


Novel Azole-Modified Porphyrins for Mitochondria-Targeted Photodynamic Therapy

 , , , , , ,
, , , , , ,  ,
,  ,
,  and
and
Abstract

1. Introduction
2. Results and Discussion
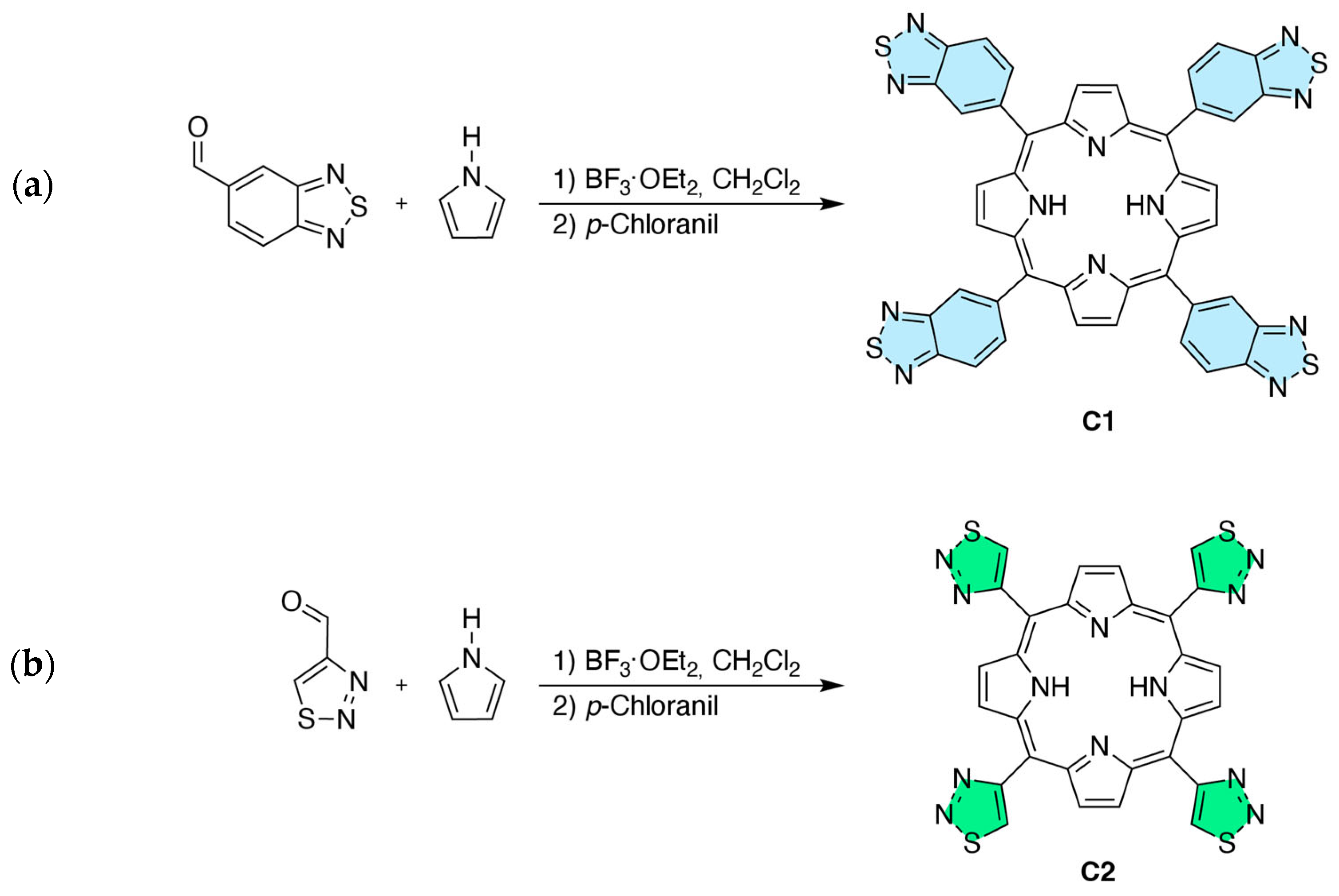
2.1. Synthetic Procedure and Structural Characterization
2.1.1. Synthesis
2.1.2. NMR Analysis
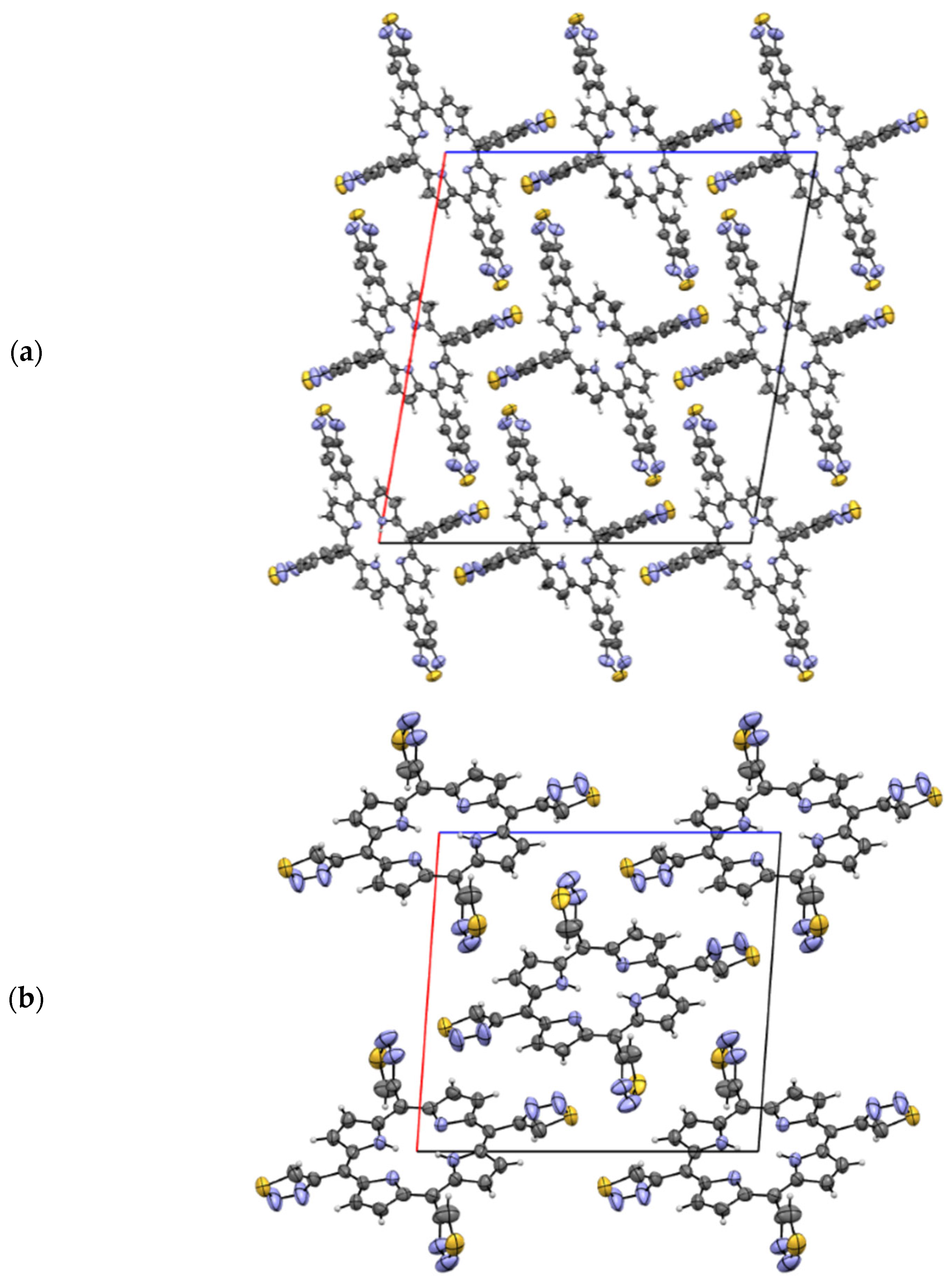
2.1.3. X-Ray Analysis
2.2. Photophysical Characterization
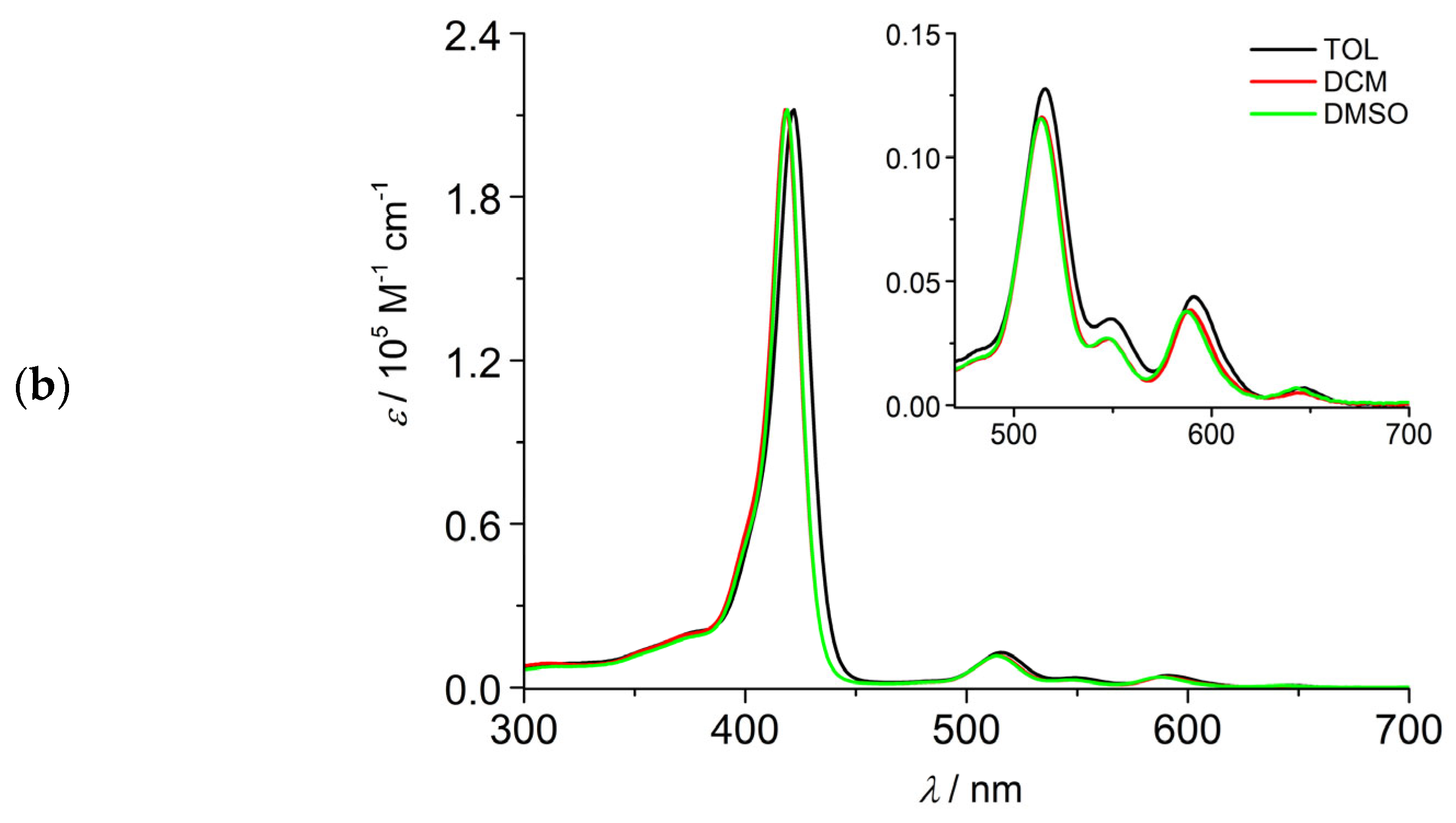
2.2.1. Absorption and Emission Properties, Singlet Oxygen Production
2.2.2. Theoretical Modeling
2.3. In Vitro Studies
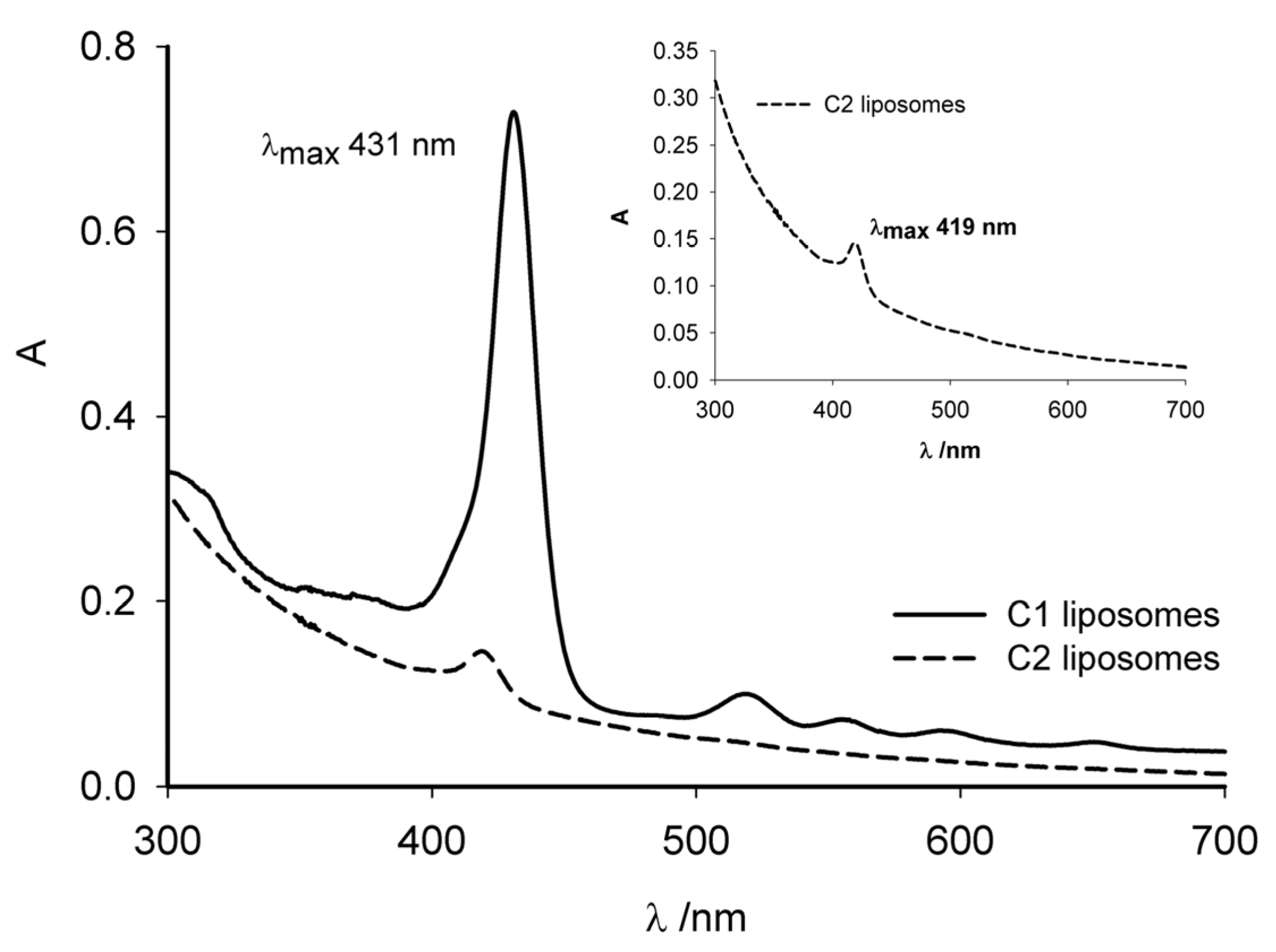
2.3.1. Cellular Internalization
2.3.2. Intracellular Uptake of C1 and C2
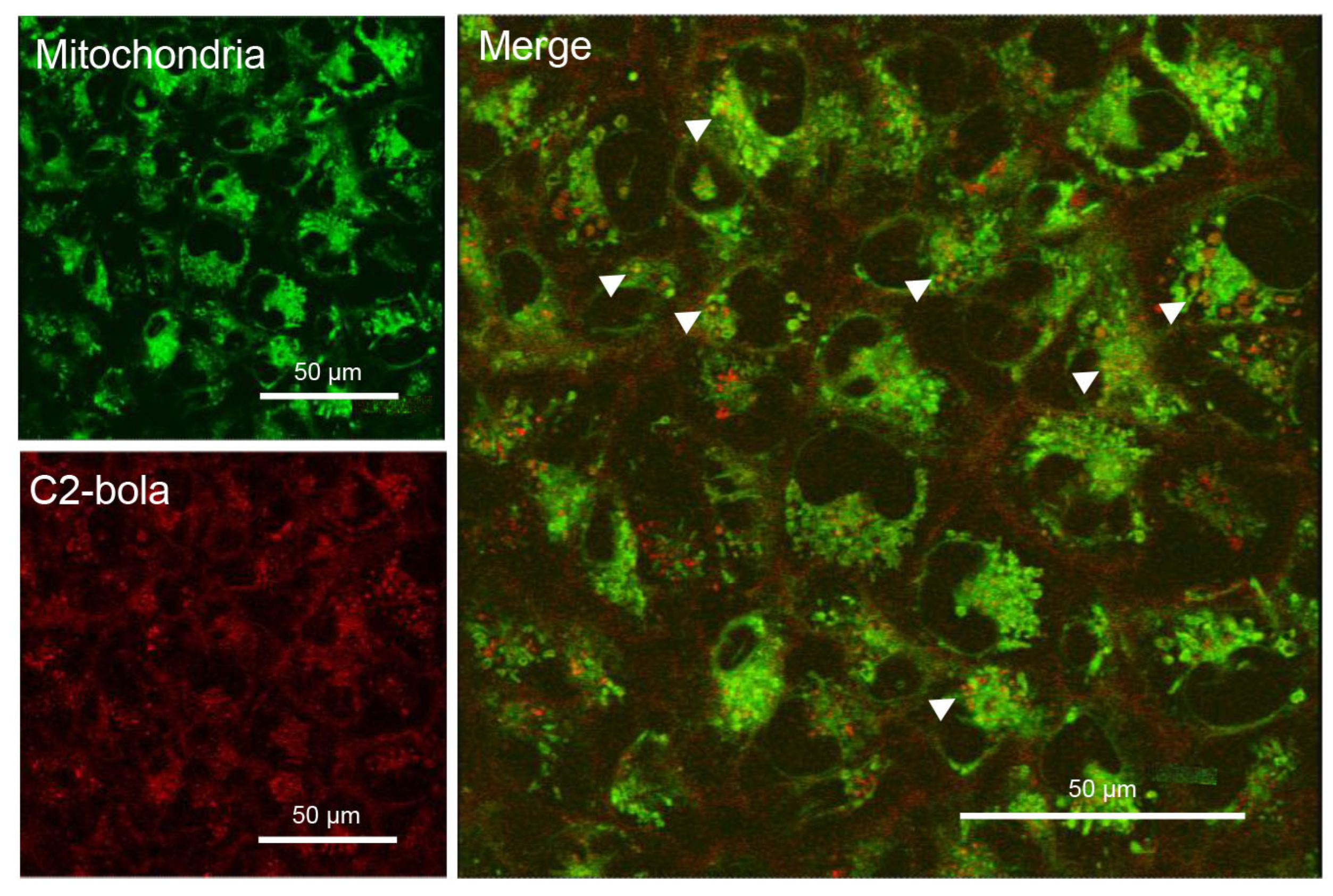
2.3.3. Intracellular Localization of C1 and C2
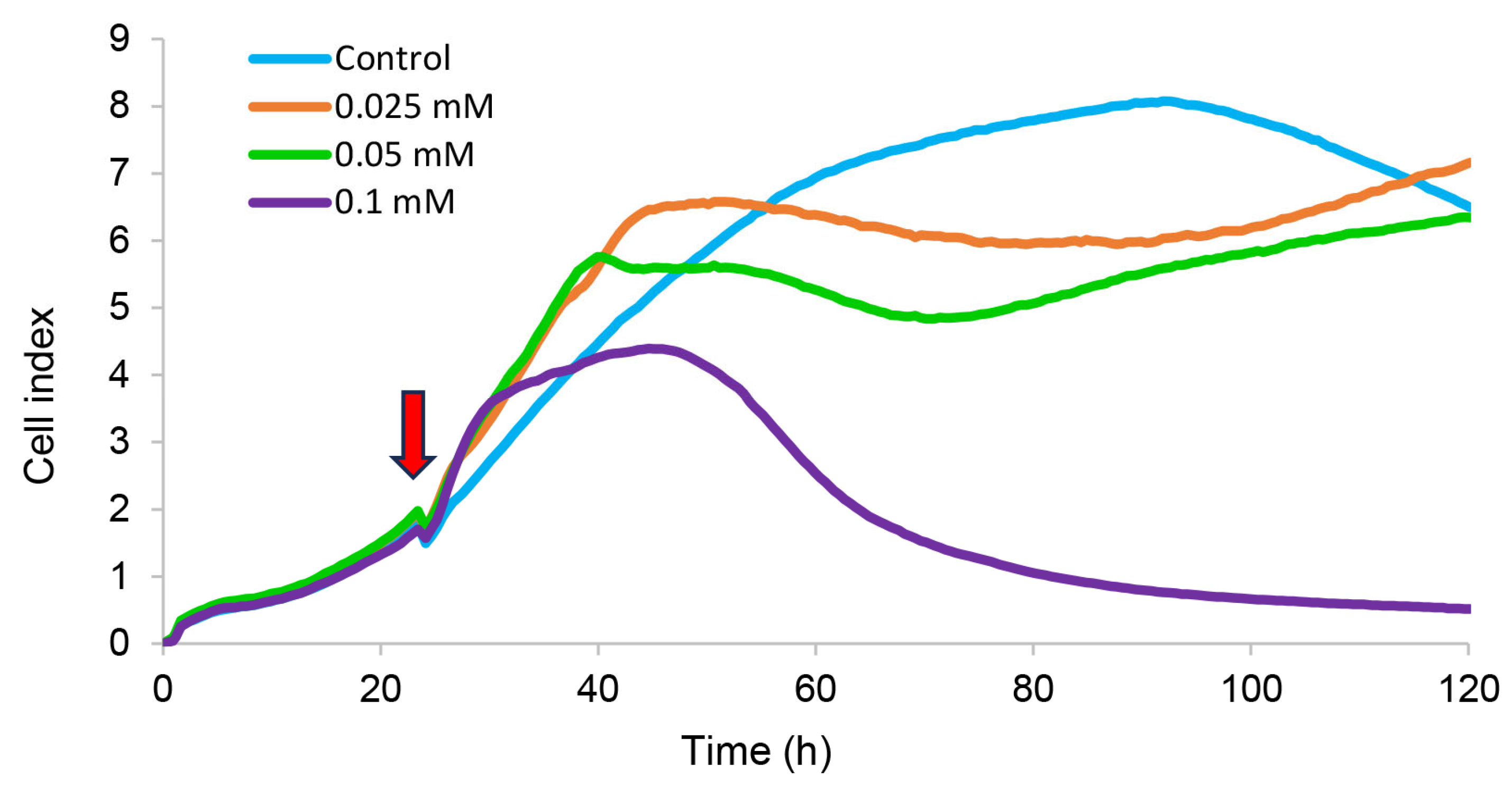
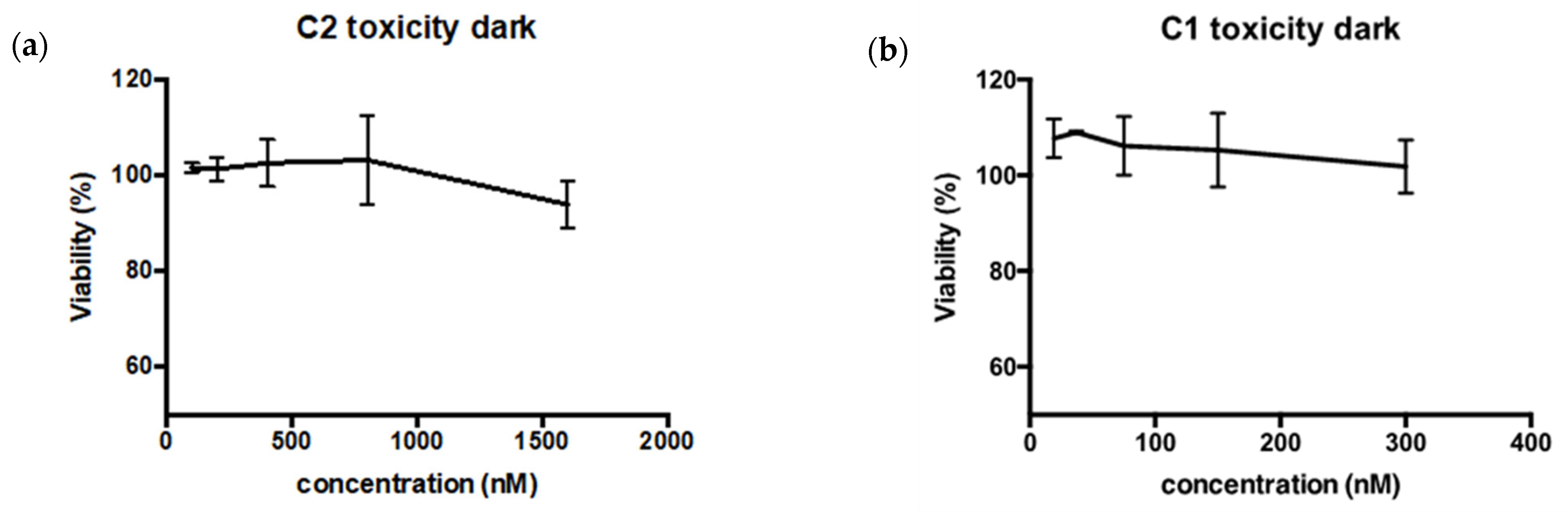
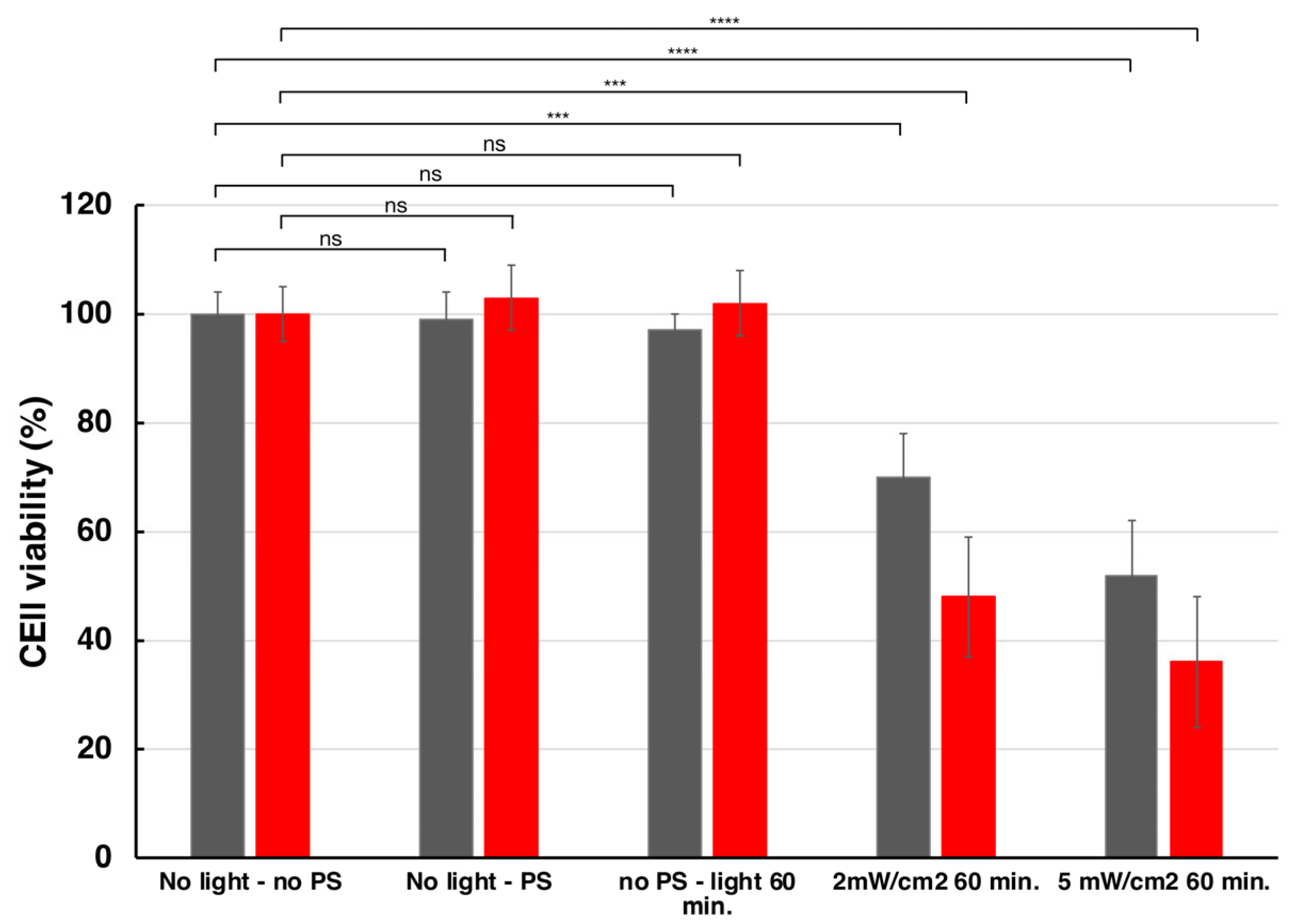
2.3.4. Dark Toxicity and Phototoxicity
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of 5,10,15,20-Tetra(2,1,3-benzothiadiazol-5-yl)porphyrin (C1)
3.1.2. Synthesis of 5,10,15,20-Tetra(1,2,3-thiadiazol-4-yl)porphyrin (C2)
3.2. X-Ray Analysis
3.3. Spectroscopy and Photophysics
3.4. Theoretical Parameters
3.5. Cells Studies on Free and Liposome-Loaded PSs
3.5.1. Materials
3.5.2. Instrumentation
3.5.3. Preparation of C1 and C2 Solutions for LSCM and Flow Cytometry
3.5.4. Preparation of Mitochondriotropic Liposomes
3.5.5. Determination of C1 and C2 Entrapment Efficacy (EE%)
3.5.6. Calibration Curves
3.5.7. Determination of EE%
3.5.8. Cell Cultures
3.5.9. Flow Cytometry
3.5.10. Electrical Impedance Assay
