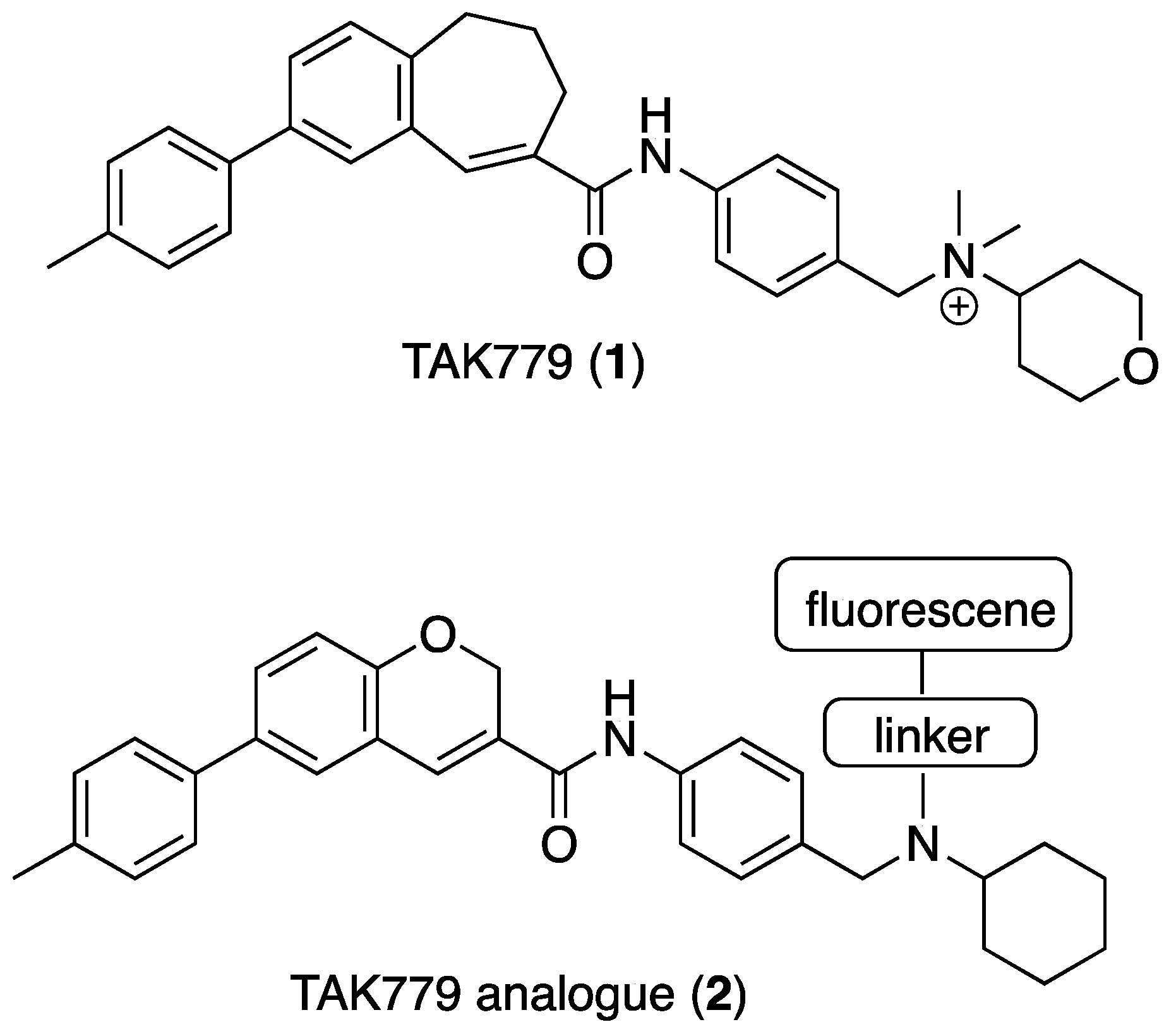
