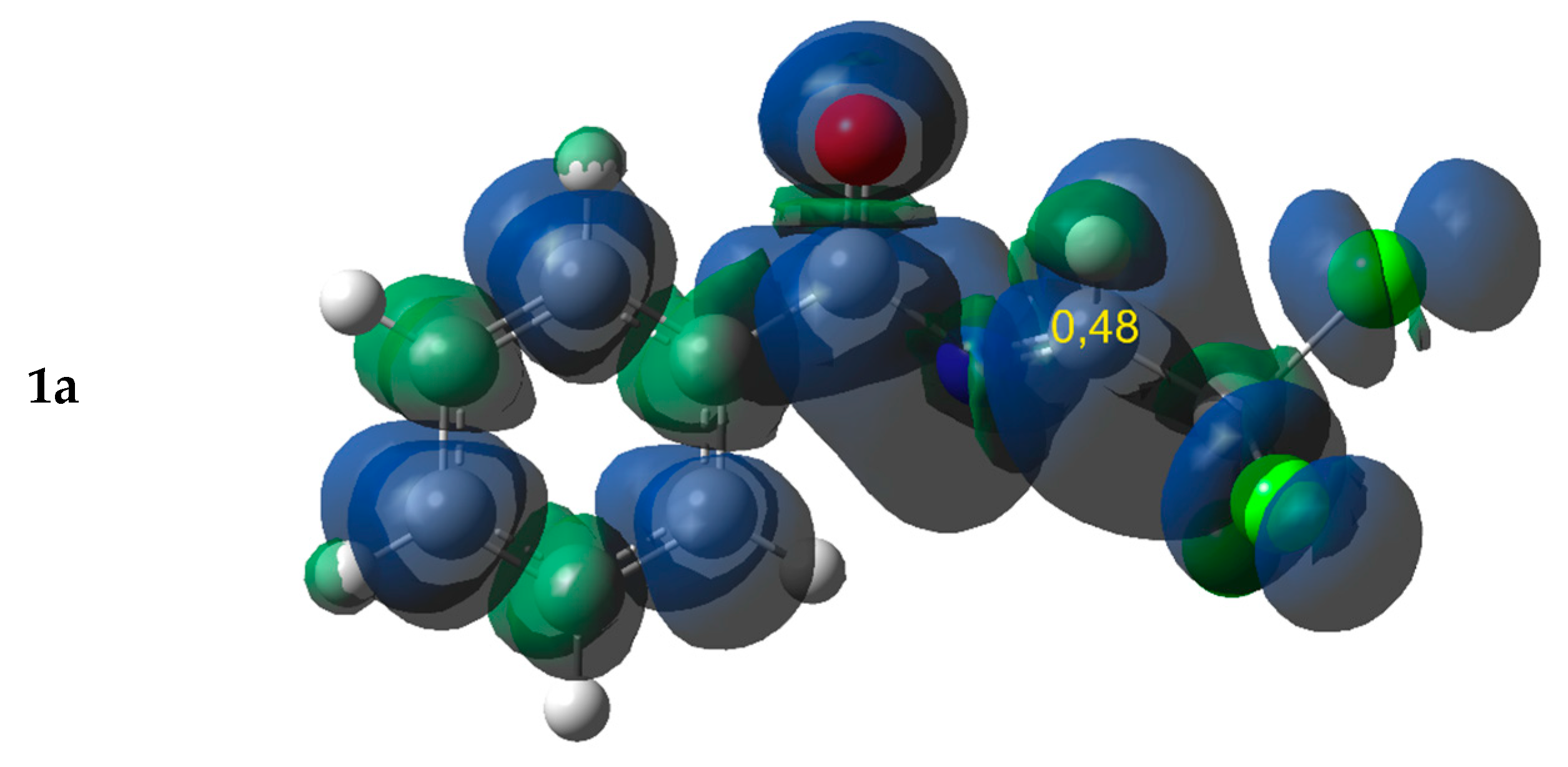
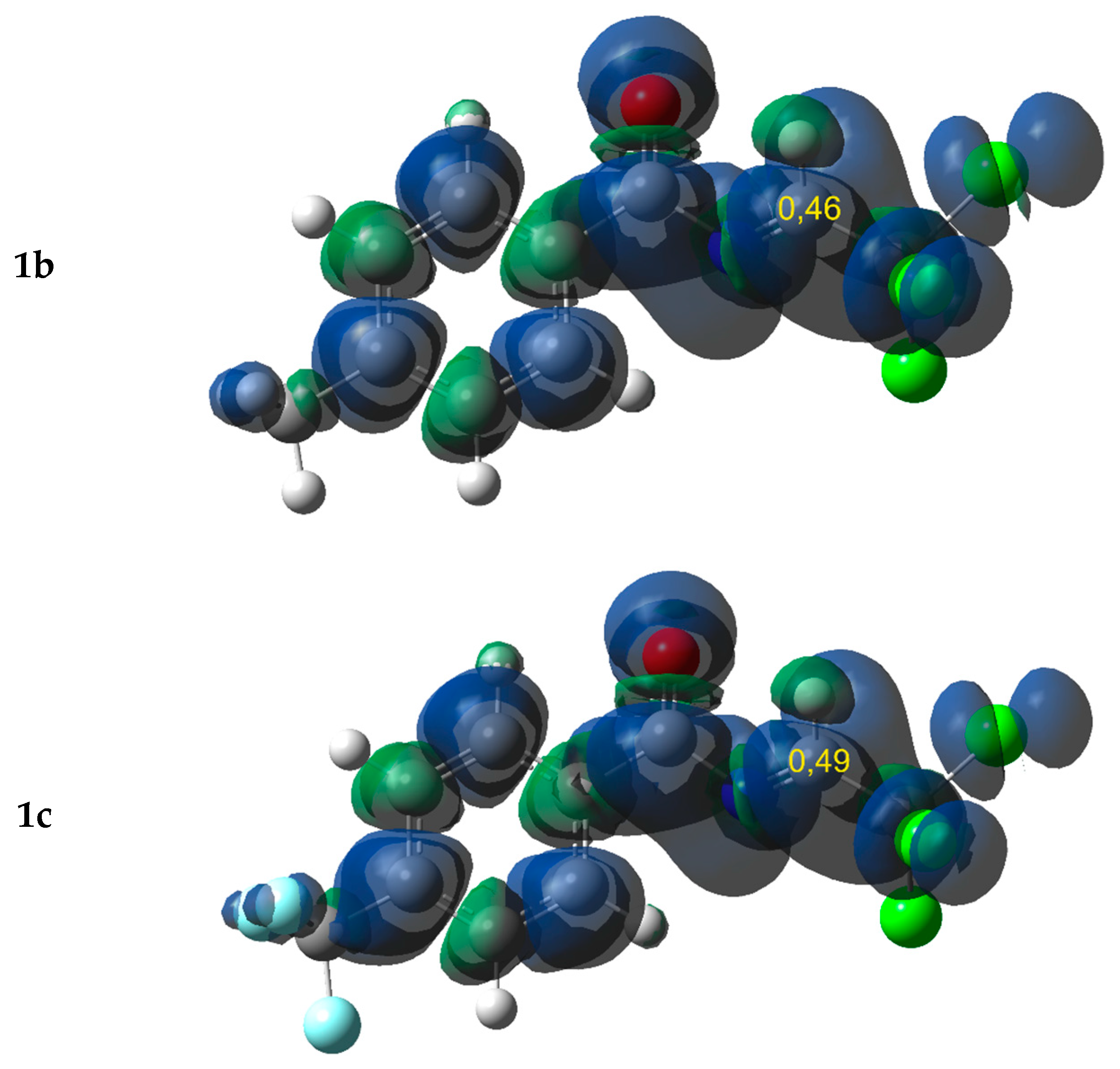
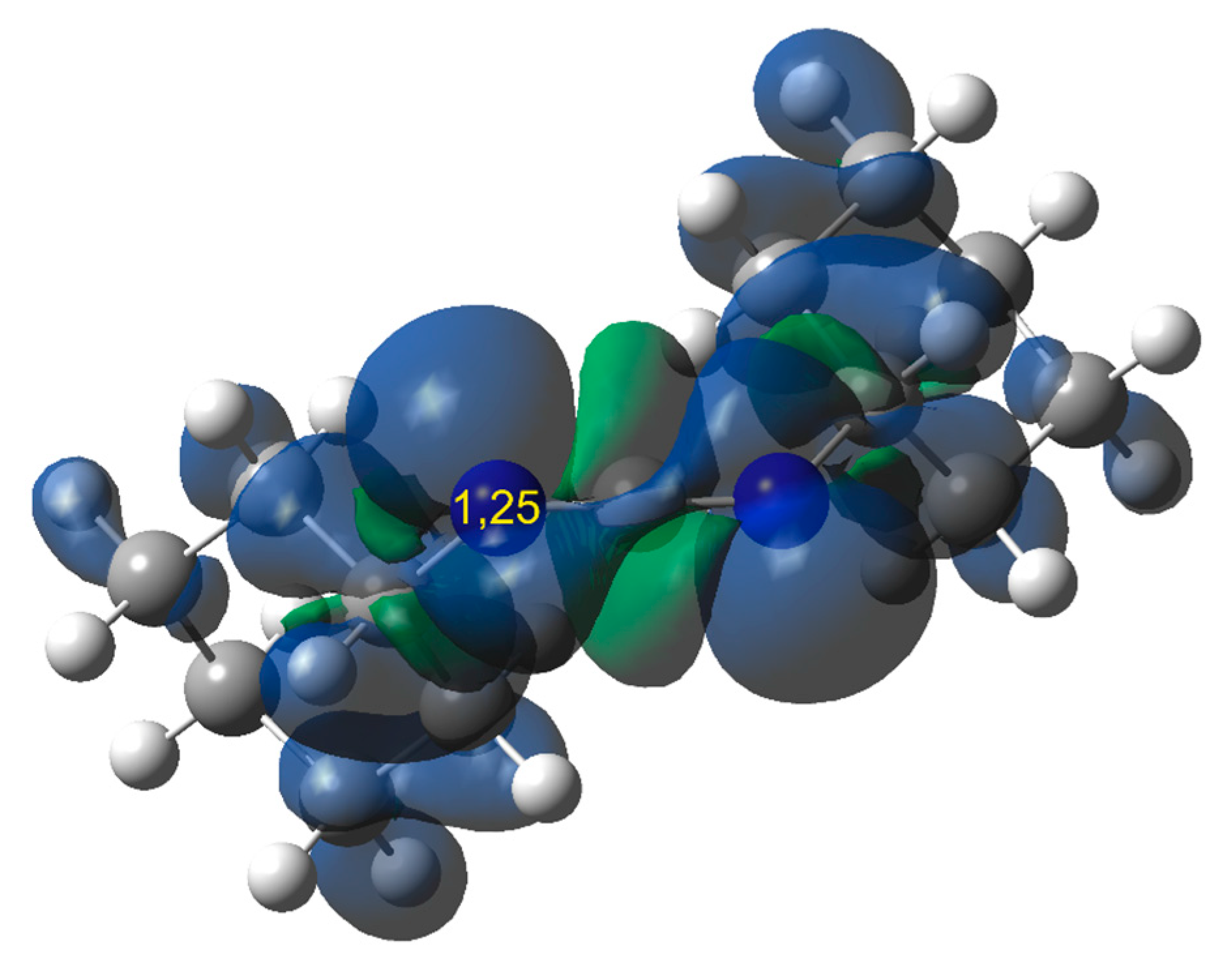
2.1. Electronic Properties and the Nature of Intermolecular Interactions of Cycloaddition Components
2.2. Critical Points on Reaction Profiles and Critical Structures
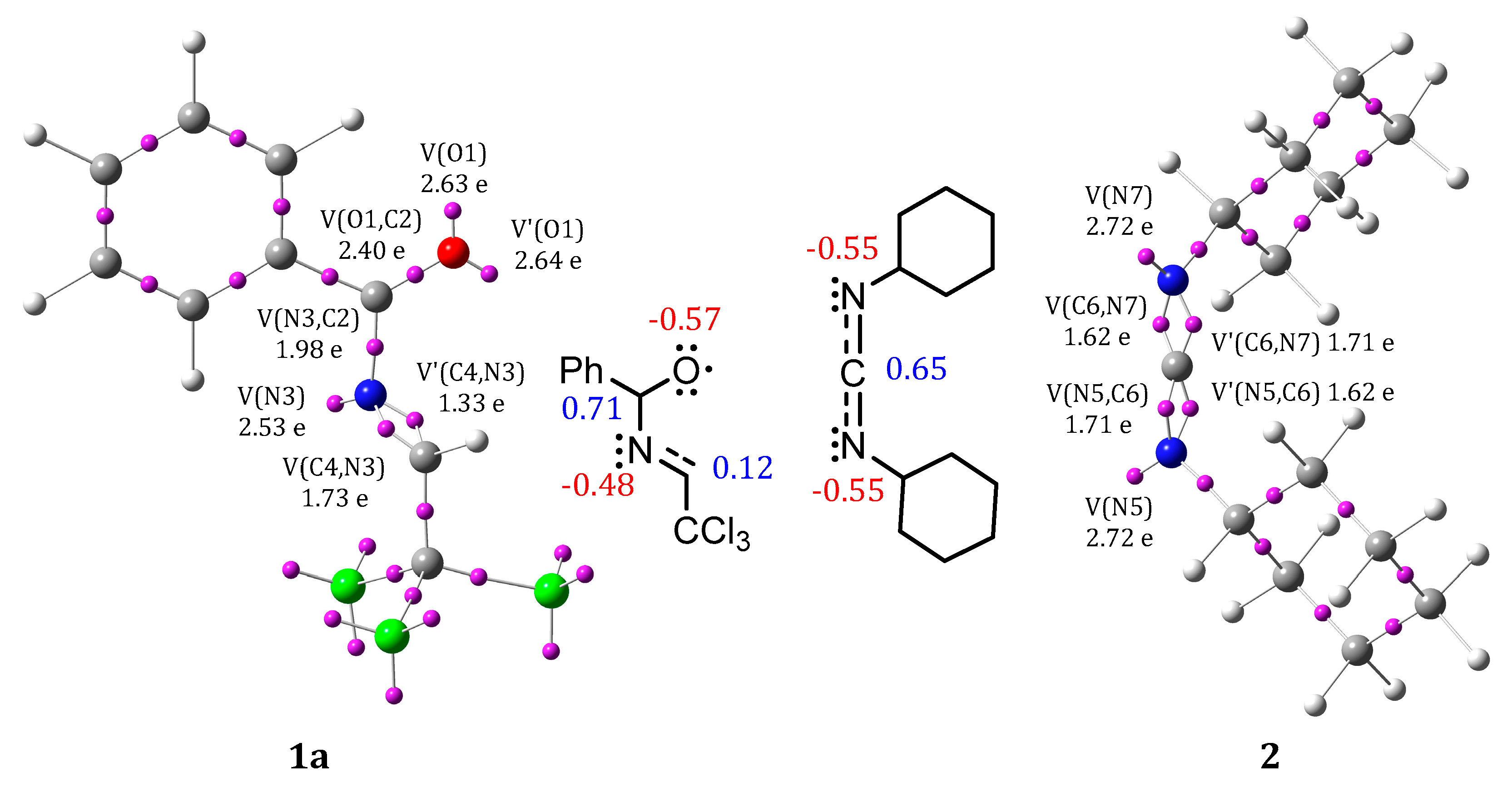
2.3. The ELF Study of the Reagents and BET Study of Mechanism of the Model Reaction of 1a and 2
3. Computational Details
η ≈ ELUMO − EHOMO
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koubi, Y.; Moukhliss, Y.; Hajji, H.; Abdessadak, O.; Alaqarbeh, M.; Ajana, M.A.; Maghat, H.; Lakhlifi, T.; Bouachrine, M. Computational structure—Biological activity and retrosynthesis investigations of 1,2,3-triazole-quinoline hybrid molecules as potential respiratory virus inhibitors. Chem. Heterocycl. Comp. 2024, 60, 491–504. [Google Scholar] [CrossRef]
- Abbasi, M.; Farnia, S.M.F.; Tahghighi, A. The possibility of applying some heteroatom-decorated g-C3N4 heterocyclic nanosheets for delivering 5-aminosalicylic acid anti-inflammatory agent. Chem. Heterocycl. Comp. 2024, 60, 655–662. [Google Scholar] [CrossRef]
- Elbouhi, M.; Tabti, K.; Ouabane, M.; Alaqarbeh, M.; Elkamel, K.; Lakhlifi, T.; Sbai, A.; Bouachrine, M. A computational exploration of the antioxidant potential of conjugated quinazolinone Schiff bases. Chem. Heterocycl. Comp. 2024, 60, 627–638. [Google Scholar] [CrossRef]
- Gassim, H.B.M.A.; Hassan, M.; Abadi, R.S.M.; Mustafa, Y.A.A. Phytochemical constituents and antioxidant activity of Ricinus communis Linn leaf and seeds extracts. Sci.Rad. 2024, 3, 74–88. [Google Scholar] [CrossRef]
- Sadowski, M.; Kula, K. Nitro-functionalized analogues of 1,3-Butadiene: An overview of characteristic, synthesis, chemical transformations and biological activity. Curr. Chem. Lett. 2024, 13, 15–30. [Google Scholar] [CrossRef]
- Alobaide, Z.N.; Habel, A.; Awin, T.; Buzgaia, N.; Elamari, A.; Elteera, F.; Elabbar, F. Arum Cyrenaicum: A comprehensive review of Phytochemistry and Bioactivity. Sci.Rad. 2024, 3, 218–227. [Google Scholar] [CrossRef]
- Shiri, P.; Roosta, A.; Ramezanpour, S.; Amani, A.M. Editorial: Six-membered heterocycles: Their synthesis and bio applications. Front. Chem. 2023, 11, 1229825. [Google Scholar] [CrossRef]
- Jasiński, R. Synthesis of 1,2-oxazine N-oxides vianoncatalyzedhetero-Diels–Alder reactions of nitroalkenes (microreview). Chem. Heterocycl. Comp. 2024, 60, 121–123. [Google Scholar] [CrossRef]
- Poonam, N.J.; Dhadda, S.; Jangid, D.K. Recent advances in the synthesis of five- and six-membered heterocycles as bioactive skeleton: A concise overview. Chem. Select 2022, 7, e202103139. [Google Scholar]