3.5.11. Laser Scanning Confocal Microscopy
3.5.12. Dark Toxicity and Phototoxicity
3.5.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PDT | Photodynamic therapy |
| PS | Photosensitizer |
| ROS | Reactive oxygen species |
| DNA | Deoxyribonucleic acid |
| MDA-MB-231 | M D Anderson—Metastatic Breast—231 |
| NMR | Nuclear magnetic resonance |
| COSY | Correlation spectroscopy |
| HSQC | Heteronuclear single quantum coherence |
| TOL | Toluene |
| DCM | Dichloromethane |
| DMSO | Dimethyl sulfoxide |
| DPBF | 1,3-diphenylisobenzofuran |
| ZnPc | Zn-phthalocyanine |
| TPP | meso tetraphenylporphyrin |
| DFT | Density functional theory |
| TD-DFT | Time-dependent density functional theory |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| TPP3 | Triphenylphosphonium bolaamphiphile |
| DMEM | Dulbecco’s Modified Eagle Medium |
| PBS | Phosphate-buffered saline |
| PDI | Polydispersity index |
| EE | Entrapment efficiency |
| CI | Cell Index |
| RTCA | Real-time cell analysis |
| LSCM | Laser scanning confocal microscopy |
| CCD | Charged coupled device |
| CCDC | Cambridge Crystallographic Data Center |
| MTS | 3-(4,5-dimetiltiazol-2-il)-5-(3-metilfenil)-2-(4-metilfenil)-2H-tetrazolio |
| RPMI | Roswell Park Memorial Institute |
References and Note
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Gegechkori, N.; Haines, L.; Lin, J.J. Long-Term and Latent Side Effects of Specific Cancer Types. Med. Clin. North Am. 2017, 101, 1053–1073. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef]