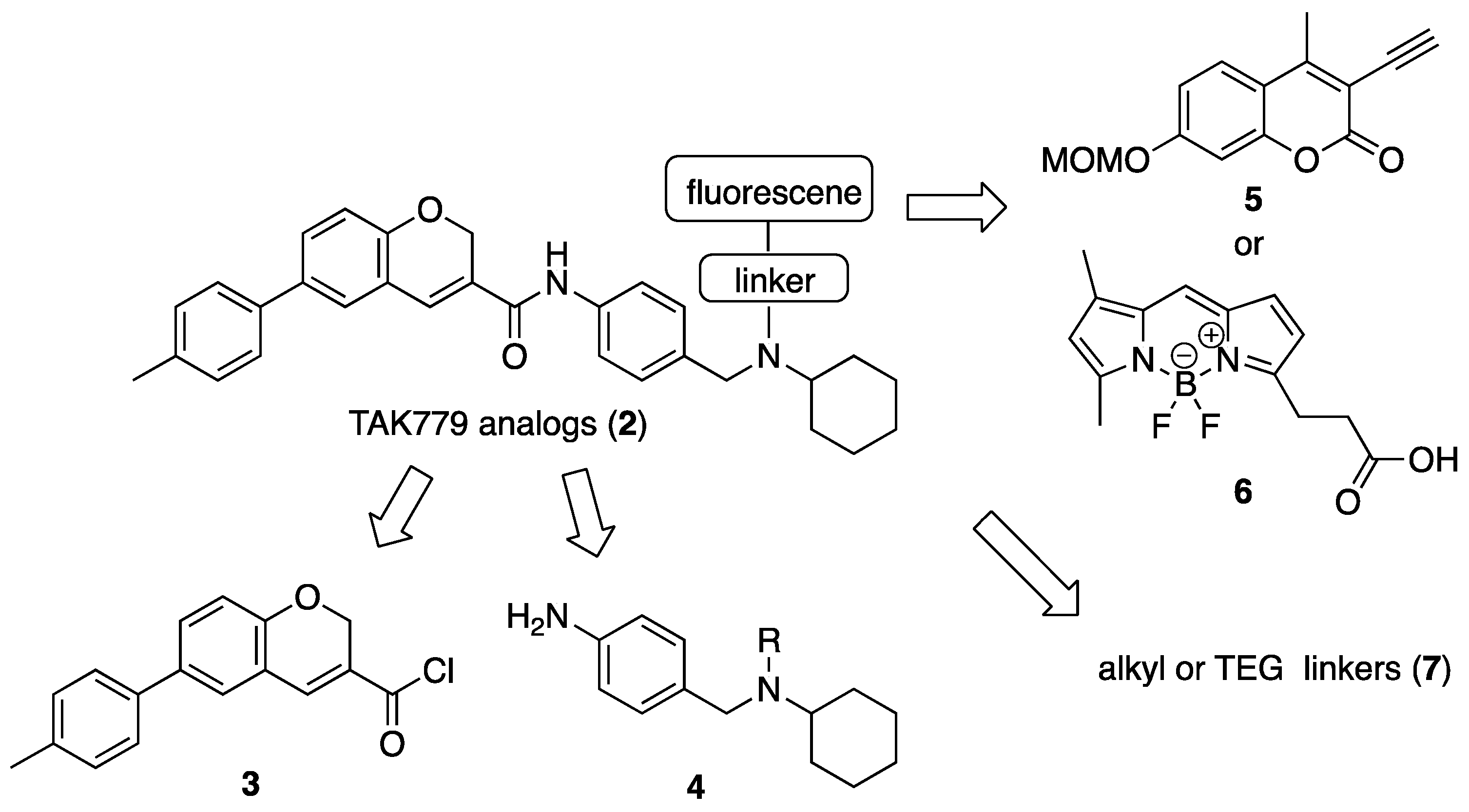