- Sadowski, M.; Synkiewicz-Musialska, B.; Kula, K. (1E,3E)-1,4-Dinitro-1,3-butadiene—Synthesis, Spectral Characteristics and Computational Study Based on MEDT, ADME and PASS Simulation. Molecules 2024, 29, 542. [Google Scholar] [CrossRef]
- Wen, C.; Dechsupa, N.; Yu, Z.; Zhang, X.; Liang, S.; Lei, X.; Xu, T.; Gao, X.; Hu, Q.; Innuan, P.; et al. Pentagalloyl Glucose: A Review of Anticancer Properties, Molecular Targets, Mechanisms of Action, Pharmacokinetics, and Safety Profile. Molecules 2023, 28, 4856. [Google Scholar] [CrossRef] [PubMed]
- Hellel, D.; Chafaa, F.; Nacereddine, A.K. Synthesis of tetrahydroquinolines and quinoline derivatives through the Lewis acid catalysed Povarov reaction: A comparative study between multi step and multi-component methods. Sci. Rad. 2023, 2, 295–308. [Google Scholar] [CrossRef]
- Ameur, S.; Barhoumi, A.; Abdallaoui, H.E.A.; Syed, A.; Belghiti, M.E.; Elgorban, A.M.; Wong, L.S.; Wang, S.; El Idrissi, M.; Zeroual, A.; et al. Molecular docking, exploring diverse selectivities and mechanistic insights in the cycloaddition reaction between 3-benzoylpyrrolo-[1,2-a]quinoxaline-1,2,4(5H)-triones and butyl vinyl ether. Chem. Heterocycl. Comp. 2024, 60, 584–591. [Google Scholar] [CrossRef]
- Bodnar, B.S.; Miller, M.J. The Nitrosocarbonyl Hetero-Diels–Alder Reaction as a Useful Tool for Organic Syntheses. Angew. Chem. Int. Ed. 2011, 50, 5630–5647. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K. Hetero-Diels−Alder Reactions of Ketones—A Challenge for Chemists. Eur. J. Org. Chem. 2004, 2004, 2093–2102. [Google Scholar] [CrossRef]
- Waldmann, H. Asymmetric Hetero Diels-Alder Reactions. Synthesis 1994, 1994, 535–551. [Google Scholar] [CrossRef]
- Jørgensen, K. Catalytic Asymmetric Hetero-Diels–Alder Reactions of Carbonyl Compounds and Imines. Angew. Chem. Int. Ed. 2000, 39, 3558–3588. [Google Scholar] [CrossRef]
- Vermeeren, P.; Hamlin, T.A.; Fernández, I.; Bickelhaupt, F.M. How Lewis Acids Catalyze Diels–Alder Reactions. Angew. Chem. Int. Ed. 2020, 59, 6201–6206. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kesler, B.S.; Moon, K.C. Inter- and intramolecular [4+2] cycloadditions of nitroalkenes with olefins. 2-Nitrostyrenes. J. Org. Chem. 1992, 57, 4912. [Google Scholar] [CrossRef]
- Zadorozhnii, P.V.; Pokotylo, I.O.; Kiselev, V.V.; Kharchenko, A.V. Synthesis of (Z)-N,3-dicyclohexyl-6-substituted-4-(trichloromethyl)-3,4-dihydro-2H-1,3,5-oxadiazin-2-imines via [4+2] hetero Diels-Alder reaction: Their spectral characteristics and molecular structure. Chem. Data Coll. 2023, 48, 101093. [Google Scholar] [CrossRef]