- Ferreira dos Santos, A.; Queiroz de Almeida, D.R.; Ferreira Terra, L.; Baptista, M.S.; Labriola, L. Photodynamic therapy in cancer treatment—An update review. J. Cancer Metastasis Treat. 2019, 5, 25. [Google Scholar]
- Correia, J.H.; Rodrigues, J.A.; Pimenta, S.; Dong, T.; Yang, Z. Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions. Pharmaceutics 2021, 13, 1332. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Sharma, D.; Singh, S.; Kumar, P.; Jain, G.K.; Aggarwal, G.; Almalki, W.H.; Kesharwani, P. Mechanisms of photodynamic therapy. In Nanomaterials for Photodynamic Therapy; Kesharwani, P., Ed.; Woodhead Publishing: Cambridge, UK, 2023; pp. 41–54. [Google Scholar]
- Kou, J.; Dou, D.; Yang, L. Porphyrin photosensitizers in photodynamic therapy and its applications. Oncotarget 2017, 8, 81591–81603. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and nonporphyrin photosensitizers in oncology: Preclinical and clinical advances in photodynamic therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef]
- Pushpan, S.K.; Venkatraman, S.; Anand, V.G.; Sankar, J.; Parmeswaran, D.; Ganesan, S.; Chandrashekar, T.K. Porphyrins in photodynamic therapy—A search for ideal photosensitizers. Curr. Med. Chem. Anticancer Agents 2002, 2, 187–207. [Google Scholar] [CrossRef]
- Alam, M.M.; Bolze, F.; Daniel, C.; Flamigni, L.; Gourlaouen, C.; Heitz, V.; Jenni, S.; Schmitt, J.; Sour, A.; Ventura, B. pi-Extended diketopyrrolopyrrole-porphyrin arrays: One- and two-photon photophysical investigations and theoretical studies. Phys. Chem. Chem. Phys. 2016, 18, 21954–21965. [Google Scholar] [CrossRef] [PubMed]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Heitz, V.; Sour, A.; Bolze, F.; Ftouni, H.; Nicoud, J.F.; Flamigni, L.; Ventura, B. Diketopyrrolopyrrole-porphyrin