Abstract
BOPAM exhibits high fluorescence quantum yields, along with exceptional photostability, rendering it a promising platform for applications as fluorescence sensors. However, the development of BOPAM-based fluorophores with extended emission wavelengths remains limited, and the underlying mechanisms of fluorescence quenching via the population of dark twisted intramolecular charge transfer (1TICT) excited states are not yet fully understood. To address these gaps, we synthesized a series of BOPAM derivatives by incorporating electron-donating groups at the boron atoms and the phenyl rings of the BOPAM core. The introduction of bromide, phenyl, and naphthyl groups preserved the intrinsic locally excited (1LE) emission of BOPAM. In contrast, the incorporation of diphenylamine (BP-DA) and triphenylamine (BP-TA) moieties resulted in a red-shifted emission, attributed to an enhanced intramolecular charge transfer (ICT) process. Notably, in acetonitrile, BP-DA exhibited weak fluorescence originating from a 1TICT state, which was populated via the S2 → 1TICT transition. Furthermore, the emission observed from BP-TA was associated with a higher-lying excited state, likely the initially populated S2 state possessing a 1LE character. These findings not only introduce novel red-emissive BOPAM-based fluorophores, but also offer valuable insights into the role of the S2 state in governing fluorescence quenching mechanisms in BOPAM derivatives.
1. Introduction
In recent years, fluorophores have shown broad application in fields such as bioimaging, sensors, and organic optoelectronics, significantly driving the exploration and development of novel organic fluorescent dyes [1,2,3,4,5,6]. Particularly, organic small-molecule fluorescent dyes, with their high synthetic flexibility, exhibit remarkable advantages in molecular structure design and functional modifications. Since boron dipyrromethene (BODIPY) fluorescent dyes were first reported by Treibs and Kreuzer in 1968, research on boron-chelated small-molecule dyes has continued to flourish [7,8,9,10]. The success of BODIPYs can be attributed not only to their excellent photophysical properties, but also to their rich potential for functional modifications [11,12]. The core structure of BODIPY has multiple modification sites, allowing for functionalization at each position of the central skeleton through straightforward chemical reactions such as coupling reactions, Knoevenagel condensation, and halogenation [13,14]. These modifications enable the improvement in various physicochemical properties including water solubility and biocompatibility [13,14,15,16,17,18,19].
A recently reported novel asymmetric diboron complex small-molecule dye, bis(difluoroboron)pyrrole amidrazone (BOPAM), features a central core composed of a pyrrole unit, a six-membered and a five-membered heterocyclic ring, maintaining excellent overall planarity [20]. BOPAM demonstrates highly desirable photophysical properties, including a high fluorescence quantum yield (FLQY) in both organic solvents and the solid state, along with a straightforward synthesis, structural tunability, and significant stability [20,21]. These promising characteristics enhance the potential of BOPAM for practical applications such as acid sensing [21]. However, for broader applicability, particularly in optoelectronic and bioimaging purposes, its emission should be close to the red or near-infrared (NIR) region while achieving a large Stokes shift. This spectral tuning can be realized through molecular modifications, specifically by introducing strong electron-donating groups (EDGs). Thanks to the BOPAM core’s intrinsic strength as an electron-withdrawing group (EWG), establishing a donor–acceptor framework is straightforward in achieving the desired, red-shifted emission and increased Stokes shift.
Therefore, we systematically introduced phenyl (Ph) and naphthyl (Na) groups onto the boron center, yielding BP-Ph and BP-Na, respectively. Subsequently, bromine (Br), diphenylamine (DA), and triphenylamine (TA) groups were incorporated into the phenyl ring of the BOPAM core to generate BP-Br, BP-DA, and BP-TA, respectively (see Figure 1). Single-crystal X-ray diffraction (SC-XRD) analysis was conducted, revealing distinct emission characteristics in the solid state including the intrinsic localized excited (1LE) emission of BOPAM as well as twisted configurations and charge transfer-induced emissions. In organic solvents, BP-Ph, BP-Na, and BP-Br predominantly exhibit 1LE emissions with high FLQY comparable to the parent BOPAM (BP). In contrast, BP-DA and BP-TA display characteristic intramolecular charge transfer (ICT) emissions in toluene and tetrahydrofuran, resulting in a red-shifted emission relative to BP. Notably, BP-TA exhibits a broad-band emission centered at 608 nm with a high FLQY of 51% in tetrahydrofuran. These findings provide a new molecular design of BOPAM-based fluorophores bearing red-shifted emissions combined with large Stokes shifts, demonstrating their potential for practical bioimaging applications.
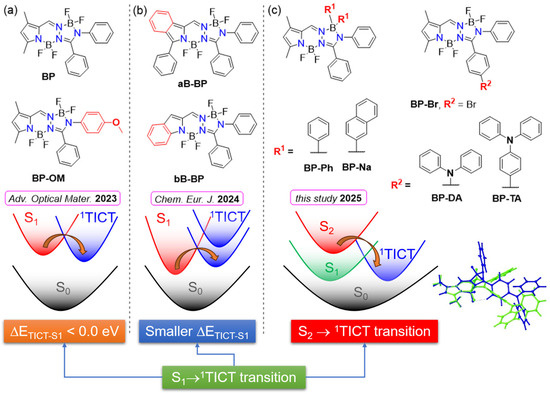
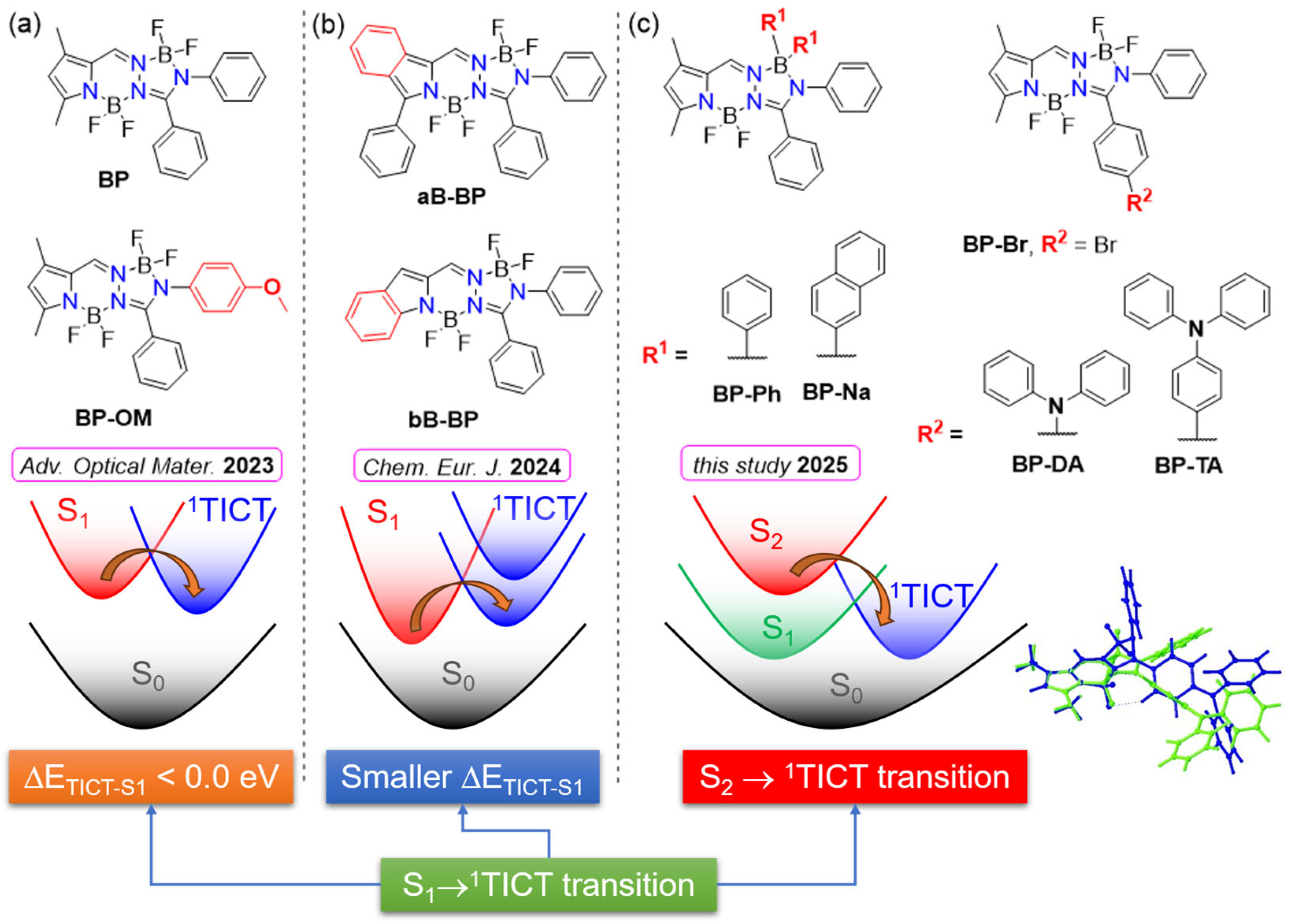
Figure 1.
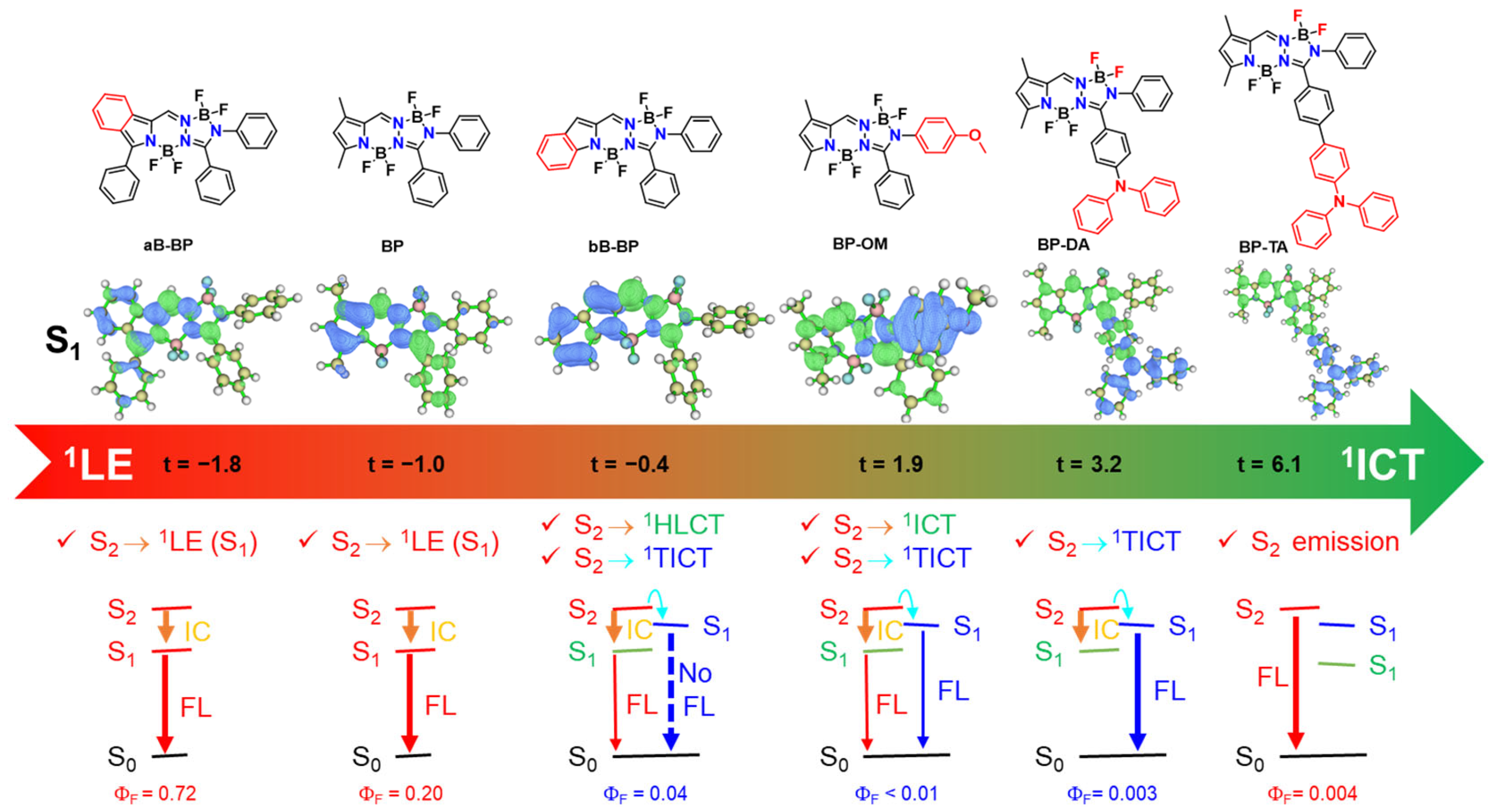
Structures of BOPAM and excited state decay models highlighting the role of the 1TICT state in fluorescence quenching: (a) BP (5a) and BP-OM (5i) [20]; (b) aB-BP (5a) and bB-BP (5) [21]; (c) in the current study: BP-R1 and BP-R2. Overlay of the optimized geometries of 1ICT (green) and 1TICT (blue) states of BP-DA are shown in the right-bottom of the figure.