- Jasiński, R. On the Question of Stepwise [4+2] Cycloaddition Reactions and Their Stereochemical Aspects. Symmetry 2021, 13, 1911. [Google Scholar] [CrossRef]
- Kącka-Zych, A. Participation of Phosphorylated Analogues of Nitroethene in Diels–Alder Reactions with Anthracene: A Molecular Electron Density Theory Study and Mechanistic Aspect. Organics 2020, 1, 36–48. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Chermette, H.; Morell, C. Revisiting nucleophilicity: An index for chemical reactivity from a CDFT approach. J. Mol. Model. 2024, 30, 232. [Google Scholar] [CrossRef] [PubMed]
- Rincón, L.; Rodríguez, W.M.; Mora, J.R.; Zambrano, C.; Seijas, L.E.; Reyes, A.; Torres, F.J. A redefinition of global conceptual density functional theory reactivity indexes by means of the cubic expansions of the energy. Phys. Chem. Chem. Phys. 2025, 27, 8174–8185. [Google Scholar] [CrossRef] [PubMed]
- CDFT Sadowski, M.; Dresler, E.; Zawadzińska, K.; Wróblewska, A.; Jasiński, R. Syn-Propanethial S-Oxide as an Available Natural Building Block for the Preparation of Nitro-Functionalized, Sulfur-Containing Five-Membered Heterocycles: An MEDT Study. Molecules 2024, 29, 4892. [Google Scholar] [CrossRef]
- Dresler, E.; Wróblewska, A.; Jasiński, R. On the energetic aspects and molecular mechanism of 3-nitro-substituted 2-isoxazolines formation via nitrile N-oxide [3+2] cycloaddition: An MEDT computational study. Molecules 2024, 29, 3042. [Google Scholar] [CrossRef]
- Zawadzińska, K.; Gadocha, Z.; Pabian, K.; Wróblewska, A.; Wielgus, E.; Jasiński, R. The First Examples of [3+2] Cycloadditions with the Participation of (E)-3,3,3-Tribromo-1-Nitroprop-1-Ene. Materials 2022, 15, 7584. [Google Scholar] [CrossRef]
- Kula, K.; Łapczuk, A.; Sadowski, M.; Kras, J.; Zawadzińska, K.; Demchuk, O.M.; Gaurav, G.K.; Wróblewska, A.; Jasiński, R. On the Question of the Formation of Nitro-Functionalized 2,4-Pyrazole Analogs on the Basis of Nitrylimine Molecular Systems and 3,3,3-Trichloro-1-Nitroprop-1-Ene. Molecules 2022, 27, 8409. [Google Scholar] [CrossRef]
- Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the Regioselectivity and the Molecular Mechanism of [3+2] Cycloaddition Reactions between Nitrous Oxide and Conjugated Nitroalkenes: A DFT Computational Study. Molecules 2022, 27, 8441. [Google Scholar] [CrossRef]