conjugates with high two-photon absorption and singlet oxygen generation for two-photon photodynamic therapy. Angew. Chem. Int. Ed. Engl. 2015, 54, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.J.; Dougherty, T.J. Basic principles of photodynamic therapy. J. Porph. Phthaloc. 2001, 5, 105–129. [Google Scholar] [CrossRef]
- Josefsen, L.B.; Boyle, R.W. Photodynamic therapy: Novel third-generation photosensitizers one step closer? Br. J. Pharmacol. 2008, 154, 1–3. [Google Scholar] [CrossRef]
- Jenni, S.; Sour, A.; Bolze, F.; Ventura, B.; Heitz, V. Tumour-targeting photosensitisers for one- and two-photon activated photodynamic therapy. Org. Biomol. Chem. 2019, 17, 6585–6594. [Google Scholar] [CrossRef]
- Phillips, D. Toward targeted photodynamic therapy. Pure Appl. Chem. 2011, 83, 733–748. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y. Mitochondria as a target in cancer treatment. MedComm 2020, 1, 129–139. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef]
- Radpour, R.; Fan, A.X.; Kohler, C.; Holzgreve, W.; Zhong, X.Y. Current understanding of mitochondrial DNA in breast cancer. Breast J. 2009, 15, 505–509. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Beroud, C.; Demont, J.; Bouvier, R.; Schagger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef]
- Ikeda, H.; Kawase, K.; Nishi, T.; Watanabe, T.; Takenaga, K.; Inozume, T.; Ishino, T.; Aki, S.; Lin, J.; Kawashima, S.; et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature 2025, 638, 225–236. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Zhang, T.; Xing, D. Mitochondria-Specific Agents for Photodynamic Cancer Therapy: A Key Determinant to Boost the Efficacy. Adv. Healthc. Mater. 2021, 10, 2001240. [Google Scholar] [CrossRef]