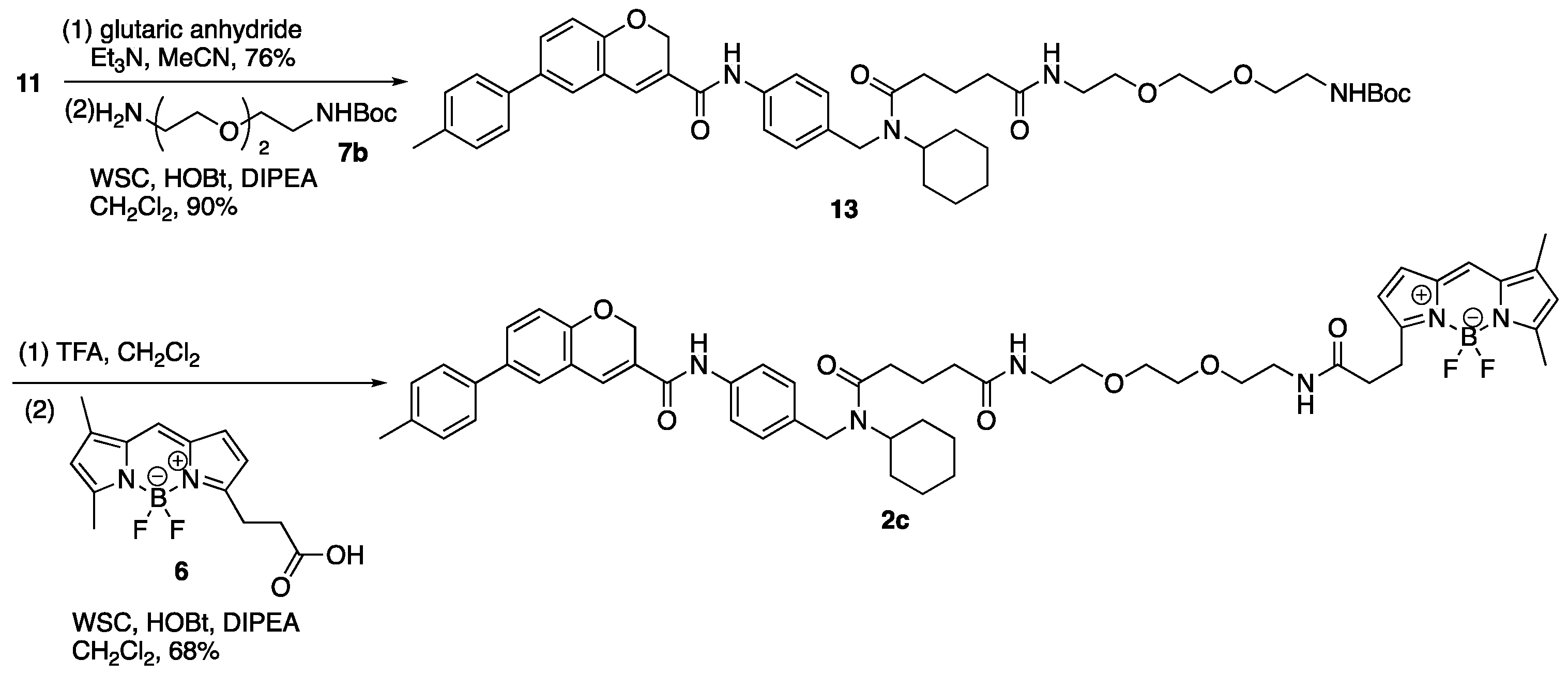
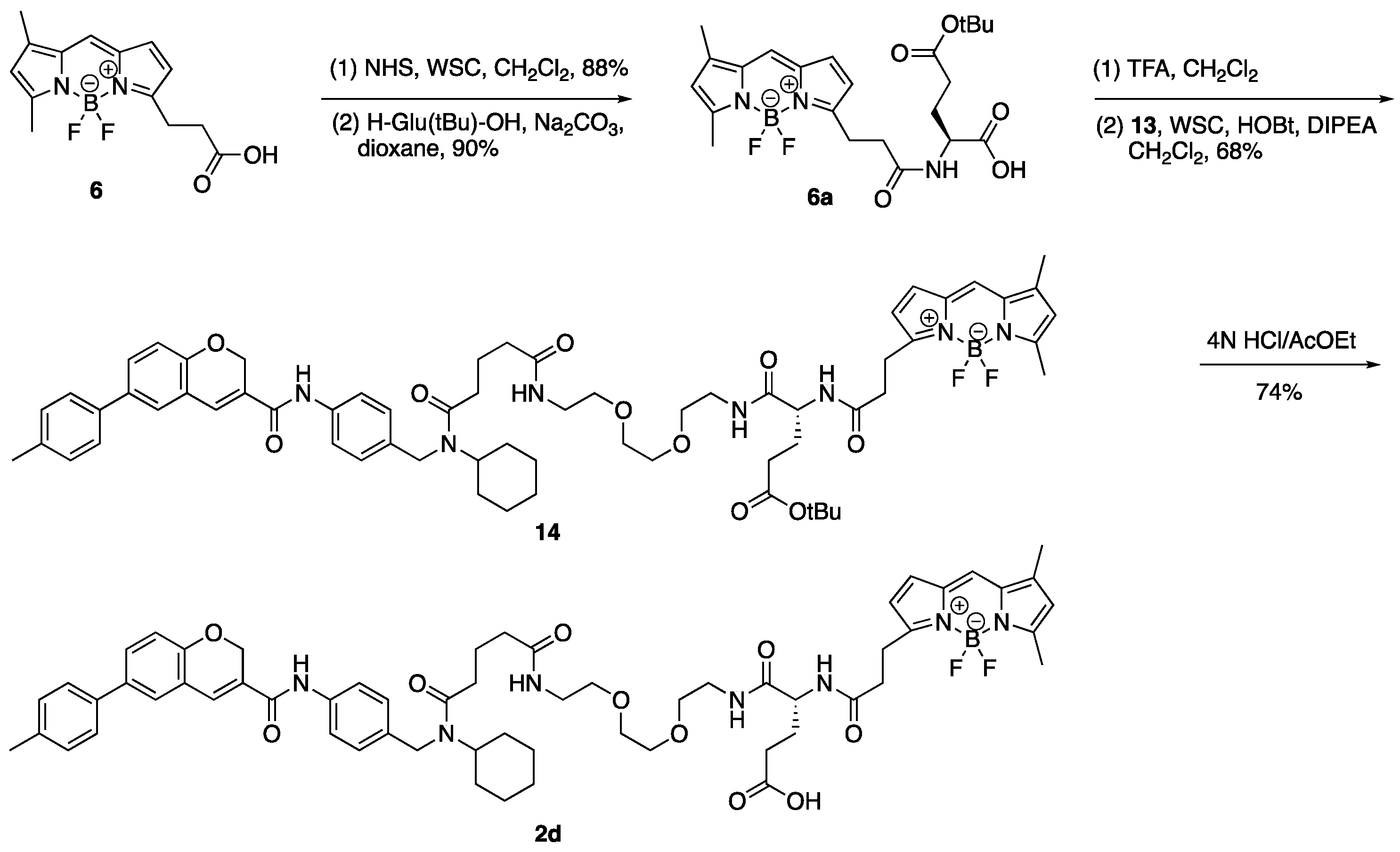