Previous studies [20,21] have highlighted the role of a twisted intramolecular charge transfer (1TICT) state, corresponding to a local minimum in the S1 potential energy surface, as a non-emissive state and have identified its pivotal role in the fluorescence quenching mechanisms of BP-OM [20] (Figure 1a) and bB-BP [21] (Figure 1b). Importantly, the optimized geometry of the 1TICT state adopts a twisted configuration, in contrast to the planar configurations typically associated with other excited states such as the 1ICT or 1LE states (see the 1TICT configuration of BP-DA in Figure 1c). Moreover, the 1TICT state is characterized by its small oscillator strength (f < 0.02) and red-shifted energy. The S1 → 1TICT transition is readily accessible due to either a negative energy gap [20] (Figure 1a) or a smaller energy gap [21] (Figure 1b) between the S1 and 1TICT states, both of which critically influence the observed quenching mechanisms. For the systems shown in Figure 1a,b, S1 is of 1LE character.
Within the series of the new investigated compounds (see Figure 1c), some of them still adhere to this model (e.g., BP-Br, BP-Ph, and BP-Na). However, in the case of BP-DA, bearing an 1ICT state as S1, the calculated S1-1TICT energy gap (0.49 eV) is substantially larger than those calculated for BP, BP-Br, BP-Ph, BP-Na, and bB-BP (0.10–0.28 eV). Despite the large energy gap, emission from the 1TICT state is still observed for BP-DA. Furthermore, the emission observed from BP-TA is associated with a higher-lying excited state, likely the initially populated S2 state. These findings suggest that a more complex photophysical scenario is obtained for BP-DA and BP-TA. Alternative and complementary models were devised here for BOPAM’s excited state decay based on our joint experimental/computational investigations.
2. Results and Discussion
2.1. Synthesis
In the synthetic route (see Scheme 1), we implemented the previously reported optimized one-pot method [20,21], starting from thioamide to prepare the corresponding amidrazone. Subsequently, the intermediate 1 obtained by condensation of the amidrazone with 3,5-dimethyl-1H-pyrrole-2-carbaldehyde was further complexed with boron trifluoride etherate to obtain the BOPAM molecule. This method allows for the facile introduction of substitutions on the boron center, not only avoiding careful control of the harsh reaction conditions employed in previous literature, but also preventing the occurrence of multiple substitutions within the system [22,23,24]. To elucidate the influence of substituents on the photophysical properties, structures were designed and modified based on two sites on the BOPAM skeleton, namely the boron and the carbon-linked aryl group of the amidrazone.
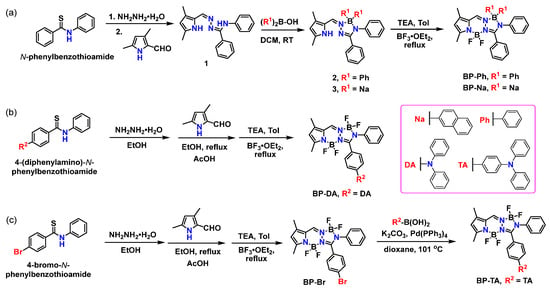
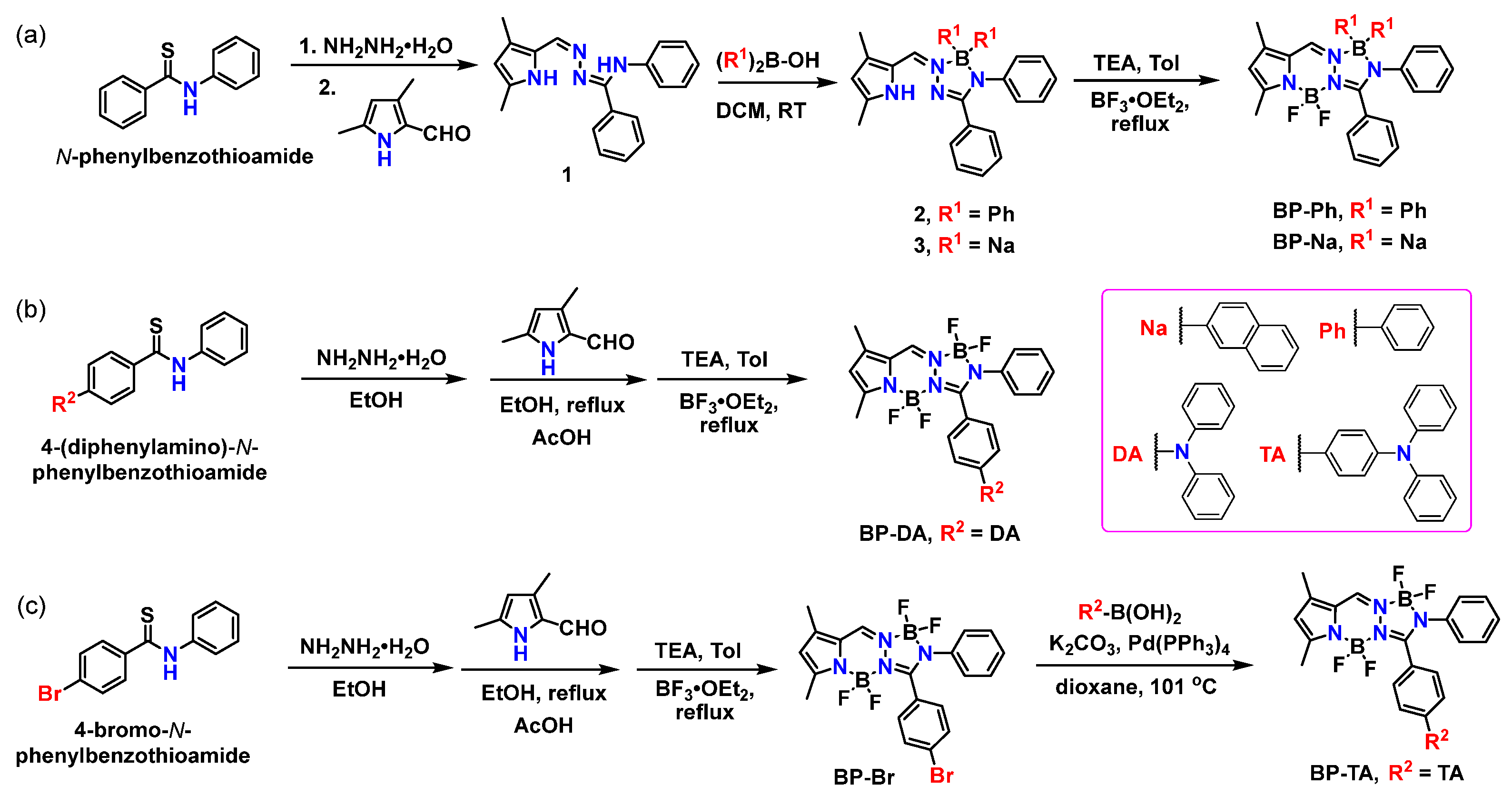
Scheme 1.
Synthetic route of (a) BP-Ph and BP-Na, (b) BP-DA, and (c) BP-Br and BP-TA.
First, two BOPAMs (BP-Ph and BP-Na) were designed through the introduction of phenyl and naphthyl groups to the boron atom (of the 5-membered ring), respectively. The ligand 1 selectively reacts with diphenylborinic acid or di(2-naphthyl)borinic acid under mild conditions (room temperature) to form monochelated arylboron complex 2 or 3, which is further complexed with boron trifluoride etherate to obtain the final compound BP-Ph or BP-Na, respectively (Scheme 1a). Second, a diphenylamine (DA) group was introduced into the phenyl ring of the BOPAM skeleton. Starting with 4-(diphenylamino)benzaldehyde, 4-(diphenylamino)-N-phenylbenzothioamide was synthesized with a high yield through a three-step reaction sequence (see Scheme S1), followed by our three-step one-pot method to obtain the target product BP-DA (Scheme 1b). Finally, another modification involved attaching a bromine atom onto the phenyl group of the BOPAM skeleton to obtain BP-Br. Thus, the triphenylamine group (TA) was introduced into BP-Br through the Suzuki coupling reaction to obtain BP-TA (Scheme 1c).
Products were characterized through NMR spectroscopy (1H, 13C, 11B, and 19F) and TOF-HRMS, and their molecular geometry was further confirmed by single-crystal X-ray diffraction analysis (SC-XRD) (more details are summarized in the Supplementary Materials). The analysis of their SC-XRD structures is discussed in the next section.
2.2. Structural Characterization by SC-XRD
In this study, the solid state emission properties of BOPAM were rationalized in terms of the single crystal X-ray diffraction (SC-XRD) analyses (Figure 2). Specific intermolecular interactions are discussed below and support the investigation of their emission in a solid state (see Section 2.6).
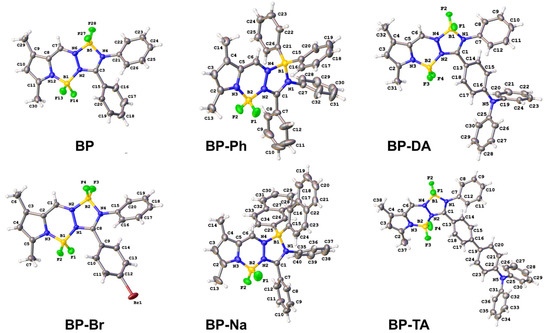
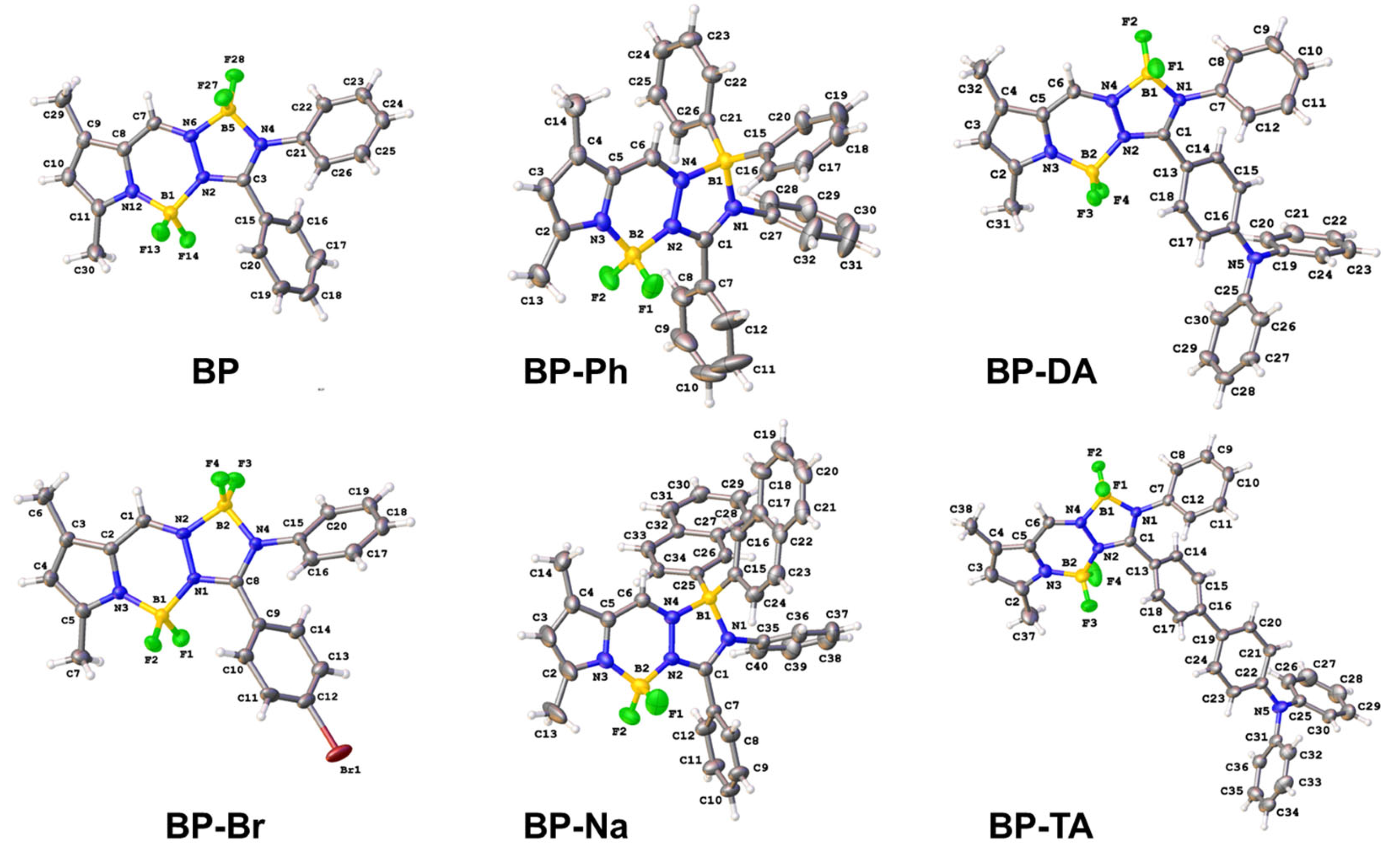
Figure 2.