- Hilf, R. Mitochondria are targets of photodynamic therapy. J. Bioenerg. Biomembr. 2007, 39, 85–89. [Google Scholar] [CrossRef]
- Li, M.; Zheng, K.; Liu, X. Mitochondria-Targeting Phthalocyanines and Porphyrins for Enhanced Photodynamic Tumor Therapy. ChemistrySelect 2023, 8, e202205022. [Google Scholar] [CrossRef]
- Kabi, A.K.; Gujjarappa, R.; Vodnala, N.; Tyagi, U.; Kaldhi, D.; Malakar, C.C. An Introduction on Evolution of Azole Derivatives in Medicinal Chemistry. In Nanostructured Biomaterials. Materials Horizons: From Nature to Nanomaterials; Swain, B.P.E., Ed.; Springer: Singapore, 2022; pp. 79–99. [Google Scholar]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Shingu-Vazquez, M.; Traven, A. Mitochondria and fungal pathogenesis: Drug tolerance, virulence, and potential for antifungal therapy. Eukaryot. Cell 2011, 10, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Khan, M.K.A.; Baig, M.H.; Imran, M.; Gupta, G.K. Role of Azoles in Cancer Prevention and Treatment: Present and Future Perspectives. Anticancer Agents Med. Chem. 2018, 18, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Sharma, A. Pharmacological Significance of Triazoles and Tetrazoles in Neurodegenerative Disease: An Overview. In N-Heterocycles, Ameta, K.L., Kant, R., Penoni, A., Maspero, A., Scapinello, L., Eds.; Sprimger: Singapore, 2022; Volume 10. [Google Scholar]