- Dresler, E.; Woliński, P.; Wróblewska, A.; Jasiński, R. On the Question of Zwitterionic Intermediates in the [3+2] Cycloaddition Reactions between Aryl Azides and Ethyl Propiolate. Molecules 2023, 28, 8152. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Useful Classification of Organic Reactions Based on the Flux of the Electron Density. Sci. Rad. 2023, 2, 1–24. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Electrophilicity w and Nucleophilicity N Scales for Cationic and Anionic Species. Sci. Rad. 2025, 4, 1–17. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the mechanism of polar Diels–Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Kula, K. Unexpected Course of Reaction Between (1E,3E)-1,4-Dinitro-1,3-butadiene and N-Methyl Azomethine Ylide—A Comprehensive Experimental and Quantum-Chemical Study. Molecules 2024, 29, 5066. [Google Scholar] [CrossRef]
- Aitouna, A.O.; Syed, A.; Alfagham, A.T.; Mazoir, N.; de Julián-Ortiz, J.V.; Elgorban, A.M.; El idrissi, M.; Wong, L.S.; Zeroual, A. Investigating the chemical reactivity and molecular docking of 2-diazo-3,3,3-trifluoro-1-nitropropane with phenyl methacrylate using computational methods. Chem. Heterocycl. Comp. 2024, 60, 592–599. [Google Scholar] [CrossRef]
- Mtiraou, H.; Ghabi, A.; Al-Ghulikah, H.; Habib, M.A.; Hajji, M. Structural and chemical reactivity insights of a benzimidazolidinone-based N-heterocycle: A multiapproach quantum-chemical study. Chem. Heterocycl. Comp. 2024, 60, 611–616. [Google Scholar] [CrossRef]
- Sadowski, M.; Dresler, E.; Wróblewska, A.; Jasiński, R. A New Insight into the Molecular Mechanism of the Reaction between 2-Methoxyfuran and Ethyl (Z)-3-phenyl-2-nitroprop-2-enoate: An Molecular Electron Density Theory (MEDT) Computational Study. Molecules 2024, 29, 4876. [Google Scholar] [CrossRef]
- Dresler, E.; Wróblewska, A.; Jasiński, R. Understanding the Molecular Mechanism of Thermal and LA-Catalysed Diels–Alder Reactions between Cyclopentadiene and Isopropyl 3-Nitroprop-2-Enate. Molecules 2023, 28, 5289. [Google Scholar] [CrossRef]
- Łapczuk, A.; Ríos-Gutiérrez, M. Mechanistic Aspects of [3+2] Cycloaddition Reaction of Trifluoroacetonitrile with Diarylnitrilimines in Light of Molecular Electron Density Theory Quantum Chemical Study. Molecules 2025, 30, 85. [Google Scholar] [CrossRef]
- Pintér, B.; De Proft, F.; Veszprémi, T.; Geerlings, P. Theoretical Study of the Orientation Rules in Photonucleophilic Aromatic Substitutions. J. Org. Chem. 2008, 73, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Gillies, C.W.; Gillies, J.Z.; Suenram, D.R.; Lovas, F.J.; Kraka, E.; Cremer, D. Van der Waals complexes in 1,3-dipolar cycloaddition reactions: Ozone-ethylene. J. Am. Chem. Soc. 1991, 113, 2412–2421. [Google Scholar] [CrossRef]
- Jasiński, R. On the question of selective protocol for the preparation of juglone via (4+2) cycloaddition involving 3-hydroxypyridazine: DFT mechanistic study. Chem. Heterocycl. Comp. 2023, 59, 179–182. [Google Scholar] [CrossRef]