X-ray structures of BP [20], BP-Br, BP-Ph, BP-Na, BP-DA, and BP-TA. Displacement ellipsoids drawn at the 30% probability level.
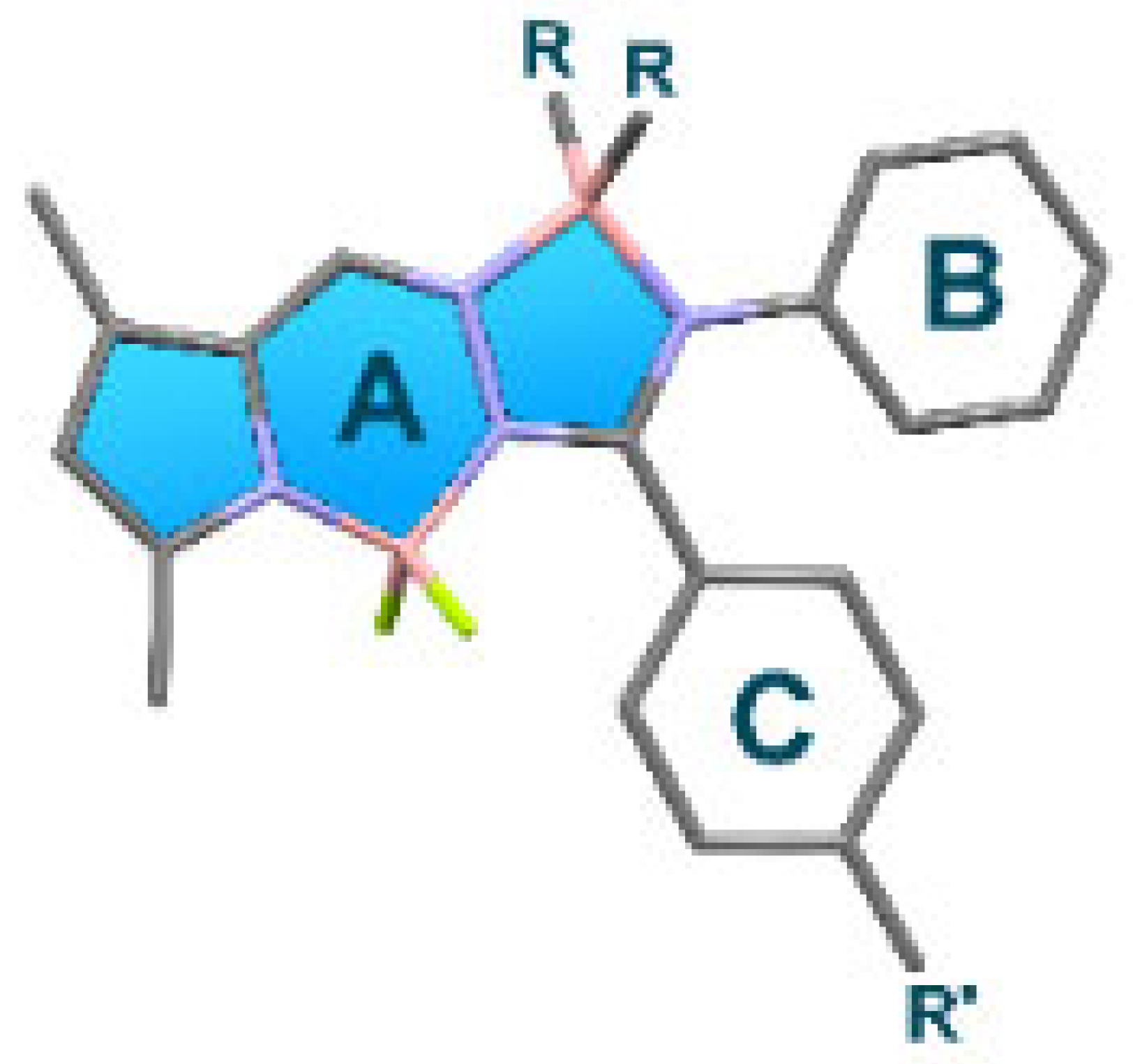
In the first observation, the central 12-membered ring system was planar with root mean square (r.m.s) deviations from planarity between 0.048 Å (for molecule B in BP-Na) and 0.162 Å (for BP-DA). BP-Br crystallized in the orthorhombic space group Pbca, while the parent BP crystallized in the monoclinic space group I2/a [20]. The r.m.s. deviation of fitting the central 12-membered ring of both structures was 0.127 Å (Figure S1a). Both molecules were characterized by an intramolecular C-H⋯F interaction (H22⋯F28 = 2.53 Å in BP, H10⋯F2 = 2.45 Å in BP-Br). The crystal packing of both molecules showed C-H⋯F interactions for BP, and C-H⋯F, C-H⋯π and C-Br⋯π interactions for BP-Br (Table S1). Notably, BP exhibited an even and uniform arrangement with head-to-tail stacking between two (plane A) aromatic system cores (Figure 6b). Upon the introduction of a bromine atom in BP-Br, alterations in the orientation of the phenyl rings were observed (refer to Table 1), resulting in a more disordered spatial arrangement (Figure S2a). The phenyl group in plane C of BP demonstrated a greater degree of twisting compared with that of BP-Br, whereas the phenyl group in plane B in BP was less twisted than the corresponding group in BP-Br, relative to the aromatic core (plane A) (see Table 1).

Table 1.
Dihedral angles (°) between planes A, B, and Cin BOPAM dyes BP [20], BP-Br, BP-Ph, BP-Na, BP-DA, and BP-TA.
The spatial arrangement of BP-Ph and BP-Na in the solid state was predicted to be disordered, requiring a direct observation via SC-XRD analysis. BP-Ph crystallized in the monoclinic space group P21/n, whereas BP-Na crystallized in the triclinic space group P-1, with two molecules in the asymmetric unit. Both molecules in the asymmetric unit of BP-Na differed in the orientation of one of the naphthyl groups (r.m.s. deviation of fitting the central 12-membered ring of both molecules is 0.110 Å, Figure S1b). The substitution of both fluorine atoms on boron by two phenyl rings in BP-Ph or two naphthyl groups in BP-Na had a major effect on the orientation of the adjacent phenyl groups (in planes B and C) (Table 1). Only one example of C-H⋯F interaction was observed in the crystal packing of BP-Ph, and numerous C-H⋯F and C-H⋯π interactions were present in the crystal packing of BP-Na (Table S2). However, there was no specific packing arrangement, such as herring-bone motif or head-to-tail interactions, and displayed a notably disordered spatial arrangement (Figure S2b,c). This structural disorder suggests that the emission of BP-Ph and BP-Na in the solid state originates from the intrinsic 1LE emission of the BOPAM core or 1ICT emission.
Both compounds BP-DA and BP-TA crystallized in the triclinic space group P-1. The extension of the conjugated system did not cause major changes in the dihedral angles between the best plane through the 12-membered ring and the phenyl substituents (Table 1). The extra nitrogen was planar with a deviation from the best plane through three neighboring C atoms of 0.107(2) Å for BP-DA and 0.082(4) Å for BP-TA. Phenyl rings C13–C18 and C19–C24 in BP-TA formed a dihedral angle of 27.41(15)°. An intramolecular C-H⋯F interaction was present (H18⋯F3 = 2.53 Å for BP-TA and 2.40 Å for BP-DA). Both crystal packings were stabilized by C-H⋯F and C-H⋯π interactions, but for BP-DA, B-F⋯π interactions also contributed (Table S3). Notably, both BP-DA and BP-TA displayed an even and uniform molecular arrangement, characterized by a head-to-tail stacking pattern between adjacent triphenylamine groups (see Figure 6c,d). This structural feature facilitates charge transfer processes in the solid state.
2.3. UV–Vis Absorption and Emission Properties
The photophysical characteristics of BOPAM were investigated through steady-state spectroscopic measurements. The UV–Vis absorption and emission spectra were recorded in toluene (Tol), tetrahydrofuran (THF), acetonitrile (ACN), and ethanol (EtOH) at a concentration of 10 µM. Complementary to the experimental analyses, their absorption and emission properties were determined with TD-DFT calculations using the PBE0/6-31+G(d) level of theory within the integral equation formalism of the polarizable continuum model (IEFPCM) to model solvent effects in Tol and ACN (see the Section 3 for the full computational details).
The absorption spectra of all BOPAM derivatives exhibited their most intense absorption bands at 389–403 nm, characterized by a similar spectral shape and strong absorbance (ε = 35.8–42.5 × 103 cm−1 M−1) (Figure 3a–c and Figure S3, Table 2). Their absorption maxima demonstrated a relatively weak dependence on solvent polarity, minimal solvatochromic shifts (<10 nm) across Tol, THF, ACN, and EtOH. The absorption spectra of BP, BP-Br, BP-Ph, and BP-Na were quite broad (FWHM = 3225–5150 cm−1, see Table S4) compared with that of the BODIPY [25,26] nearly structureless band, which showed a red shift from ACN over THF to Tol, and can be correlated with the increase in the polarizability of the solvent [25]. This suggests that the investigated BOPAMs had no or only a very small dipole moment in their ground states. Upon closer inspection, their absorption bands showed a weak shoulder at the blue side (BP, BP-Br) or the red side (BP-Ph, BP-Na), which can be attributed to the 0-1 or 0-0 vibronic bands, respectively. This means that while for BP and BP-Br, the maximum corresponded to the 0-0 vibronic band, it corresponded to the 0-1 transition in BP-Ph and BP-Na. Furthermore, for BP-DA and BP-TA, a shoulder with low intensity could be observed between 500 and 600 nm, especially in EtOH. The 0-0 vibronic band of BP-Ph and BP-Na was red shifted at about 1200 cm−1 versus that of BP, BP-Br, and BP-TA.
Table 2.
Photophysical properties of BP, BP-Br, BP-Ph, BP-Na, BP-DA, and BP-TA (10 µM) in toluene (Tol), tetrahydrofuran (THF), acetonitrile (ACN), ethanol (EtOH), and the solid state. Maximum absorption wavelength (λabs) (nm); molar absorption coefficient (ε) at maximum absorption wavelength (cm−1 M−1 10−3); maximum emission wavelength (λems) (nm); fluorescence emission quantum yield (ΦF) (%) [λex = 390 nm].
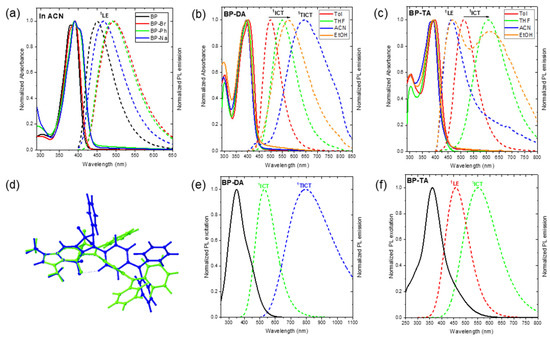
Figure 3.
Normalized UV–Vis absorption (solid line) and fluorescence emission (dot line) (λex = 390 nm) spectra of (a) BP, BP-Br, BP-Ph, and BP-Na (10 µM) in acetonitrile (ACN); (b) BP-DA (10 µM) in toluene (Tol), tetrahydrofuran (THF), (ACN), and ethanol (EtOH); (c) BP-TA (10 µM) in Tol, THF, ACN, and EtOH; (d) overlay of the optimized geometries of 1ICT (green) and 1TICT (blue) states of BP-DA. Calculated absorption (solid line); and emission (dot line) spectra from the 1ICT, 1LE, and 1TICT states of (e) BP-DA and (f) BP-TA using TD-PBE0/6-31+G(d) in IEFPCM for ACN.
Figure 3.
Normalized UV–Vis absorption (solid line) and fluorescence emission (dot line) (λex = 390 nm) spectra of (a) BP, BP-Br, BP-Ph, and BP-Na (10 µM) in acetonitrile (ACN); (b) BP-DA (10 µM) in toluene (Tol), tetrahydrofuran (THF), (ACN), and ethanol (EtOH); (c) BP-TA (10 µM) in Tol, THF, ACN, and EtOH; (d) overlay of the optimized geometries of 1ICT (green) and 1TICT (blue) states of BP-DA. Calculated absorption (solid line); and emission (dot line) spectra from the 1ICT, 1LE, and 1TICT states of (e) BP-DA and (f) BP-TA using TD-PBE0/6-31+G(d) in IEFPCM for ACN.