- Head, S.A.; Shi, W.; Zhao, L.; Gorshkov, K.; Pasunooti, K.; Chen, Y.; Deng, Z.; Li, R.J.; Shim, J.S.; Tan, W.; et al. Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7276–E7285. [Google Scholar] [CrossRef] [PubMed]
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol. Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef]
- Geier, G.R., 3rd; Ciringh, Y.; Li, F.; Haynes, D.M.; Lindsey, J.S. A survey of acid catalysts for use in two-step, one-flask syntheses of meso-substituted porphyrinic macrocycles. Org. Lett. 2000, 2, 1745–1748. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthesis of meso-Substituted Porphyrins. In The Porphyrin Handbook; Kadish, K., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 1, pp. 45–118. [Google Scholar]
- Abraham, R.J.; Hawkes, G.E.; Hudson, M.F.; Smith, K.M. The nuclear magnetic resonance spectra of porphyrins. Part X. Carbon-13 nuclear magnetic resonance spectra of some meso-tetraarylporphyrins and their metal chelates. J. Chem. Soc. Perkin Trans. 1975, 2, 204–211. [Google Scholar] [CrossRef]
- Abraham, R.J.; Hawkes, G.E.; Smith, K.M. N-H tautomerism in porphyrins: An NMR study. Tetrahedron Lett. 1974, 15, 1483–1486. [Google Scholar] [CrossRef]
- Dominguez, D.D.; Snow, A.W.; Shirk, J.S.; Pong, R.G.S. Polyethyleneoxide-capped phthalocyanines: Limiting phthalocyanine aggregation to dimer formation. J. Porphyr. Phthalocyanines 2001, 5, 582–592. [Google Scholar] [CrossRef]
- The absorption spectrum of C1 could be precisely determined in TOL and DCM while the same determination in DMSO was precluded by solubility reasons. For the same solubility issues, the molar absorption coefficient of C2 could be measured only in DCM.