- Kula, K.; Sadowski, M. Regio- and Stereoselectivity of [3+2] Cycloaddition Reactions between (Z)-1-(Anthracen-9-Yl)-N-Methyl Nitrone and Analogs of Trans-β-Nitrostyrene on the Basis of MEDT Computational Study. Chem. Heterocycl. Comp. 2023, 59, 138–144. [Google Scholar] [CrossRef]
- Fryźlewicz, A.; Olszewska, A.; Zawadzińska, K.; Woliński, P.; Kula, K.; Kącka-Zych, A.; Łapczuk-Krygier, A.; Jasiński, R. On the Mechanism of the Synthesis of Nitrofunctionalised Δ2-Pyrazolines via [3+2] Cycloaddition Reactions between α-EWG-Activated Nitroethenes and Nitrylimine TAC Systems. Organics 2022, 3, 59–76. [Google Scholar] [CrossRef]
- Kącka-Zych, A. The Molecular Mechanism of the Formation of Four-Membered Cyclic Nitronates and Their Retro (3+2) Cycloaddition: A DFT Mechanistic Study. Molecules 2021, 26, 4786. [Google Scholar] [CrossRef]
- Karaś, A.; Łapczuk, A. Computational model of the formation of novel nitronorbornene analogs via Diels–Alder process. Reac. Kinet. Mec. Cat. 2025. [Google Scholar] [CrossRef]
- Fałowska, A.; Grzybowski, S.; Kapuściński, D.; Sambora, K.; Łapczuk, A. Modeling of the General Trends of Reactivity and Regioselectivity in Cyclopentadiene–Nitroalkene Diels–Alder Reactions. Molecules 2025, 30, 2467. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Topmod Package; Universite Pierre et Marie Curie: Paris, France, 1997. [Google Scholar]
- Krokidis, K.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Domingo, L.R. A New C–C Bond Formation Model Based on the Quantum Chemical Topology of Electron Density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative Characterization of the Global Electrophilicity Power of Common Diene/Dienophile Pairs in Diels–Alder Reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R. 1999–2024, a Quarter Century of the Parr’s Electrophilicity ω Index. Sci. Rad. 2024, 157–186. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Global Properties | Local Properties | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μ [eV] | η [eV] | ω [eV] | N [eV] | P−N | P−C | NN [eV] | NC [eV] | P+C | P+O | ωC [eV] | ωO [eV] | |
| 1a | −5.06 | 8.86 | 1.45 | 1.91 | 0.333 | 0.176 | 0.48 | 0.25 | ||||
| 1b | −4.87 | 8.61 | 1.38 | 2.23 | 0.335 | 0.175 | 0.46 | 0.24 | ||||
| 1c | −5.49 | 8.98 | 1.68 | 1.42 | 0.295 | 0.197 | 0.49 | 0.33 | ||||
| 2 | −3.05 | 11.38 | 0.41 | 2.66 | 0.473 | −0.163 | 1.25 | −0.43 | ||||
| Solvent | Path | Transition | ΔH | ΔS | ΔG |
|---|---|---|---|---|---|
| CCl4 | A | 1a + 2 → MCA | −12.1 | −43.8 | 0.9 |
| 1a + 2 → TSA | 1.5 | −54.0 | 17.6 | ||
| 1a + 2 → IA | −1.6 | −55.2 | 14.8 | ||
| 1a + 2 → TS2A | −0.9 | −61.8 | 17.5 | ||
| 1a + 2 → 3a | −20.4 | −63.0 | −1.6 | ||