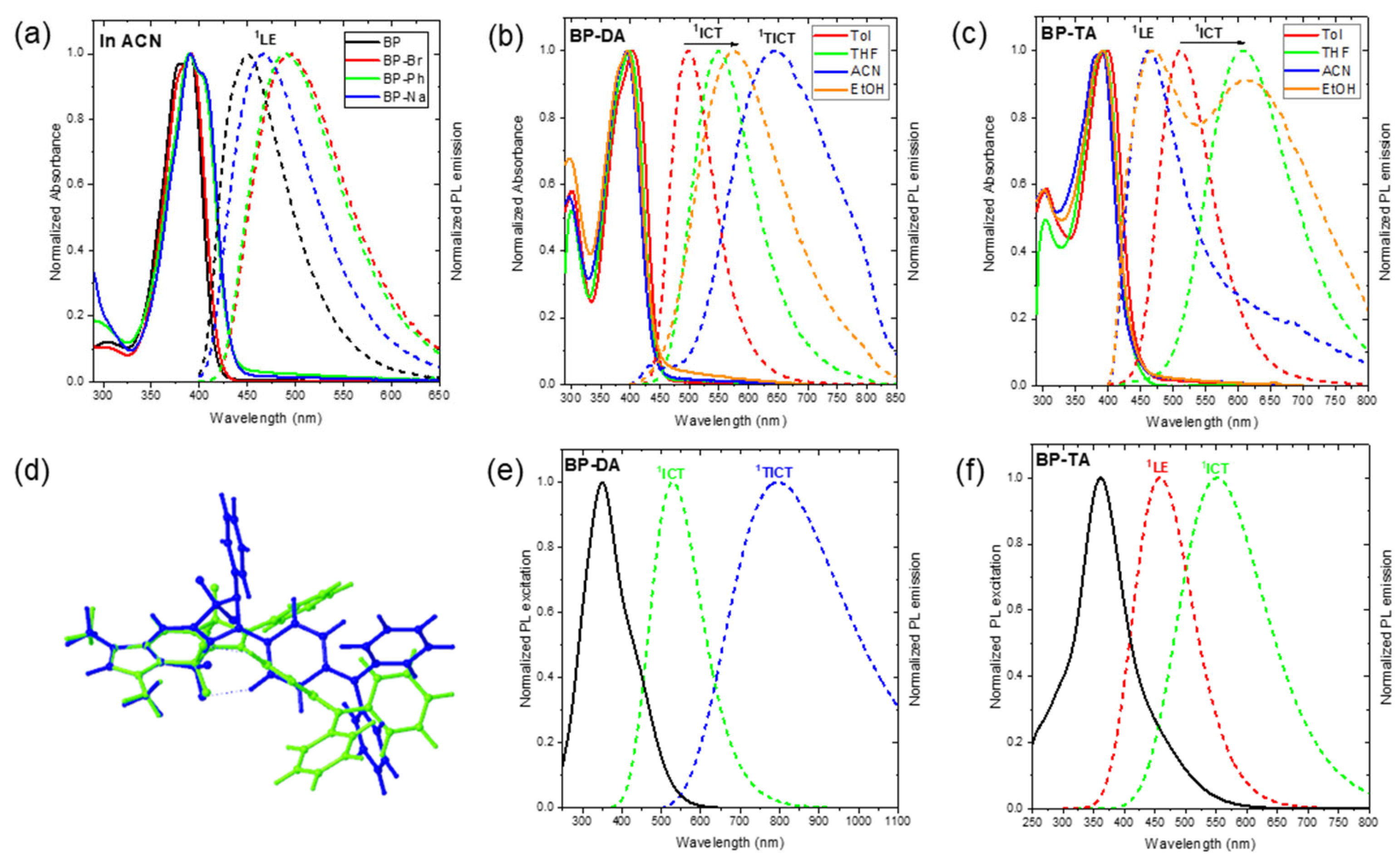
Notably, the most intense absorption band in BP, BP-Br, BP-Ph, and BP-Na was assigned to the S0 → S1 transition (f > 0.78), predominantly characterized by a highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) transition (>97%) of 1LE character (Table S7). Conversely, for BP-DA and BP-TA, the strongest absorption was attributed to the S0 → S2 transition (f > 0.70), mainly governed by the HOMO-1 → LUMO transition (Table S7) of 1LE character. The HOMO and LUMO in BP, BP-Br, BP-Ph, and BP-Na as well as the HOMO-1 and LUMO in BP-DA and BP-TA were primarily localized within the BOPAM core (Figure S4). Furthermore, the associated electronic transitions exhibited highly negative hole-electron distribution indices (t < −1.3) (Figure S5), indicating that the absorption band with a peak around 395 nm of all of the studied BOPAMs corresponded to a purely 1LE transition.
Notably, the S0 → S1 transition in BP-DA and BP-TA was primarily contributed by the HOMO → LUMO transition (Table S7), with the HOMO residing in the triphenylamine moiety and the LUMO localized within the BOPAM core (Figure S4). The associated electronic transition exhibited highly positive hole-electron distribution indices (t-index = 3.08 and 6.58 Å, for BP-DA and BP-TA), indicative of an 1ICT transition. However, the oscillator strength (f) of this transition (0.29–0.43) was notably lower than that of the 1LE absorption (0.70–0.91). The 1ICT transition could be responsible for the long wavelength shoulder observed for BP-DA and BP-TA. Additionally, a higher-energy absorption band around 300 nm was observed in BP and BP-Br (see Figure 3a and Figure S3), which was assigned to the S0 → S3 transition (see Table S7), exhibiting an 1LE character (see Figure S5). In BP-Ph and BP-Na, the absorption band in this region (see Figure 3a and Figure S3) was attributed to the S0 → S4 transition (see Table S7), primarily associated with 1ICT characteristics (see Figure S5). Meanwhile, BP-DA and BP-TA (see Figure 3b,c) exhibited an S0 → S4 transition (see Table S7), which was assigned to the 1LE transition (see Figure S5) of the triphenylamine moiety.
2.4. The Presence of a Dark State
BP, BP-Br, BP-Ph, and BP-Na exhibited a strong unstructured fluorescence emission (ΦF = 45.0–33.5% in Tol) (Figure 3a). The Stokes shift of BP and BP-Br amounted to 3230 and 3710 cm−1 in Tol, while that of BP-Ph and BP-Na was apparently much larger (5010 cm−1 and 4630 cm−1, respectively). However, one should take into account that while for BP and BP-Br, the maximum of the absorption spectrum corresponded to the 0-0 vibronic band, it corresponded to the 0-1 vibronic band for Bp-Ph and BP-Na. For BP-Ph and BP-Na, when the shift between the 0-0 vibronic band of the absorption spectrum (shoulder) and the fluorescence maximum was calculated, this quantity was reduced to 3850 and 3320 cm−1, respectively, which was of the same magnitude as the Stokes shift observed for BP and BP-Br.
For BP, BP-Br, BP-Ph, and BP-Na, the Stokes shift increased slightly (by 190 to 520 cm−1) between Tol and ACN (Figure S3a, Table 2 and Table S5). This increase in the Stokes shift was accompanied by an increase in the FWMH of their corresponding emission spectra. This indicates that the excited state is characterized by a small dipole moment. One should note that in analogy to the earlier investigated BOPAM derivatives [20], the observed Stokes shift was much larger than that found for the BODIPY derivatives [25]. This suggests a significant change in some bond lengths or angles upon excitation, which was also reflected in the larger FWHM of the emission spectra. For BP, BP-Br, BP-Ph, and BP-Na, the ΦF decreased by a factor of two to three when increasing the solvent polarity from Tol to ACN. This possibly suggests that a charge transfer state is involved in the internal conversion to the ground state and that its accessibility is determined by the polarity of the solvent (i.e., we recall that while the energy of the 1ICT state will be strongly affected by the solvent polarity, this is not the case with the 1LE state). The incorporation of a bromine atom into BP induced a red-shifted emission (Table 2), which can be attributed to a decrease in the LUMO energy level (Figure S7). The ΦF value of BP-Br remained comparable to that of BP, suggesting a negligible heavy-atom effect in organic solvents. The introduction of phenyl (BP-Ph) and naphthyl (BP-Na) substituents at the boron atom resulted in a reduction in the HOMO-LUMO energy gap (Figure S7), thereby leading to a red-shifted emission compared with BP in organic solvents (Table 2). Consistent with their S1 absorption characteristics, the S1 emission of BP, BP-Br, BP-Ph, and BP-Na was primarily attributed to a pure 1LE transition, as supported by the computed results presented in Table S8 (for emission characteristic) and Figure S7 (for MO) and Figure S8 (for hole-electron analysis).
Conversely, BP-DA in Tol already had a larger Stokes shift of 4810 cm−1, which was increased in EtOH to 7890 cm−1 (Figure 3b). This suggests that the emission occurs from a highly polar excited state. The increase in the Stokes shift (4810–7890 cm−1) was accompanied by an increase in the FWHM of the emission band (3380–5000 cm−1) and a decrease in the ΦF values (49.1–3.6%) as the solvent polarity increased from Tol to THF and EtOH, displaying the characteristic behavior of an 1ICT (Figure 3b and Table 2). This behavior was further corroborated by the computational results, consistent with its assigned S1 emission characteristics (i.e., we recall the computed 1ICT character of S1, see Figure 3 and Figure S7, Table S8). However, in ACN, BP-DA exhibited a broad emission band with a peak centered at 644 nm and a significantly low quantum yield (ΦF = 0.3%). This observation further suggests the presence of an additional emissive species in BP-DA (e.g., 1TICT state).
Previous investigations of BOPAM derivatives [20,21] identified a dark and stable twisted intramolecular charge-transfer (1TICT) state/configuration (see Figure 3d) through an integrated computational and experimental photophysical characterization. This 1TICT state is known to facilitate non-radiative decay after the S1 → 1TICT transition because of its typically small oscillator strength and red-shifted emission energy, thus, leading to a significant reduction in fluorescence emission. Accordingly, the optimized geometry of the 1TICT state was calculated for compound BP-DA (see Figure 3d). Computational results predicted an emission wavelength of 796 nm (in ACN) for the 1TICT state of BP-DA, which was notably shorter than that of previously reported BOPAM derivatives such as BP-OM (1216 nm) and bB-BP (883 nm). Moreover, the oscillator strength of the 1TICT → S0 transition of BP-DA (0.10) was significantly larger than that of BP-OM (0.01) and bB-BP (0.02), suggesting a greater likelihood of observing the 1TICT emission experimentally (see Figure 3b,e). Moreover, the oscillator strength of the 1TICT state in BP-DA (0.1) was markedly lower than that of its 1ICT state in Tol (0.3) and ACN (0.6) (Table S8), which was consistent with the reduced ΦF value observed for BP-DA in ACN relative to the Tol, THF, and EtOH solvents. Based on these findings, we propose that a broad emission band with a peak centered at 644 nm of BP-DA in ACN can be assigned from the weak-emissive 1TICT state/configuration (see Figure 4b). This observation further reinforces the existence of non-emissive (dark) or weak-emissive 1TICT states in BOPAM derivatives.
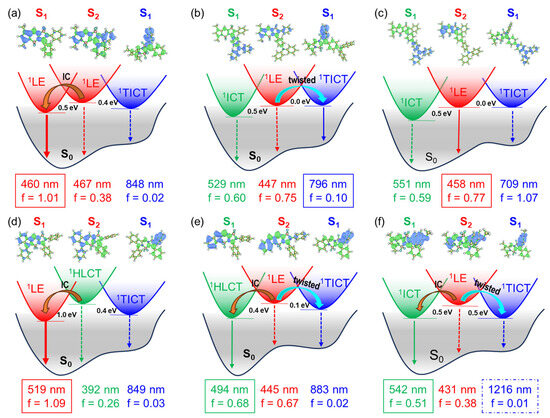
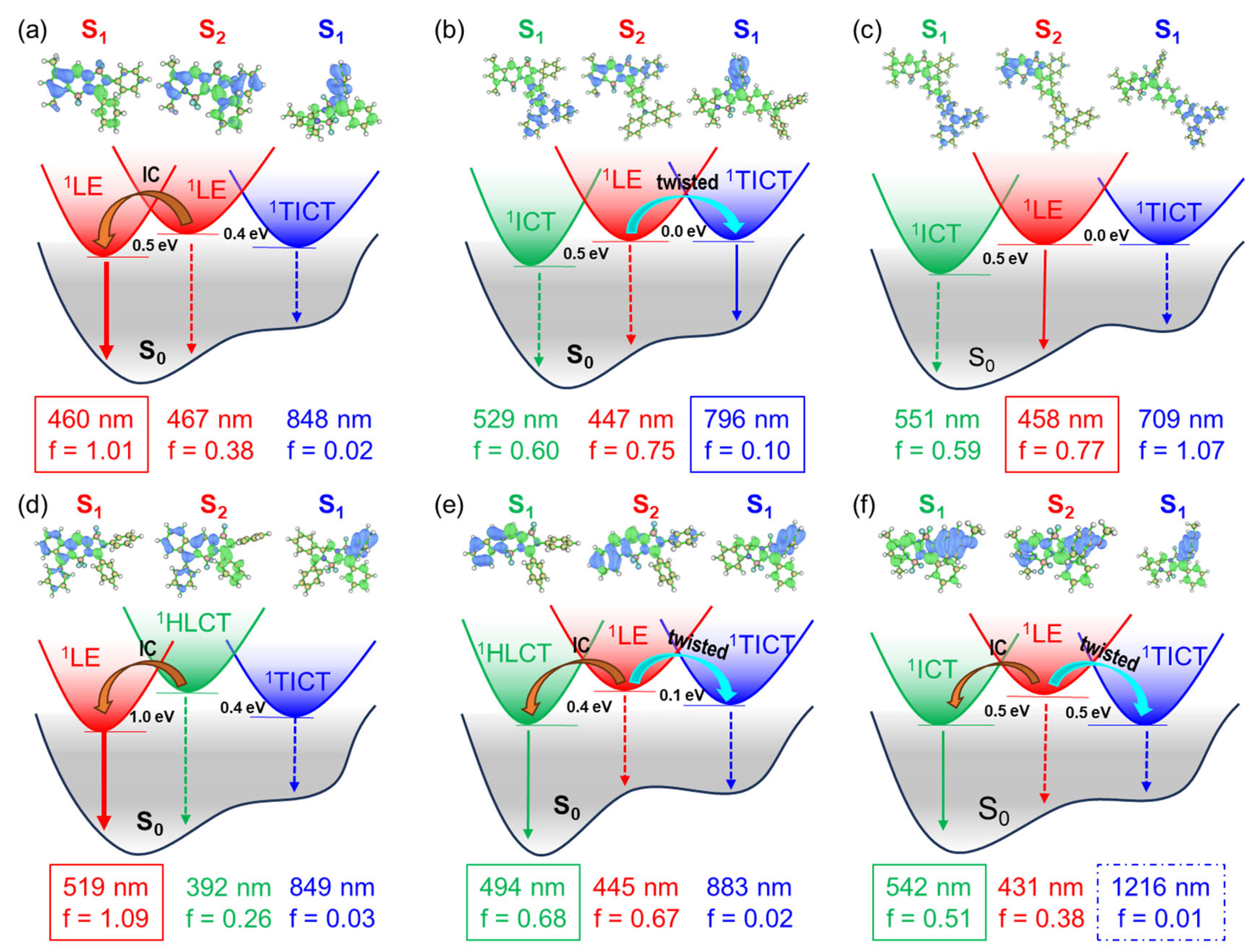
Figure 4.