- Ventura, B.; Flamigni, L.; Marconi, G.; Lodato, F.; Officer, D.L. Extending the porphyrin core: Synthesis and photophysical characterization of porphyrins with π-conjugated β-substituents. New J. Chem. 2008, 32, 166–178. [Google Scholar] [CrossRef]
- Wilkinson, F.; Brummer, J.G. Rate Constants for the Decay and Reactions of the Lowest Electronically Excited Singlet-State of Molecular-Oxygen in Solution. J. Phys. Chem. Ref. Data 1981, 10, 809–1000. [Google Scholar] [CrossRef]
- Ostasnka, E.; Aebisher, D.; Bartusik-Aebisher, D. The potential of photodynamic therapy in current breast cancer treatment methodologies. Biomed. Pharmacother. 2021, 137, 111302. [Google Scholar]
- Satrialdi; Munechika, R.; Biju, V.; Takano, Y.; Harashima, H.; Yamada, Y. The optimization of cancer photodynamic therapy by utilization of a pi-extended porphyrin-type photosensitizer in combination with MITO-Porter. Chem. Commun. 2020, 56, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, F.; Sennato, S.; Rossi, E.; Proroga, R.; Sarti, S.; Diociaiuti, M.; Casciardi, S.; Mussi, V.; Ciogli, A.; Bordi, F.; et al. Aggregation behaviour of triphenylphosphonium bolaamphiphiles. J. Colloid. Interface Sci. 2018, 531, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, F.; Sennato, S.; Rossi, E.; Proroga, R.; Sarti, S.; Diociaiuti, M.; Casciardi, S.; Mussi, V.; Ciogli, A.; Bordi, F.; et al. Synthesis and Characterization of Mitochondria-Targeted Triphenylphosphonium Bolaamphiphiles. In Mitochondrial Medicine, 2nd ed.; Weissig, V., Edeas, M., Eds.; Methods in Molecular Biology vol. 2275; Humana Press: New York, NY, USA, 2021; Volume 1: Targeting Mitochondria, pp. 27–47. [Google Scholar]
- Ceccacci, F.; Ciranna, F.; Sennato, S.; Ciogli, A.; Gallina, S.; Migani, S.; Calcabrini, A.; Salvio, R.; Calicchia, E.; Bozzuto, G.; et al. Triphenylphosphonium bolaamphiphile-liposomes: Loading of Resveratrol and Trolox and evaluation of mithocondrial targeting. Int. J. Nanomed. 2021; 23, 112. [Google Scholar]
- Bonechi, C.; Martini, S.; Ciani, L.; Lamponi, S.; Rebmann, H.; Rossi, C.; Ristori, S. Using Liposomes as Carriers for Polyphenolic Compounds: The Case of Trans-Resveratrol. PLoS ONE 2012, 7, e41438. [Google Scholar] [CrossRef]
- Pagano, L.; Gkartziou, F.; Aiello, S.; Simonis, B.; Ceccacci, F.; Sennato, S.; Ciogli, A.; Mourtas, S.; Spiliopoulou, I.; Antimisiaris, S.G.G.; et al. Resveratrol loaded in cationic glucosylated liposomes to treat Staphylococcus epidermidis infections. Chem. Phys. Lipids 2022, 243, 105174. [Google Scholar] [CrossRef]
- Kuznetsova, D.A.; Gaynanova, G.A.; Vasileva, L.A.; Sibgatullina, G.V.; Samigullin, D.V.; Sapunova, A.S.; Voloshina, A.D.; Galkina, I.V.; Petrov, K.A.; Zakharova, L.Y. Mitochondria-targeted cationic liposomes modified with alkyltriphenylphosphonium bromides loaded with hydrophilic drugs: Preparation, cytotoxicity and colocalization assay. J. Mater. Chem. B 2019, 7, 7351–7362. [Google Scholar] [CrossRef]
- Shimamura, Y.; Tamatani, D.; Kuniyasu, S.; Mizuki, Y.; Suzuki, T.; Katsura, H.; Yamada, H.; Endo, Y.; Osaki, T.; Ishizuka, M.; et al. 5-Aminolevulinic Acid Enhances Ultrasound-mediated Antitumor Activity via Mitochondrial Oxidative Damage in Breast Cancer. Anticancer Res. 2016, 36, 3607–3612. [Google Scholar]
- Bozzuto, G.; D’Avenio, G.; Condello, M.; Sennato, S.; Battaglione, E.; Familiari, G.; Molinari, A.; Grigioni, M. Label-free cell based impedance measurements of ZnO nanoparticles-human lung cell interaction: A comparison with MTT, NR, Trypan blue and cloning efficiency assays. J. Nanobiotechnol. 2021, 19, 306. [Google Scholar] [CrossRef]
- Fan, D.; Cao, Y.; Cao, M.; Wang, Y.; Cao, Y.; Gong, T. Nanomedicine in cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 293. [Google Scholar] [CrossRef]
- Feng, J.; Tang, Y.; Xu, Y.; Sun, Q.; Liao, F.; Han, D. Substrate stiffness influences the outcome of antitumor drug screening in vitro. Clin. Hemorheol. Microcirc. 2013, 55, 121–131. [Google Scholar] [CrossRef]
- Schmitt, J.; Jenni, S.; Sour, A.; Heitz, V.; Bolze, F.; Pallier, A.; Bonnet, C.S.; Toth, E.; Ventura, B. A Porphyrin Dimer-GdDOTA Conjugate as a Theranostic Agent for One- and Two-Photon Photodynamic Therapy and MRI. Bioconjug. Chem. 2018, 29, 3726–3738. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Herbst-Irmer, R.; Sheldrick, G.M. Refinement of Twinned Structures with SHELXL97. Acta Crystallogr. Sect. B 1998, 54, 443–449. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Seybold, P.G.; Gouterman, M. Porphyrins: 13: Fluorescence Spectra and Quantum Yields. J. Mol. Spectrosc. 1969, 31, 1–13. [Google Scholar] [CrossRef]