| B | 1a + 2 → MCB | −4.9 | −43.5 | 8.0 | |
| 1a + 2 → TSB | 62.6 | −58.4 | 79.9 | ||
| 1a + 2 → 4a | 15.4 | −61.4 | 33.7 | ||
| MeCN | A | 1a + 2 → MCA | −10.9 | −45.1 | 2.5 |
| 1a + 2 → TSA | 2.7 | −53.8 | 18.7 | ||
| 1a + 2 → IA | −2.0 | −54.2 | 14.2 | ||
| 1a + 2 → TS2A | −0.1 | −62.7 | 18.6 | ||
| 1a + 2 → 3a | −18.2 | −63.9 | 0.8 | ||
| B | 1b + 2 → MCB | −4.6 | −47.0 | 9.4 | |
| 1b + 2 → TSB | 64.1 | −59.5 | 81.8 | ||
| 1b + 2 → 4a | 17.2 | −61.8 | 35.6 | ||
| CCl4 | A | 1b + 2 → MCA | −12.3 | −46.1 | 1.4 |
| 1b + 2 → TSA | 0.4 | −52.2 | 15.9 | ||
| 1b + 2 → IA | −1.5 | −62.4 | 17.1 | ||
| 1b + 2 → TS2A | −0.5 | −59.0 | 17.1 | ||
| 1b + 2 → 3b | −20.3 | −61.5 | −2.0 | ||
| B | 1b + 2 → MCB | −5.4 | −47.5 | 8.8 | |
| 1b + 2 → TSB | 62.7 | −58.5 | 80.2 | ||
| 1b + 2 → 4b | 15.6 | −60.4 | 33.6 | ||
| CCl4 | A | 1c + 2 → MCA | −13.2 | −46.5 | 0.7 |
| 1c + 2 → TSA | −2.2 | −62.1 | 16.4 | ||
| 1c + 2 → IA | −3.9 | −55.8 | 12.8 | ||
| 1c + 2 → TS2A | −2.7 | −61.0 | 15.4 | ||
| 1c + 2 → 3c | −20.8 | −62.3 | −2.2 | ||
| B | 1c + 2 → MCB | −5.7 | −49.2 | 9.0 | |
| 1c + 2 → TSB | 61.4 | −55.2 | 77.9 | ||
| 1c + 2 → 4c | 14.4 | −69.5 | 35.2 |
| Solvent | Reaction | Structure | Interatomic Distances [Å] | GEDT [e] | |||||
|---|---|---|---|---|---|---|---|---|---|
| O1-C2 | C2-N3 | N3-C4 | C4-N5 | N5-C6 | C6-O1 | ||||
| CCl4 | 1a + 2 | 1a | 1.217 | ||||||
| 2 | 1.206 | 1.420 | 1.253 | ||||||
| MCA | 1.207 | 1.410 | 1.254 | 3.207 | 1.221 | 3.481 | |||
| TSA | 1.233 | 1.365 | 1.318 | 1.867 | 1.244 | 2.837 | −0.33 | ||
| IA | 1.254 | 1.334 | 1.385 | 1.548 | 1.269 | 2.625 | −0.58 | ||
| TS2A | 1.274 | 1.316 | 1.404 | 1.509 | 1.294 | 2.127 | −0.51 | ||
| 3a | 1.357 | 1.262 | 1.441 | 1.468 | 1.376 | 1.389 | |||
| MeCN | 1a + 2 | 1a | 1.218 | ||||||
| 2 | 1.208 | 1.413 | 1.252 | ||||||
| MCA | 1.209 | 1.410 | 1.254 | 3.209 | 1.219 | 3.503 | |||
| TSA | 1.231 | 1.370 | 1.314 | 1.908 | 1.242 | 2.910 | −0.32 | ||
| IA | 1.252 | 1.334 | 1.392 | 1.532 | 1.271 | 2.765 | −0.63 | ||
| TS2A | 1.278 | 1.314 | 1.411 | 1.499 | 1.301 | 2.065 | −0.52 | ||
| 3a | 1.356 | 1.263 | 1.442 | 1.469 | 1.376 | 1.389 | |||
| CCl4 | 1b + 2 | 1b | 1.217 | ||||||
| MCA | 1.208 | 1.411 | 1.254 | 3.236 | 1.221 | 3.519 | |||
| TSA | 1.233 | 1.371 | 1.317 | 1.860 | 1.244 | 2.938 | −0.34 | ||
| IA | 1.254 | 1.335 | 1.384 | 1.549 | 1.269 | 2.622 | −0.57 | ||
| TS2A | 1.274 | 1.317 | 1.403 | 1.510 | 1.294 | 2.134 | −0.51 | ||
| 3b | 1.359 | 1.262 | 1.441 | 1.468 | 1.376 | 1.389 | |||
| CCl4 | 1c + 2 | 1c | 1.217 | ||||||
| MCA | 1.206 | 1.406 | 1.254 | 3.221 | 1.221 | 3.526 | |||
| TSA | 1.229 | 1.371 | 1.313 | 1.905 | 1.242 | 2.985 | −0.32 | ||