Proposed schematic mechanism for the S1 → 1TICT transition of (a) BP, (b) BP-DA, (c) BP-TA, (d) aB-BP, (e) bB-BP, and (f) BP-OM illustrated by the hole (blue)-electron (green) distribution and oscillator strength (f) and adiabatic energy gaps. Internal conversion (IC).
Similar to the emission behavior of BP-DA in Tol, BP-TA exhibited a significantly larger Stokes shift than BP, BP-Br, BP-Ph, or BP-Na, which was further increased by 3470 cm−1 and 3590 cm−1 in THF and EtOH, respectively (see Table S5); a behavior typical for 1ICT emissions. This increased Stokes shift was accompanied by an increase in the FWHM of the emission band and a decrease in the FLQY from 62.1% (in Tol) to 51.0% and 1.2% (for the 1ICT emission) in THF and EtOH, respectively (Figure 3c). In ACN, the 1ICT emission was below the detection limit, although a residual 1LE emission was observed (cfr. infra). Considering the larger increase in the Stokes shift (or the larger red shift) when going from toluene to THF or EtOH, the 1ICT state of BP-TA had a larger dipole moment than that of BP-DA. Computational analysis further corroborated the presence of the 1ICT state in BP-TA (see Figure 3f and Figure S7, and Table S8). In a solvent with higher polarity, it was anticipated that the emission peak of BP-TA in ACN would occur at wavelengths longer than 608 nm. However, the observed emission of BP-TA in ACN was centered at approximately 462 nm (Figure 3c), which was only slightly longer than the emission peak of BP in ACN (450 nm). This deviation suggests the presence of a higher-lying excited state (see Figure 4c), indicative of an anti-Kasha emission with an FLQY of 0.4% [27,28]. This anti-Kasha emission can be attributed to the initially populated S2 state, which has a 1LE character, and was further confirmed by the computational results in Table S8 and Figure S10f. The similarity of the main absorption band and its oscillator strength of, on the one hand, BP-TA, and on the other hand, BP, BP-Br, BP-Ph, and BP-Na, suggests that this main absorption band must be attributed to a transition of 1LE character. One should note that in EtOH, besides the main emission maximum at 610 nm, a weaker band (PLQY of about 0.4%) at 468 nm was also observed. In Tol and THF, this weak anti-Kasha emission was hidden by the much stronger and less red-shifted 1ICT emission.
2.5. Revisiting the Excited State Decay of BOPAMs
In this study, we have demonstrated the presence of a 1TICT state in BP-DA. This behavior is reminiscent of previously reported systems such as BP-OM (compound 5i in ref. [20]) and bB-BP (compound 5 in ref. [21]). In the former, the S1 → 1TICT transition in BP-OM was found to occur when the energy gap between these states was negative (see Figure 3c in Ref. [20]). In the latter, the smaller energy gap of bB-BP compared with aB-BP facilitated the possible S1 → 1TICT transition in bB-BP (see Figure 5 in Ref. [21]). Among the series of newly investigated compounds, several (e.g., BP-Br, BP-Ph, and BP-Na) conformed to this energy gap-based model.
However, BP-DA deviated from this trend. Despite possessing a 1TICT state as the lowest singlet excited state (S₁), the calculated S1 → 1TICT energy gap for BP-DA was significantly larger (0.49 eV) than those for BP, BP-Br, BP-Ph, BP-Na, and bB-BP (ranging from 0.10 to 0.28 eV). Nevertheless, an emission from the 1TICT state was still observed in BP-DA, indicating that the energy gap criterion does not universally apply across this series of compounds.
To ensure methodological consistency, the excited-state properties of BP-OM, aB-BP, and bB-BP were recalculated using the computational protocol employed in this study. The revised S1-1TICT energy gap for BP-OM remained negative (−0.04 eV), whereas those for aB-BP and bB-BP were significantly positive (0.52 eV and 0.28 eV, respectively). These findings suggest that the population of the 1TICT state in BP-DA and potentially other BOPAM derivatives proceeds via a mechanism distinct from a direct S1 → 1TICT transition.
To elucidate the nature of the S1 state in aB-BP, bB-BP, and BP-OM, we conducted an analysis of the hole-electron distribution. The S1 state of aB-BP exhibited clear 1LE characteristics, as both the hole and electron were localized within the same [a]benzo-fused BOPAM core (t = −1.8 Å) (Figure 4d, Figure 5 and Figure S8). Conversely, the S1 state of BP-OM exhibited distinct 1ICT characteristics, with the hole localized in the methoxy phenyl group and the electron in the BOPAM core (t = 1.9 Å) (Figure 4f, Figure 5 and Figure S9). These assignments were further corroborated by the experimental UV–Vis absorption and fluorescence spectra [20,21]. For bB-BP, the hole was localized within the benzo fragment, while the electron resided in the BOPAM core (Figure 4e, Figure 5 and Figure S9). The t-index for this state was slightly negative (−0.4 Å), which was significantly higher than the values observed for the 1LE states of BP (−1.0 Å) and aB-BP (−1.8 Å). Additionally, the Stokes shift between the absorption and emission peaks of bB-BP (134 nm) was significantly larger than that of aB-BP (35 nm) in ACN [21]. The emission peak of bB-BP was red shifted from Tol (505 nm) to ACN (531 nm), indicating that the S1 state of bB-BP is best characterized as a hybridized local and charge-transfer (1HLCT) excited state.
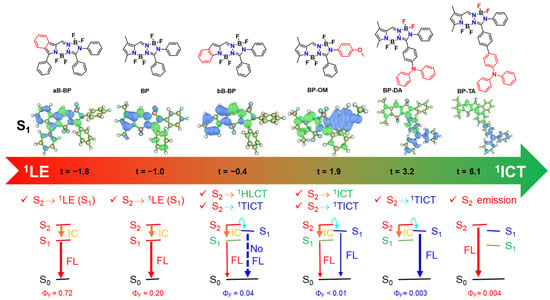
Figure 5.
Illustration of the possibility of the transition from S2 (1LE and 1HLCT) to S1 (1LE, 1HLCT, 1ICT and 1TICT) state of BOPAM. Nature of the states is manifested through the hole (blue)-electron (green) distribution and its t-index (Å). A negative t-index stands for the 1LE state while a positive (and larger) t-index correlates with a larger 1ICT character.
Our findings indicate that the population of the 1TICT state of BOPAM is suppressed when the S1 state of BP, BP-Br, BP-Ph, and BP-Na has an 1LE character, even in cases where the 1LE-1TICT energy gap is relatively low (e.g., 0.10–0.18 eV for BP, BP-Br, BP-Ph, and BP-Na). However, the 1TICT state of bB-BP, BP-OM, and BP-DA is populated when the S1 state exhibits 1HLCT and 1ICT characteristics (Figure 4 and Figure 5). These excited states possess charge-transfer features similar to the 1TICT state but adopt distinct configurations. The fluorescence emission from the 1TICT configuration correlates with the charge-transfer contribution in the S1 state. For instance, the fluorescence emission from the 1TICT state of bB-BP is non-observable [21], whereas the emission from BP-OM is ambiguous due to potential overlap with its 1ICT emissions [20]. BP-DA, which possesses stronger charge-transfer characteristics, exhibited observable 1TICT emissions in the fluorescence spectra (see illustration in Figure 5).
Notably, BP-TA, which predominantly exhibited ICT characteristics (t-index = 6.1 Å), did not display an observable 1TICT emission. However, an emission from the S2 state with 1LE character was detected in BP-TA, indicating a potential role of the S2 state in facilitating the population of the 1TICT state in BOPAM derivatives. To further investigate this, the second singlet excited states (S2) of the BOPAM compounds were optimized using the TD-PBE0/6-31+G(d) level, incorporating the IEFPCM solvent model for ACN. Analysis of the hole-electron distribution revealed that the S2 states of most of the studied BOPAM derivatives predominantly exhibited an 1LE character, as indicated by the negative t-index values. An exception was the S2 state of aB-BP, which displayed a slight positive t-index (0.37 Å), suggesting an intramolecular charge-transfer (ICT) character. This state may be classified as an 1HLCT state, analogous to the S1 state observed in bB-BP.
The adiabatic energy of the S₂ state was higher than that of the 1TICT state (see Figure 4 and Figure S11), suggesting the potential of an S2 → 1TICT internal conversion. However, in the cases of BP, BP-Br, BP-Ph, and BP-Na, the internal conversion from S2 (1LE) to S1 (1LE) dominated, resulting in population of the S1 state. Similarly, in aB-BP, the primary relaxation pathway involved the internal conversion from S2 (1HLCT) to S1 (1LE). In contrast, for bB-BP, BP-OM, and BP-DA, the S2 (1LE) → S1 (1TICT) internal conversion competed effectively with the S2 (1LE) → S1 (1ICT or 1HLCT) internal conversion, leading to the population of the 1TICT state. Notably, the 1TICT state adopted a twisted geometry in contrast to the planar configuration of the S2 and other S1 states. In BP-TA, the S2 state exhibited a purely LE character, as evidenced by the lowest t-index (−1.9 Å) among the BOPAM derivatives studied, indicating a high stability. This enhanced stability renders the S2 emission more favorable than the S2 → S1 (1LE, 1HLCT, 1ICT, and 1TICT) transitions [27,28]. Collectively, these findings highlight the significant role of the S2 state in facilitating the population of the 1TICT state via the S2 → 1TICT internal conversion. This, in turn, provides insights into the formation and behavior of the weakly emissive or non-emissive 1TICT (dark) state in BOPAM derivatives.
2.6. Solid State Spectra
The synthesized BOPAM compounds exhibited a bright emission in the solid state when irradiated with 365 nm light (Figure S12b), which was subsequently quantified via photoluminescence spectroscopy (see results in Figure 6a and Table 2). The emissions displayed a yellow-to-orange hue, consistent with the initial observations of the BOPAMs in our previous study [20]. However, the precise nature and mechanisms underlying their emission behavior in the solid state have yet to be thoroughly elucidated.
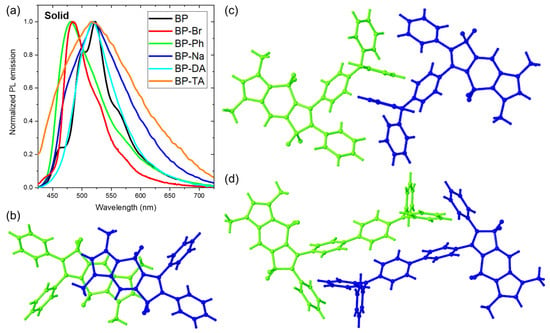
Figure 6.