- Lamberts, J.J.M.; Schumacher, D.R.; Neckers, D.C. Novel Rose-Bengal Derivatives—Synthesis and Quantum Yield Studies. J. Am. Chem. Soc. 1984, 106, 5879–5883. [Google Scholar] [CrossRef]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Quantum Yields for the Photosensitized Formation of the Lowest Electronically Excited Singlet-State of Molecular-Oxygen in Solution. J. Phys. Chem. Ref. Data 1993, 22, 113–262. [Google Scholar] [CrossRef]
- Ogunsipe, A.; Chen, J.Y.; Nyokong, T. Photophysical and photochemical studies of zinc(II) phthalocyanine derivatives—Effects of substituents and solvents. New J. Chem. 2004, 28, 822–827. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Bloino, J.; Baiardi, A.; Biczysko, M. Aiming at an accurate prediction of vibrational and electronic spectra for medium-to-large molecules: An overview. Int. J. Quantum Chem. 2016, 116, 1543–1574. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Ketola, T.M.; Muñoz-Losa, A.; Esposito, L.; Markovitsi, D.; Improta, R. Excited State Pathways Leading to Formation of Adenine Dimers. J. Phys. Chem. Lett. 2016, 7, 2020–2023. [Google Scholar] [CrossRef] [PubMed]
- Koppel, D.E. Analysis of Macromolecular Polydispersity in Intensity Correlation Spectroscopy—Method of Cumulants. J. Chem. Phys. 1972, 57, 4814–4820. [Google Scholar] [CrossRef]
- Tucker, I.M.; Corbett, J.C.W.; Fatkin, J.; Jack, R.O.; Kaszuba, M.; MacCreath, B.; McNeil-Watson, F. Laser Doppler Electrophoresis applied to colloids and surfaces. Curr. Opin. Colloid. Interface Sci. 2015, 20, 215–226. [Google Scholar] [CrossRef]
- Bombelli, C.; Stringaro, A.; Borocci, S.; Bozzuto, G.; Colone, M.; Giansanti, L.; Sgambato, R.; Toccaceli, L.; Mancini, G.; Molinari, A. Efficiency of liposomes in the delivery of a photosensitizer controlled by the stereochemistry of a gemini surfactant component. Mol. Pharm. 2010, 7, 130–137. [Google Scholar] [CrossRef]
- Hope, M.J.; Nayar, R.; Mayer, L.D.; Cullis, P.R. Reduction of Liposomes Size and Preparation of Unila-mellar Vesicles by Extrusion Techniques. In Liposome Technology, 2nd ed.; Gregoriadis, G.E., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 123–139. [Google Scholar]
- Fahmy, S.A.; Azzazy, H.M.E.; Schaefer, J. Liposome Photosensitizer Formulations for Effective Cancer Photodynamic Therapy. Pharmaceutics 2021, 13, 1345. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solvent | λmax (nm) a | ϕfl b | τ (ns) c | kr (s−1) | knr (s−1) | ϕΔ d |
|---|---|---|---|---|---|---|---|
| C1 | TOL | 664, 726 | 0.13 | 9.5 | 1.4 × 107 | 9.1 × 107 | 0.71 |
| DCM | 660, 722 | 0.11 | 8.5 | 1.3 × 107 | 1.0 × 108 | 0.75 | |
| DMSO | 658, 722 | 0.14 | 10.5 | 1.3 × 107 | 8.2 × 107 | - e | |
| C2 | TOL | 656, 718 | 0.027 | 3.7 | 7.3 × 106 | 2.6 × 108 | - e |
| DCM | 654, 718 | 0.019 | 3.3 | 5.8 × 106 | 3.0 × 108 | 0.73 | |
| DMSO | 652, 716 | 0.026 | 3.6 | 7.2 × 106 | 2.7 × 108 | 0.74 |
| Liposome | DOPC/TPP3 | Total Lipids/PS | R ± sd (nm) | PDI ± sd | ζ pot ± sd (mV) | EE% |
|---|---|---|---|---|---|---|
| DOPC/TPP3/C1 | 9.75:0.25 | 250:1 | 50.2 ± 0.2 | 0.114 ± 0.004 | 10.5 ± 0.4 | 40% |
| DOPC/TPP3/C2 | 9.75:0.25 | 250:1 | 63.0 ± 0.3 | 0.12 ± 0.03 | 8 ± 1 | 18% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangasamy, S.; Bandini, E.; Venturini, A.; Bozzuto, G.; Migani, S.; Calcabrini, A.; Sennato, S.; Zuffa, C.; Maini, L.; Brion, A.; et al. Novel Azole-Modified Porphyrins for Mitochondria-Targeted Photodynamic Therapy. Molecules 2025, 30, 2688. https://doi.org/10.3390/molecules30132688
Rangasamy S, Bandini E, Venturini A, Bozzuto G, Migani S, Calcabrini A, Sennato S, Zuffa C, Maini L, Brion A, et al. Novel Azole-Modified Porphyrins for Mitochondria-Targeted Photodynamic Therapy. Molecules. 2025; 30(13):2688. https://doi.org/10.3390/molecules30132688
Chicago/Turabian StyleRangasamy, Sabarinathan, Elisa Bandini, Alessandro Venturini, Giuseppina Bozzuto, Sofia Migani, Annarica Calcabrini, Simona Sennato, Caterina Zuffa, Lucia Maini, Anaïs Brion, and et al. 2025. "Novel Azole-Modified Porphyrins for Mitochondria-Targeted Photodynamic Therapy" Molecules 30, no. 13: 2688. https://doi.org/10.3390/molecules30132688
APA StyleRangasamy, S., Bandini, E., Venturini, A., Bozzuto, G., Migani, S., Calcabrini, A., Sennato, S., Zuffa, C., Maini, L., Brion, A., Bolze, F., Bombelli, C., & Ventura, B. (2025). Novel Azole-Modified Porphyrins for Mitochondria-Targeted Photodynamic Therapy. Molecules, 30(13), 2688. https://doi.org/10.3390/molecules30132688