| IA | 1.252 | 1.332 | 1.388 | 1.542 | 1.269 | 2.649 | −0.59 | ||
| TS2A | 1.275 | 1.312 | 1.408 | 1.504 | 1.298 | 2.094 | −0.51 | ||
| 3c | 1.354 | 1.260 | 1.442 | 1.468 | 1.377 | 1.391 | |||
| Solvent | Reaction | Structure | Interatomic Distances [Å] | GEDT [e] | |||||
|---|---|---|---|---|---|---|---|---|---|
| O1-C2 | C2-N3 | N3-C4 | C4-C5 | C5-N6 | N6-O1 | ||||
| CCl4 | 1a + 2 | MCB | 1.210 | 1.401 | 1.251 | 3.785 | 1.219 | 5.086 | |
| TSB | 1.251 | 1.341 | 1.338 | 1.880 | 1.288 | 2.128 | −0.22 | ||
| 4a | 1.349 | 1.270 | 1.444 | 1.531 | 1.397 | 1.413 | |||
| MeCN | 1a + 2 | MCB | 1.212 | 1.398 | 1.251 | 3.819 | 1.219 | 5.215 | |
| TSB | 1.250 | 1.342 | 1.340 | 1.878 | 1.289 | 2.138 | −0.23 | ||
| 4a | 1.348 | 1.271 | 1.446 | 1.531 | 1.397 | 1.413 | |||
| CCl4 | 1b + 2 | MCB | 1.210 | 1.402 | 1.251 | 3.791 | 1.219 | 5.083 | |
| TSB | 1.251 | 1.342 | 1.338 | 1.879 | 1.288 | 2.127 | −0.21 | ||
| 4b | 1.350 | 1.271 | 1.444 | 1.531 | 1.396 | 1.412 | |||
| CCl4 | 1c + 2 | MCB | 1.208 | 1.397 | 1.252 | 3.780 | 1.219 | 5.062 | |
| TSB | 1.249 | 1.339 | 1.340 | 1.877 | 1.289 | 2.138 | −0.24 | ||
| 4c | 1.347 | 1.269 | 1.445 | 1.531 | 1.399 | 1.414 | |||
| Structures | 1a | 2 | MCA | PA1 | TSA | PA2 | PA3 | IA |
|---|---|---|---|---|---|---|---|---|
| Phases | - | - | I | II | III | IV | - | |
| d(C4-N5) | - | - | 3.207 | 2.872 | 1.867 | 1.796 | 1.738 | 1.548 |
| GEDT | - | - | 0.01 | 0.01 | 0.35 | 0.41 | 0.45 | 0.56 |
| dE | - | - | −13.62 | −8.43 | 0.42 | 0.27 | −0.09 | −3.96 |
| V(O1) | 2.63 | - | 2.67 | 2.63 | 2.70 | 2.71 | 2.73 | 2.74 |
| V’(O1) | 2.64 | - | 2.61 | 2.65 | 2.77 | 2.79 | 2.82 | 2.90 |
| V(O1,C2) | 2.40 | - | 2.39 | 2.40 | 2.20 | 2.16 | 2.13 | 2.01 |
| V(N3,C2) | 1.98 | - | 2.00 | 2.00 | 2.23 | 2.27 | 2.32 | 2.44 |
| V(N3) | 2.53 | - | 2.52 | 2.64 | 3.12 | 3.19 | 3.23 | 3.33 |
| V(C4,N3) | 1.73 | - | 1.66 | 2.95 | 2.36 | 2.24 | 2.15 | 1.93 |
| V’(C4,N3) | 1.33 | - | 1.39 | - | - | - | - | - |
| V(N5,C6) | - | 1.71 | 1.68 | 1.68 | 1.76 | 1.80 | 3.18 | 2.99 |
| V’(N5,C6) | - | 1.62 | 1.61 | 1.63 | 1.39 | 1.33 | - | - |
| V(N5) | - | 2.72 | 2.78 | 2.74 | 2.74 | 0.96 | 1.08 | 1.35 |
| V(C6,N7) | - | 1.62 | 1.60 | 1.54 | 1.73 | 1.75 | 1.78 | 1.90 |
| V’(C6,N7) | - | 1.71 | 1.73 | 1.78 | 1.88 | 1.89 | 1.91 | 1.93 |
| V(N7) | - | 2.72 | 2.73 | 2.76 | 2.44 | 2.41 | 2.38 | 2.22 |
| V(C4,N5) | - | - | - | - | - | 1.83 | 1.66 | 1.60 |
| Structures | IA | TS2A | PB1 | PB2 | PB3 | PB4 | PB5 | 3a |
|---|---|---|---|---|---|---|---|---|
| Phases | I | II | III | IV | V | VI | - | |
| d1(C4-N5) | 1.548 | 1.509 | 1.486 | 1.478 | 1.474 | 1.469 | 1.455 | 1.468 |