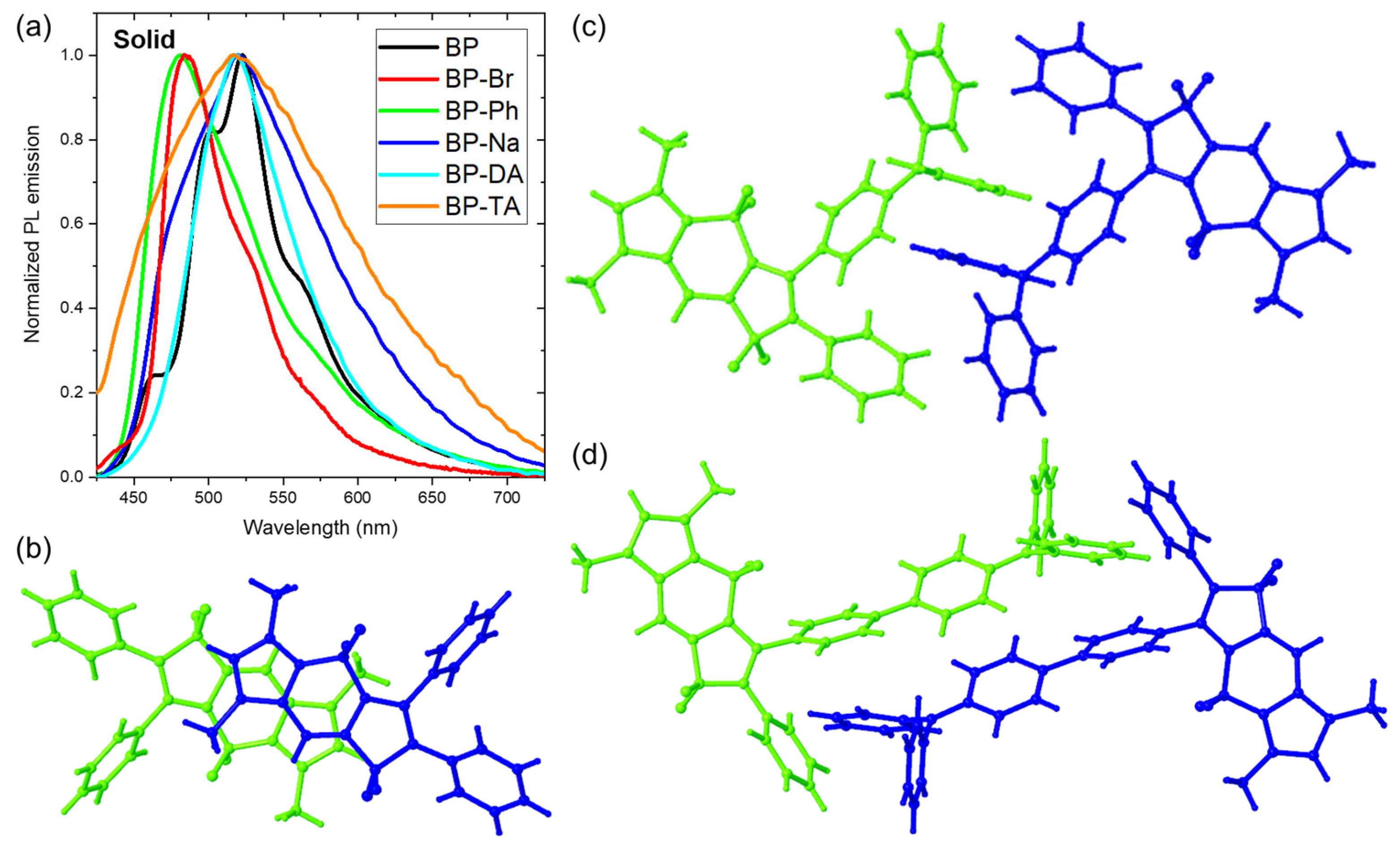
(a) Normalized fluorescence emission of BOPAMs in the solid state; partial crystal packing of (b) BP, (c) BP-DA, and (d) BP-TA.
Prior to that, their absorption spectra were measured in the solid state. Those of BP-Ph, BP-Na, BP-DA and BP-TA consisted of a broad structureless band whose maximum was red shifted by 2000 ± 200 cm−1 versus toluene (Figure S12a). For BP [20] and BP-Br, the red shift of the maximum was less than 400 cm−1, but the absorption spectra showed on their red edge a shoulder at 460 and 455 nm, respectively, corresponding to a red shift of 3320 and 3150 cm−1 versus toluene. The shoulder and maximum are probably related to vibronic progression. While in the solid state the onset of all absorption spectra was red shifted, suggesting exciton interaction [29,30,31] between neighboring molecules, this red shift was less outspoken for the emission spectra. This can be related to the rigidity of the environment in the solid state or to exciton interaction between neighboring molecules, both leading to less structural relaxation in the excited state [32,33].
The emission spectrum of BOPAM 5a (BP), which exhibited 1LE emissions in both Tol and ACN, consisted of a shoulder and maximum red shifted over 1710 and 2710 cm−1, respectively, versus toluene [20]. In contrast, BOPAM 5i (BP-OM), which showed ICT contributions in ACN, experienced a smaller Stokes shift relative to Tol (Δν = 1270 cm−1) [20]. As the absorption spectrum (long wavelength shoulder) was red shifted over 3320 cm−1 versus toluene, the Stokes shift compared with toluene decreased by 600 and 1600 cm−1 for the shoulder and the maximum of the emission spectrum, respectively. In addition, the red shift of the absorption suggests the formation of J-type aggregation, which corresponds to the crystal structure of BP, where the A planes of two neighboring molecules were parallel but shifted along their long axis (Figure 6b). BP exhibited additional shoulder peaks, which can be rationalized by the presence of strong vibronic coupling. Thus, the emission of BP has an 1LE character and was red shifted due to the head-to-tail parallel arrangement of the A planes at a short distance (3.7 Å), leading to a strong exciton coupling.
The emission spectra of BP-Br and BP-Ph consisted of a maximum at 484 nm and 481, respectively, compared with toluene of a red shift of 750 cm−1 or a blue shift of 380 cm−1, respectively. The shoulder and the maximum are possibly part of vibronic progression. This means that compared with toluene, the Stokes shift was reduced by 2400 cm−1 (using the long wavelength shoulder of the absorption spectrum) and 2700 cm−1 for BP-Br and BP-Ph, respectively. The observation of BP-Br parallels the intrinsic emission of the benzo-fused BOPAM via its brominated derivative in our previous study [21]. Upon the introduction of a bromine atom in BP-Br, alterations in the orientation of the phenyl rings were observed (refer to Table 1), resulting in a more disordered spatial arrangement (Figure S2a). This could lead to a weaker exciton interaction, and hence a smaller red shift of the emission between toluene and the solid state. In BP-Ph, the exciton interaction was reduced to such an extent that the effect of the reduced excited state relaxation exceeded that of the exciton interaction, resulting in a blue shift of the emission between toluene and the solid state. In addition, the FLQY of BP-Br (9.0%) was significantly lower than that of BP (32.4%), which can be ascribed to an intermolecular heavy-atom effect. This is consistent with the behavior of the bromide-substituted benzo-fused BOPAM (5b in Ref. [21]). BP-Ph demonstrated a significant FLQY of 42.7%, suggesting that its emission originates from the intrinsic 1LE luminescence of the BOPAM core in the solid state.
The emission spectrum of BP-Na in the solid state again resembled that of BP and consisted of a shoulder at 502 nm and a maximum at 519 nm, which were red shifted over 570 and 1220 cm−1, respectively, versus toluene. The shoulder and the maximum are possibly part of a vibronic progression. As the absorption spectrum was red shifted over 2140 cm−1 versus toluene, the Stokes shift was, compared with toluene, decreased by 910 and 1910 cm−1 for the shoulder and the maximum of the emission spectrum, respectively. While one would, based on the absence of π–π interactions between neighboring 12 membered rings (A-planes), expect a similar emission spectrum as observed for BP-Ph, the emission spectrum of BP-Na in the solid state was red shifted by 1220 cm−1 (maximum) versus toluene. Furthermore, it was significantly broader than that of BP, BP-Br, and BP-Ph, and its features resembled more than those of BP-DA and BP-TA (Figure 6a). While BP and BP-Ph had a FLQY of 32.4% and 42.7%, respectively, that of BP-Na was only 15.7%. This suggests that while the emission of BP, BP-Br, and BP-Ph originate in the solid state from a 1LE state of the BOPAM, that of BP-Na should be attributed to 1ICT.
The emission spectra of BP-DA and BP-TA consisted in the solid state of a broad band with maxima at 518 nm and 517 nm, corresponding to a red shift of 700 cm−1 and 190 cm−1 compared with toluene. As the absorption spectrum was red shifted by 1830 cm−1 and 2140 cm−1, respectively, the Stokes shift was again reduced by 1130 cm−1 and 1930 cm−1. The broad emission bands of both BP-DA and BP-TA, accompanied by a relatively low FLQY of 11.2% and 6.4%, respectively, suggest that their emission in the solid state is also primarily influenced by charge-transfer processes. The head-to-tail stacking pattern between adjacent triphenylamine groups of BP-DA and BP-TA (Figure 6c,d) will, besides the intramolecular charge transfer occurring in solution, facilitate intermolecular charge transfer processes in the solid state. It is noteworthy that BP-TA exhibited a broader emission band and a lower FLQY compared with BP-DA, suggesting a more pronounced charge-transfer process in its emission.
3. Materials and Methods
3.1. General Synthesis of BOPAM
The synthesis of 4-(diphenylamino)-N-phenylbenzothioamide is specifically provided in the Supplementary Material File. 4-Bromo-N-phenylbenzothioamide, 4-(diphenylamino)-N-phenylbenzothioamide, diphenylborinic acid, and di(2-naphthyl)borinic acid were obtained according to the previous literature, and the other synthetic steps are similar to those described in previously reported works [34,35,36].
Compound 1: N-phenylbenzothioamide (2.07 mmol, 441 mg) was dissolved in 20 mL of anhydrous ethanol and stirred at room temperature. Subsequently, hydrazine monohydrate (0.34 mL) was added to the reaction mixture, and the mixture was heated at reflux at 78 °C for 15 min. The reaction progress was monitored using thin-layer chromatography (TLC) until the starting material was fully converted. After cooling to room temperature, the organic phase was extracted three times with ethyl acetate and washed with deionized water. The organic phase was dried over anhydrous sodium sulfate to remove residual moisture, then the solvent was removed using a rotary evaporator to obtain the intermediate for direct use in the next step. 3,5-Dimethyl-2-formylpyrrole (1.72 mmol, 212 mg) and 20 mL anhydrous ethanol were added to the flask, and the mixture heated at reflux after adding 4 drops of glacial acetic acid. The reflux was continued at 78 °C, while monitoring the reaction by TLC until the 3,5-dimethyl-2-formylpyrrole had been completely consumed. The solvent was evaporated under reduced pressure to proceed to the next step.
Compound 2: Ligand 1 was dissolved in 15 mL of dry dichloromethane (DCM), and diphenylborinic acid (0.8 eq, 1.72 mmol, 313 mg) was added to the solution. The reaction mixture was stirred overnight under a nitrogen atmosphere at room temperature. The solvent was evaporated under reduced pressure to obtain the crude product.
BP-Ph: Compound 2 was dissolved in 15 mL of dry toluene, and triethylamine (14 eq, 4 mL) was added. The reaction mixture was stirred at room temperature for 15 min and cooled to 0 °C. Boron trifluoride diethyl etherate (BF3·OEt2, 16 mmol, 4 mL) was slowly added under nitrogen protection. Then, the mixture was heated at 110 °C overnight. After cooling to room temperature, the reaction was quenched with deionized water, the organic phase was extracted three times with DCM and washed with deionized water. The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent: petroleum ether/DCM = 1:1) and recrystallized with a DCM-pentane solvent system to obtain BP-Ph in 20% yield, mp: 235–237 °C. 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 7.3 Hz, 1H), 7.48–7.28 (m, 3H), 7.04–6.89 (m, 1H), 6.58 (d, J = 7.9 Hz, 1H), 6.06 (s, 1H), 2.37 (s, 2H), 2.14 (s, 2H).13C NMR (101 MHz, CDCl3) δ 163.41, 145.79, 138.36, 133.98, 133.56, 132.32, 131.04, 129.77, 128.44, 128.00, 127.94, 127.85, 127.45, 126.97, 126.29, 122.76, 116.65, 14.02, 11.04.11B NMR (128 MHz, CDCl3) δ 5.42, 1.21.19F NMR (376 MHz, CDCl3) δ −132.01, −132.10. HRMS (ESI, [M + H]+) for C32H28B2F2N4; calcd. 529.2541, found: 529.2573.
Compound 3: Ligand 1 was dissolved in 15 mL of dry dichloromethane (DCM), and di(2-naphthyl)borinic acid (0.8 eq) was added to the solution. The reaction mixture was stirred overnight under a nitrogen atmosphere at room temperature. The solvent was evaporated under reduced pressure to obtain the crude product.