| d2(O1-C6) | 2.625 | 2.127 | 1.865 | 1.756 | 1.703 | 1.631 | 1.406 | 1.389 |
| GEDT | 0.56 | 0.49 | 0.37 | 0.31 | 0.28 | 0.25 | 0.14 | 0.11 |
| dE | 0.00 | 1.2 | −0.5 | −2.7 | −4.1 | −6.3 | −16.7 | −19.7 |
| V(O1) | 2.74 | 2.49 | 5.62 | 5.84 | 4.91 | 4.83 | 4.49 | 4.46 |
| V’(O1) | 2.90 | 3.15 | - | - | - | - | - | - |
| V(O1,C2) | 2.01 | 1.89 | 1.77 | 1.70 | 1.68 | 1.64 | 1.59 | 1.58 |
| V(N3,C2) | 2.44 | 2.60 | 2.77 | 2.84 | 2.88 | 1.42 | 1.52 | 1.54 |
| V’(N3,C2) | - | - | - | - | - | 1.50 | 1.50 | 1.52 |
| V(N3) | 3.33 | 3.22 | 3.09 | 3.03 | 3.00 | 2.95 | 2.89 | 2.85 |
| V(C4,N3) | 1.93 | 1.88 | 1.85 | 1.84 | 1.84 | 1.83 | 1.79 | 1.79 |
| V(N5,C6) | 2.99 | 2.67 | 2.40 | 2.22 | 2.16 | 2.11 | 2.04 | 2.07 |
| V(N5) | 1.35 | 1.64 | 1.85 | 1.92 | 1.96 | 2.00 | 1.98 | 1.58 |
| V(C6,N7) | 1.90 | 1.93 | 1.88 | 1.83 | 1.81 | 1.78 | 1.71 | 1.68 |
| V’(C6,N7) | 1.93 | 1.81 | 1.78 | 1.73 | 1.70 | 1.69 | 1.61 | 1.53 |
| V(N7) | 2.22 | 2.35 | 2.50 | 2.56 | 2.59 | 2.63 | 2.75 | 2.86 |
| V(C4,N5) | 1.60 | 1.67 | 1.70 | 1.71 | 1.72 | 1.72 | 1.75 | 1.65 |
| V(O1,C6) | - | - | - | - | 0.97 | 1.09 | 1.50 | 1.55 |
| V’(N5) | - | - | - | - | - | - | 0.27 | 0.73 |
| V(C6) | - | - | 0.04 | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woliński, P.; Zawadzińska-Wrochniak, K.; Dresler, E.; Jasiński, R. On the Question of the Course of the Hetero Diels–Alder Reactions Between N-(2,2,2-Trichloroethylidene)Carboxamides and Dicyclohexylcarbodiimide: A New Case of the Stepwise Zwitterionic Cycloaddition Process. Molecules 2025, 30, 2692. https://doi.org/10.3390/molecules30132692
Woliński P, Zawadzińska-Wrochniak K, Dresler E, Jasiński R. On the Question of the Course of the Hetero Diels–Alder Reactions Between N-(2,2,2-Trichloroethylidene)Carboxamides and Dicyclohexylcarbodiimide: A New Case of the Stepwise Zwitterionic Cycloaddition Process. Molecules. 2025; 30(13):2692. https://doi.org/10.3390/molecules30132692
Chicago/Turabian StyleWoliński, Przemysław, Karolina Zawadzińska-Wrochniak, Ewa Dresler, and Radomir Jasiński. 2025. "On the Question of the Course of the Hetero Diels–Alder Reactions Between N-(2,2,2-Trichloroethylidene)Carboxamides and Dicyclohexylcarbodiimide: A New Case of the Stepwise Zwitterionic Cycloaddition Process" Molecules 30, no. 13: 2692. https://doi.org/10.3390/molecules30132692
APA StyleWoliński, P., Zawadzińska-Wrochniak, K., Dresler, E., & Jasiński, R. (2025). On the Question of the Course of the Hetero Diels–Alder Reactions Between N-(2,2,2-Trichloroethylidene)Carboxamides and Dicyclohexylcarbodiimide: A New Case of the Stepwise Zwitterionic Cycloaddition Process. Molecules, 30(13), 2692. https://doi.org/10.3390/molecules30132692