BP-Na: Compound 3 was dissolved in 15 mL of dry toluene, and triethylamine (14 eq) was added. The reaction mixture was stirred at room temperature for 15 min and cooled to 0 °C. Boron trifluoride diethyl etherate (16 mmol) was slowly added under nitrogen protection. Then, the mixture was heated at 110 °C overnight. After cooling to room temperature, the reaction was quenched with deionized water, the organic phase was extracted three times with DCM, and washed with deionized water. The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent: petroleum ether/DCM = 1:1) and recrystallized with a DCM-pentane solvent system to obtain BP-Na in 33% yield, mp: 301–303 °C. 1H NMR (600 MHz, CDCl3) δ 7.91 (s, 2H), 7.86 (d, J = 7.7 Hz, 2H), 7.82 (d, J = 8.3 Hz, 2H), 7.80 (d, J = 7.7 Hz, 2H), 7.56 (d, J = 8.1 Hz, 4H), 7.51–7.45 (m, 4H), 7.40 (t, J = 7.5 Hz, 1H), 7.37 (s, 1H), 7.35 (t, J = 7.5 Hz, 2H), 6.98 (t, J = 7.4 Hz, 1H), 6.90 (t, J = 7.8 Hz, 2H), 6.60 (d, J = 7.3 Hz, 2H), 6.07 (s, 1H), 2.40 (s, 3H), 2.10 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 163.52, 146.10, 142.63, 138.37, 134.36, 133.48, 133.22, 133.18, 132.59, 131.25, 131.13, 129.79, 128.55, 128.44, 128.07, 127.86, 127.71, 127.15, 127.07, 126.26, 125.87, 125.69, 122.86, 116.84, 14.08, 11.08. 11B NMR (128 MHz, CDCl3) δ 5.95, 1.38. 19F NMR (376 MHz, CDCl3) δ −131.82 (d, J = 42.0 Hz). HRMS (ESI, [M + Na]+) for C40H32B2F4N4; calcd. 651.2674, found: 651.2659.
BP-DA: 4-(Diphenylamino)-N-phenylbenzothioamide (1.31 mmol, 500 mg) was dissolved in 18 mL of anhydrous ethanol and stirred at room temperature. Subsequently, hydrazine monohydrate (0.2 mL) was added to the reaction mixture, and the mixture was heated at reflux at 78 °C for 15 min. The reaction progress was monitored using TLC until the starting material was fully converted. After cooling to room temperature, the organic phase was extracted three times with ethyl acetate and washed with deionized water. The organic phase was dried over anhydrous sodium sulfate to remove the residual moisture, then the solvent was removed using a rotary evaporator to obtain the intermediate. 3,5-Dimethyl-2-formylpyrrole (1.09 mmol, 134 mg) and anhydrous ethanol (15 mL) were added to the flask, and the mixture heated at reflux after adding 3 drops of glacial acetic acid. The reflux was continued at 78 °C, while monitoring the reaction by TLC until the 3,5-dimethyl-2-formylpyrrole had been completely consumed. The solvent was evaporated under reduced pressure to proceed to the next step. Triethylamine (14 eq, 2.2 mL) and dry toluene (14 mL) were added to the intermediate bottle under nitrogen protection. The reaction mixture was stirred at room temperature for 15 min and cooled to 0 °C. BF3·OEt2 (16 mmol, 2.2 mL) was slowly added under nitrogen protection. Then, the mixture was heated at 110 °C overnight. After cooling to room temperature, the reaction was quenched with deionized water, and the organic phase was extracted three times with DCM and washed with deionized water. The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent: petroleum ether/DCM = 1:1) and recrystallized with a DCM-pentane solvent system to obtain BP-DA in 27% yield, mp: 260–262 °C. 1H NMR (400 MHz, DMSO) δ 8.72 (s, 1H), 7.40–7.31 (m, 4H), 7.28 (t, J = 7.3 Hz, 1H), 7.16 (t, J = 7.4 Hz, 1H), 7.10 (d, J = 7.0 Hz, 1H), 7.05 (d, J = 7.5 Hz, 2H), 6.72 (d, J = 8.9 Hz, 2H), 6.30 (s, 1H), 2.35 (s, 2H), 2.30 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 163.14, 150.54, 150.17, 146.34, 138.62, 137.07, 131.68, 131.03, 129.59, 129.02, 127.04, 126.37, 126.05, 124.61, 122.98, 119.05, 118.27, 116.23, 14.22, 10.97.11B NMR (128 MHz, CDCl3) δ 3.06, 0.86. 19F NMR (376 MHz, CDCl3) δ −132.13, −132.23, −145.13, −145.22. HRMS (ESI, [M + H]+) for C32H27B2F4N5; calcd. 580.2461, found: 580.2495.
BP-Br: 4-Bromo-N-phenylbenzothioamide (1.2 mmol, 292mg) was dissolved in 10 mL of anhydrous ethanol and stirred at room temperature. Subsequently, hydrazine monohydrate (0.2 mL) was added to the reaction mixture and the mixture was heated at reflux at 78 °C for 15 min. The reaction progress was monitored using TLC until the starting material was fully converted. After cooling to room temperature, the organic phase was extracted three times with ethyl acetate and washed with deionized water. The organic phase was dried over anhydrous sodium sulfate to remove the residual moisture, then the solvent was removed using a rotary evaporator to obtain the intermediate. 3,5-Dimethyl-2-formylpyrrole (1 mmol, 123 mg) and anhydrous ethanol (10 mL) were added to the intermediate, and the mixture heated at reflux after adding 1 drop of glacial acetic acid. The reflux was continued at 78 °C, while monitoring the reaction by TLC until the 3,5-dimethyl-2-formylpyrrole had been completely consumed. The solvent was evaporated under reduced pressure to proceed to the next step. Triethylamine (14 eq, 2 mL) and dry toluene (10 mL) were added to the intermediate bottle under nitrogen protection. The reaction mixture was stirred at room temperature for 15 min and cooled to 0 °C. BF3·OEt2 (16 mmol, 2 mL) was slowly added under nitrogen protection. Then, the mixture was heated at 110 °C overnight. After cooling to room temperature, the reaction was quenched with deionized water, and the organic phase was extracted three times with DCM and washed with deionized water. The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent: petroleum ether/DCM = 1:1) and recrystallized with a DCM-pentane solvent system to obtain the final product BP-Br in 35% yield, mp: 295–297 °C. 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H), 7.53–7.46 (m, 2H), 7.36 (d, J = 8.6 Hz, 2H), 7.27–7.17 (m, 3H), 7.03 (dd, J = 8.0, 1.7 Hz, 2H), 6.14 (s, 1H), 2.35 (s, 3H), 2.30 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 162.02, 151.09, 139.64, 136.22, 132.06, 131.73, 131.23 (t, J = 2.3 Hz), 129.39, 127.67, 126.63, 126.47, 124.22, 123.19, 118.68, 14.28 (d, J = 1.9 Hz), 11.08; 11B NMR (128 MHz, CDCl3) δ 3.05 (t, J = 25.3 Hz), 0.72 (t, J = 29.7 Hz); 19F NMR (376 MHz, CDCl3) δ −132.07 (dd, J = 53.1, 19.6 Hz), −144.72 (dd, J = 49.2, 21.4 Hz). HRMS (ESI, [M − F − H]+) for C20H17B2F4N4Br; calcd. 471.0780, found: 471.0776.
BP-TA: BP-Br (0.2 mmol, 1 eq), (4-(diphenylamino)phenyl)boronic acid (1.3 eq, 0.26 mmol), Tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4, 0.5 eq, 0.1 mmol), and potassium carbonate (3 eq, 0.6 mmol) were added in a dry round-bottom flask. The mixture was dissolved in dioxane that had been bubbled with nitrogen. The reaction mixture was heated at reflux under a nitrogen atmosphere for 12 h. After cooling to room temperature, the reaction was quenched with deionized water, and the organic phase was extracted three times with DCM and washed with deionized water. The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain the crude product. The crude product was purified by silica gel column chromatography (eluent: petroleum ether/DCM = 1:1) and recrystallized with a DCM-pentane solvent system to obtain BP-TA in 52% yield, mp: 268–270 °C. 1H NMR (400 MHz, CDCl3) δ 7.81 (s, 1H), 7.57–7.50 (m, 4H), 7.44 (d, J = 8.7 Hz, 2H), 7.29–7.24 (m, 4H), 7.21–7.16 (m, 2H), 7.14–7.01 (m, 11H), 6.14 (s, 1H), 2.36 (s, 3H), 2.31 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 163.09, 150.69, 148.20, 147.53, 143.56, 139.17, 136.65, 132.94, 131.96, 130.26, 129.48, 129.23, 127.93, 127.36, 126.45, 126.07, 124.82, 123.45, 123.43, 123.33, 123.15, 118.51, 14.28, 11.07; 11B NMR (128 MHz, CDCl3) δ 3.24 (t, J = 26.5 Hz), 0.84; 19F NMR (376 MHz, CDCl3) δ −132.10 (d, J = 46.2 Hz), −144.87 (d, J = 35.1 Hz). HRMS (ESI, [M + H]+) for C38H31B2F4N5; calcd. 656.2774, found: 656.2790.
3.2. Computational Details
The geometries of BOPAMs in the ground state (S0) and excited states (S1 and S2) were optimized using density functional theory (DFT) [37] and time-dependent density functional theory (TD-DFT) [38], respectively, without imaginary frequency. All calculations were performed using the PBE0 [39]/6-31+G(d) [40] level of theory with the integral equation formalism polarizable continuum model (IEFPCM) to model the solvent effects in acetonitrile and toluene [41], as implemented in the Gaussian 16 package [42]. Excitation properties were computed using TD-DFT at the optimized S0 geometry with the same level of theory and solvent model. The excitation and emission energy, computed by PBE0, exhibited the best correlation with the experimental absorption and emission spectra with low MAD (0.10–0.20 eV) compared with other functionals enabling the accurate calculation of charge transfer [43,44] such as CAM-B3LYP [45] (MAD of 0.40–0.43 eV, see Table S6). The latter functional, while describing the CT states more accurately, deteriorated the description of the excited states with predominant LE character, and thus led to a worse agreement with the experimental evidence. Additionally, the hole and electron distributions were analyzed using Multiwfn [46,47].
The total magnitude of the charge transfer length was quantified by the D-index [47], defined as:
where denotes the x/y/z coordinate of the centroid of the hole or electron distribution. The H-index characterizes the average spatial extent of the hole and electron distributions along the charge transfer direction [47] and is given by:
where σ represents the root-mean-square deviation (RMSD) of the hole or electron density. To further evaluate the degree of spatial separation between the hole and electron along the charge transfer direction, the t-index [47] was introduced:
where is the unit vector along the charge transfer direction, which can be determined from the centroids of the hole and electron distributions. A clearly positive t-index indicates significant spatial separation between the hole and electron distributions, corresponding to a strong ICT character. Conversely, a negative t-index suggests an LE state, with minimal charge separation.
4. Conclusions
We synthesized a series of novel BOPAM derivatives and conducted a comprehensive investigation of their photophysical properties in both the solid state and organic solvents. SC-XRD analysis revealed that the intrinsic 1LE emission of BOPAM in the solid state could be observed in BP-Br and BP-Ph. In contrast, the fluorescence emission of BP-Na, BP-DA, and BP-TA at longer wavelengths exhibited intermolecular and/or intramolecular charge transfer characteristics. Furthermore, the red-shifted emission of BP relative to BP-Br was associated with a combination of exciton coupling and a phenyl-twisted configuration.
In Tol, BP, BP-Br, BP-Ph, and BP-Na predominantly exhibited characteristic an 1LE emission, while BP-DA and BP-TA displayed a distinct 1ICT emission. Notably, BP-DA showed weak fluorescence originating from the 1TICT configuration, which was facilitated by the S2 (1LE) → S1 (1TICT) transition. In contrast, BP-TA exhibited emissive behavior from a higher-lying S2 state in ACN. Thus, we propose that the S2 → 1TICT transition is facilitated by conformational reorganization when the S2 and S1 states possess the appropriately locally excited and charge-transfer characteristics, respectively. Conversely, when both the S2 and S1 states are purely LE and ICT in nature, the S2 → 1TICT transition is negligible, and emissions from the higher-lying S2 (1LE) state become prominent.
In summary, this finding elucidates the fluorescence quenching mechanism of BOPAM by confirming the presence of the S2 state, the S2→1TICT transition, and the dark 1TICT state/configuration. This study not only contributes to new molecular designs toward red-shifted emissions, but also provides a deeper understanding of the fluorescence quenching of BOPAM-based fluorophores.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules30132673/s1. The Supplementary Materials are available online: general synthesis, structural spectra of compounds, SC-XRD data, computational and photophysical results [48,49,50].
Author Contributions
K.Y.: Synthesis and computational study, writing—original draft; T.C.P.: Conceptualization, computational and photophysical study, writing—review and editing; J.H.: Synthesis; Y.L.: Computational study; L.V.M.: SC-XRD study; M.V.d.A.: Writing—review and editing; D.E.: Conceptualization, supervision, writing—review and editing; W.D.: Conceptualization, resources, supervision, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
D. Escudero and W. Dehaen acknowledge the financial support from the bilateral FWO-NAFOSTED program (Project number: G0E5321N). K. Yu greatly acknowledges the financial support from the China Scholarship Council (CSC, No. 202306630009) for her doctoral scholarship. L. Van Meervelt thanks the Hercules Foundation for supporting the purchase of the diffractometer through project AKUL/09/0035. J. Huang thanks the China Scholarship Council for his doctoral scholarship (CSC, No. 201906920069). Y. Li thanks the China Scholarship Council for her doctoral scholarship (CSC, No. 202208080103).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials; further inquiries can be directed to the corresponding authors.
Acknowledgments
The authors acknowledge B. Van Huffel, G. Steurs, G. Van Haele, and D. Swerts for their technical assistance with the NMR and LR-MS spectrometers; J. Rozenski for the HR-MS measurements; B. Dieu for the steady-state UV–Vis and photoluminescence spectroscopy; and H. Vansweevelt for the HPC servers. T. C. Pham thanks M. Van der Auweraer, M. T. d. Casal, F. de Jong, E. Fron, and J. Vandenwijngaerden for their valuable comments (e.g., MECP, ADC(2), and protonation).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fan, Y.; Wu, Y.; Hou, J.; Wang, P.; Peng, X.; Ge, G. Coumarin-based near-infrared fluorogenic probes: Recent advances, challenges and future perspectives. Coord. Chem. Rev. 2023, 480, 215020. [Google Scholar] [CrossRef]
- Meares, A.; Satraitis, A.; Santhanam, N.; Yu, Z.; Ptaszek, M. Deep-Red Emissive BODIPY–Chlorin Arrays Excitable with Green and Red Wavelengths. J. Org. Chem. 2015, 80, 3858–3869. [Google Scholar] [CrossRef]
- Frath, D.; Massue, J.; Ulrich, G.; Ziessel, R. Luminescent materials: Locking pi-conjugated and heterocyclic ligands with boron (III). Angew. Chem. Int. Ed. Engl. 2014, 53, 2290–2310. [Google Scholar] [CrossRef]
- Mendive-Tapia, L.; Zhao, C.; Akram, A.R.; Preciado, S.; Albericio, F.; Lee, M.; Serrels, A.; Kielland, N.; Read, N.D.; Lavilla, R.; et al. Spacer-free BODIPY fluorogens in antimicrobial peptides for direct imaging of fungal infection in human tissue. Nat. Commun. 2016, 7, 10940. [Google Scholar] [CrossRef]
- Yuan, L.; Lin, W.; Zheng, K.; He, L.; Huang, W. Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging. Chem. Soc. Rev. 2013, 42, 622–661. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Matsuo, K.; Yamaguchi, S. Polycyclic π-Electron System with Boron at Its Center. J. Am. Chem. Soc. 2012, 134, 9130–9133. [Google Scholar] [CrossRef]
- Treibs, A.; Kreuzer, F.-H. Difluorboryl-Komplexe von Di- und Tripyrrylmethenen. Justus Liebigs Ann. Chem. 1968, 718, 208–223. [Google Scholar] [CrossRef]
- Tamgho, I.-S.; Hasheminasab, A.; Engle, J.T.; Nemykin, V.N.; Ziegler, C.J. A New Highly Fluorescent and Symmetric Pyrrole–BF2 Chromophore: BOPHY. J. Am. Chem. Soc. 2014, 136, 5623–5626. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Fang, X.; Wu, Q.; Jiao, L.; Sun, L.; Li, Z.; So, P.-K.; Wong, W.-Y.; Hao, E. A Family of BODIPY-like Highly Fluorescent and Unsymmetrical Bis(BF2) Pyrrolyl–Acylhydrazone Chromophores: BOAPY. Org. Lett. 2020, 22, 4588–4592. [Google Scholar] [CrossRef]
- Pookkandam Parambil, S.; de Jong, F.; Veys, K.; Huang, J.; Veettil, S.P.; Verhaeghe, D.; Van Meervelt, L.; Escudero, D.; Van der Auweraer, M.; Dehaen, W. BOPAHY: A doubly chelated highly fluorescent pyrrole–acyl hydrazone –BF2 chromophore. Chem. Commun. 2020, 56, 5791–5794. [Google Scholar] [CrossRef]
- Boens, N.; Verbelen, B.; Ortiz, M.J.; Jiao, L.; Dehaen, W. Synthesis of BODIPY dyes through postfunctionalization of the boron dipyrromethene core. Coord. Chem. Rev. 2019, 399, 213024. [Google Scholar] [CrossRef]
- Lu, H.; Mack, J.; Yang, Y.; Shen, Z. Structural modification strategies for the rational design of red/NIR region BODIPYs. Chem. Soc. Rev. 2014, 43, 4778–4823. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Fron, E.; Liu, Z.; Boodts, S.; Thomas, J.; Harvey, J.N.; Hofkens, J.; Dehaen, W.; Van der Auweraer, M.; Smet, M. Acid-Sensitive BODIPY Dyes: Synthesis through Pd-Catalyzed Direct C(sp3)−H Arylation and Photophysics. Chem.—Eur. J. 2017, 23, 4687–4699. [Google Scholar] [CrossRef]
- Rohand, T.; Qin, W.; Boens, N.; Dehaen, W. Palladium-Catalyzed Coupling Reactions for the Functionalization of BODIPY Dyes with Fluorescence Spanning the Visible Spectrum. Eur. J. Org. Chem. 2006, 2006, 4658–4663. [Google Scholar] [CrossRef]
- Bumagina, N.A.; Antina, E.V.; Ksenofontov, A.A.; Antina, L.A.; Kalyagin, A.A.; Berezin, M.B. Basic structural modifications for improving the practical properties of BODIPY. Coord. Chem. Rev. 2022, 469, 214684. [Google Scholar] [CrossRef]
- Boens, N.; Verbelen, B.; Dehaen, W. Postfunctionalization of the BODIPY Core: Synthesis and Spectroscopy. Eur. J. Org. Chem. 2015, 2015, 6577–6595. [Google Scholar] [CrossRef]
- Peterson, J.A.; Wijesooriya, C.; Gehrmann, E.J.; Mahoney, K.M.; Goswami, P.P.; Albright, T.R.; Syed, A.; Dutton, A.S.; Smith, E.A.; Winter, A.H. Family of BODIPY Photocages Cleaved by Single Photons of Visible/Near-Infrared Light. J. Am. Chem. Soc. 2018, 140, 7343–7346. [Google Scholar] [CrossRef]
- Slanina, T.; Shrestha, P.; Palao, E.; Kand, D.; Peterson, J.A.; Dutton, A.S.; Rubinstein, N.; Weinstain, R.; Winter, A.H.; Klán, P. In Search of the Perfect Photocage: Structure–Reactivity Relationships in meso-Methyl BODIPY Photoremovable Protecting Groups. J. Am. Chem. Soc. 2017, 139, 15168–15175. [Google Scholar] [CrossRef] [PubMed]
- Ksenofontov, A.A.; Bocharov, P.S.; Ksenofontova, K.V.; Antina, E.V. Water-Soluble BODIPY-Based fluorescent probe for BSA and HSA detection. J. Mol. Liq. 2022, 345, 117031. [Google Scholar] [CrossRef]
- Huang, J.; de Jong, F.; Elisa van Raamsdonk, D.M.; Vandenwijngaerden, J.; Inoue, A.; Escudero, D.; Van Meervelt, L.; Van der Auweraer, M.; Dehaen, W. BOPAM: Efficient Synthesis of a Bright Asymmetric Bis-Boron Complex and its Dark Side. Adv. Opt. Mater. 2024, 12, 2301328. [Google Scholar] [CrossRef]
- Huang, J.; Chung Pham, T.; Coenen, D.; Vandenwijngaerden, J.; Gong, J.; Minh Thi Nguyen, H.; Van Meervelt, L.; Van der Auweraer, M.; Escudero, D.; Dehaen, W. Benzo-Fused BOPAM Fluorophores: Synthesis, Post-functionalization, Photophysical Properties and Acid sensing Applications. Chem. Eur. J. 2024, 30, e202401837. [Google Scholar] [CrossRef]
- Ulrich, G.; Goze, C.; Guardigli, M.; Roda, A.; Ziessel, R. Pyrromethene Dialkynyl Borane Complexes for “Cascatelle” Energy Transfer and Protein Labeling. Angew. Chem. Int. Ed. 2005, 44, 3694–3698. [Google Scholar] [CrossRef]
- Goze, C.; Ulrich, G.; Mallon, L.J.; Allen, B.D.; Harriman, A.; Ziessel, R. Synthesis and Photophysical Properties of Borondipyrromethene Dyes Bearing Aryl Substituents at the Boron Center. J. Am. Chem. Soc. 2006, 128, 10231–10239. [Google Scholar] [CrossRef]
- Choi, S.H.; Pang, K.; Kim, K.; Churchill, D.G. Cu2+ Colorimetric Sensing and Fluorescence Enhancement and Hg2+ Fluorescence Diminution in “Scorpionate”-like Tetrathienyl-Substituted Boron–Dipyrrins. Inorg. Chem. 2007, 46, 10564–10577. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Rohand, T.; Baruah, M.; Stefan, A.; der Auweraer, M.V.; Dehaen, W.; Boens, N. Solvent-dependent photophysical properties of borondipyrromethene dyes in solution. Chem. Phys. Lett. 2006, 420, 562–568. [Google Scholar] [CrossRef]
- Qin, W.; Baruah, M.; Van der Auweraer, M.; De Schryver, F.C.; Boens, N. Photophysical Properties of Borondipyrromethene Analogues in Solution. J. Phys. Chem. A 2005, 109, 7371–7384. [Google Scholar] [CrossRef] [PubMed]
- Veys, K.; de Jong, F.; Adriaens, A.; Coenen, G.; Coenen, K.; De Ligt, Y.; Meysman, Q.; Paredis, T.; Stalmans, J.; Dehaen, W.; et al. Revisiting the Fluorescence of Benzothiadiazole Derivatives: Anti-Kasha Emission or Not? ChemPhotoChem 2023, 7, e202200262. [Google Scholar] [CrossRef]
- Veys, K.; Escudero, D. Anti-Kasha Fluorescence in Molecular Entities: Central Role of Electron–Vibrational Coupling. Acc. Chem. Res. 2022, 55, 2698–2707. [Google Scholar] [CrossRef]
- Kasha, M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55–70. [Google Scholar] [CrossRef]
- Szabo, A.; Ostlund, N.S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; Dover Publications: Mineola, NY, USA, 1996. [Google Scholar]
- Deshmukh, A.P.; Geue, N.; Bradbury, N.C.; Atallah, T.L.; Chuang, C.; Pengshung, M.; Cao, J.; Sletten, E.M.; Neuhauser, D.; Caram, J.R. Bridging the gap between H- and J-aggregates: Classification and supramolecular tunability for excitonic band structures in two-dimensional molecular aggregates. Chem. Phys. Rev. 2022, 3, 021401. [Google Scholar] [CrossRef]
- Kuhn, H.; Kuhn, C. Chromophore coupling effects. In J-Aggregates; World Scientific: Singapore, 1996; pp. 1–40. [Google Scholar]
- Spano, F.C. Vibronic coupling in J-aggregates. In J-Aggregates; World Scientific: Singapore, 2012; pp. 49–75. [Google Scholar]
- Li, L.; Ouyang, H.; Long, Z.; Zhang, Q.; Jiang, Y.; Cai, M.; Xiong, S.; Peng, S.; Xu, G.; He, Q. A triphenylamine-based fluorescent probe with phenylboronic acid for highly selective detection of Hg2+ and CH3Hg+ in groundwater. Org. Biomol. Chem. 2023, 21, 5560–5566. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.-H.; Chen, Y.-Z. Electrosynthesis of redox-active and electrochromic polymer films from triphenylamine-cored star-shaped molecules end-capped with arylamine groups. Eur. Polym. J. 2018, 99, 422–436. [Google Scholar] [CrossRef]
- Chen, X.; Ke, H.; Chen, Y.; Guan, C.; Zou, G. Cross-Coupling of Diarylborinic Acids and Anhydrides with Arylhalides Catalyzed by a Phosphite/N-Heterocyclic Carbene Co-Supported Palladium Catalyst System. J. Org. Chem. 2012, 77, 7572–7578. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Klamt, A.; Moya, C.; Palomar, J. A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach. J. Chem. Theory Comput. 2015, 11, 4220–4225. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Baharfar, M.; Hillier, A.C.; Mao, G. Charge-Transfer Complexes: Fundamentals and Advances in Catalysis, Sensing, and Optoelectronic Applications. Adv. Mater. 2024, 36, 2406083. [Google Scholar] [CrossRef]
- Coppola, F.; Cimino, P.; Petrone, A.; Rega, N. Evidence of Excited-State Vibrational Mode Governing the Photorelaxation of a Charge-Transfer Complex. J. Phys. Chem. A 2024, 128, 1620–1633. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Le Bahers, T.; Adamo, C.; Ciofini, I. A Qualitative Index of Spatial Extent in Charge-Transfer Excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Section A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).