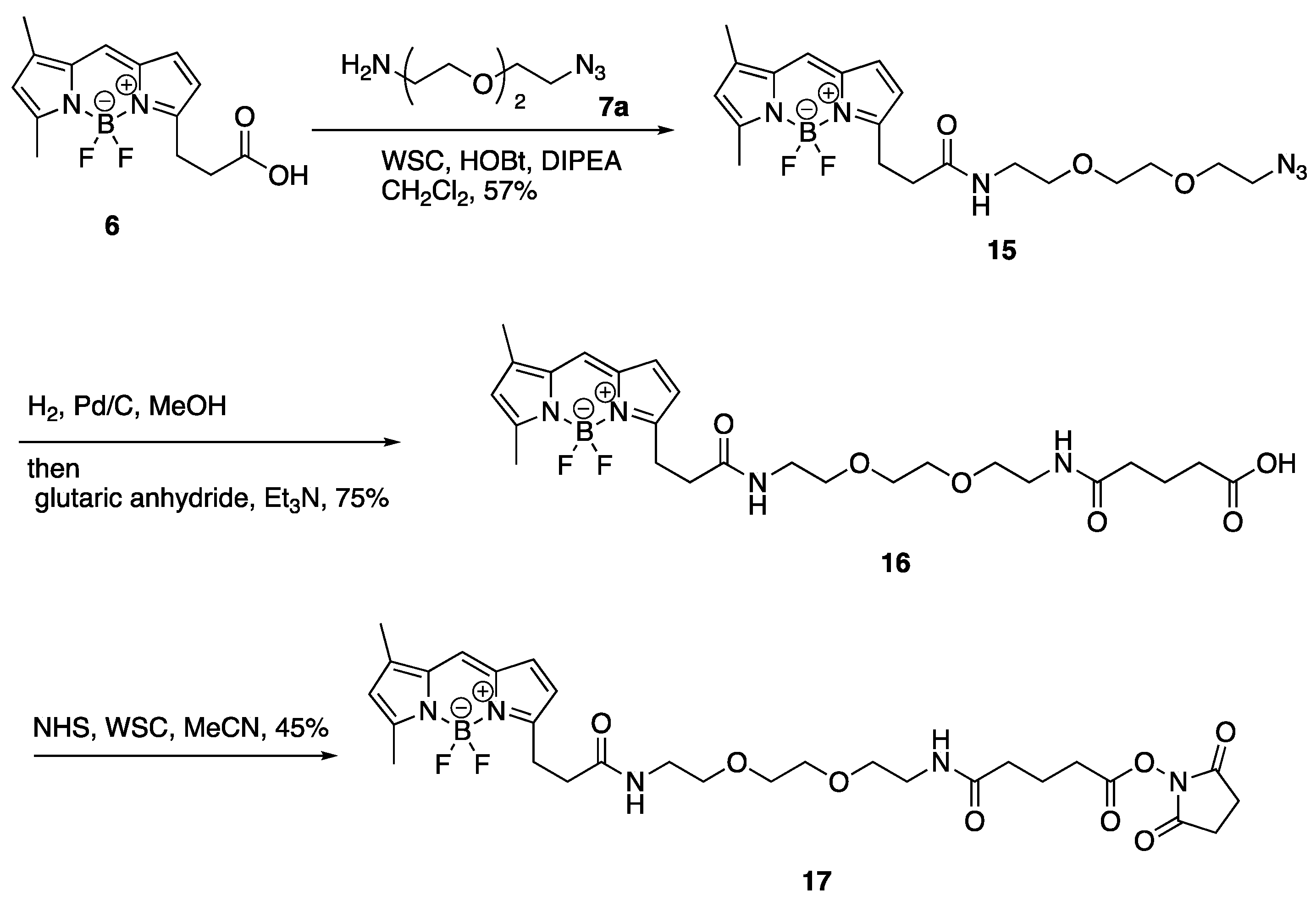
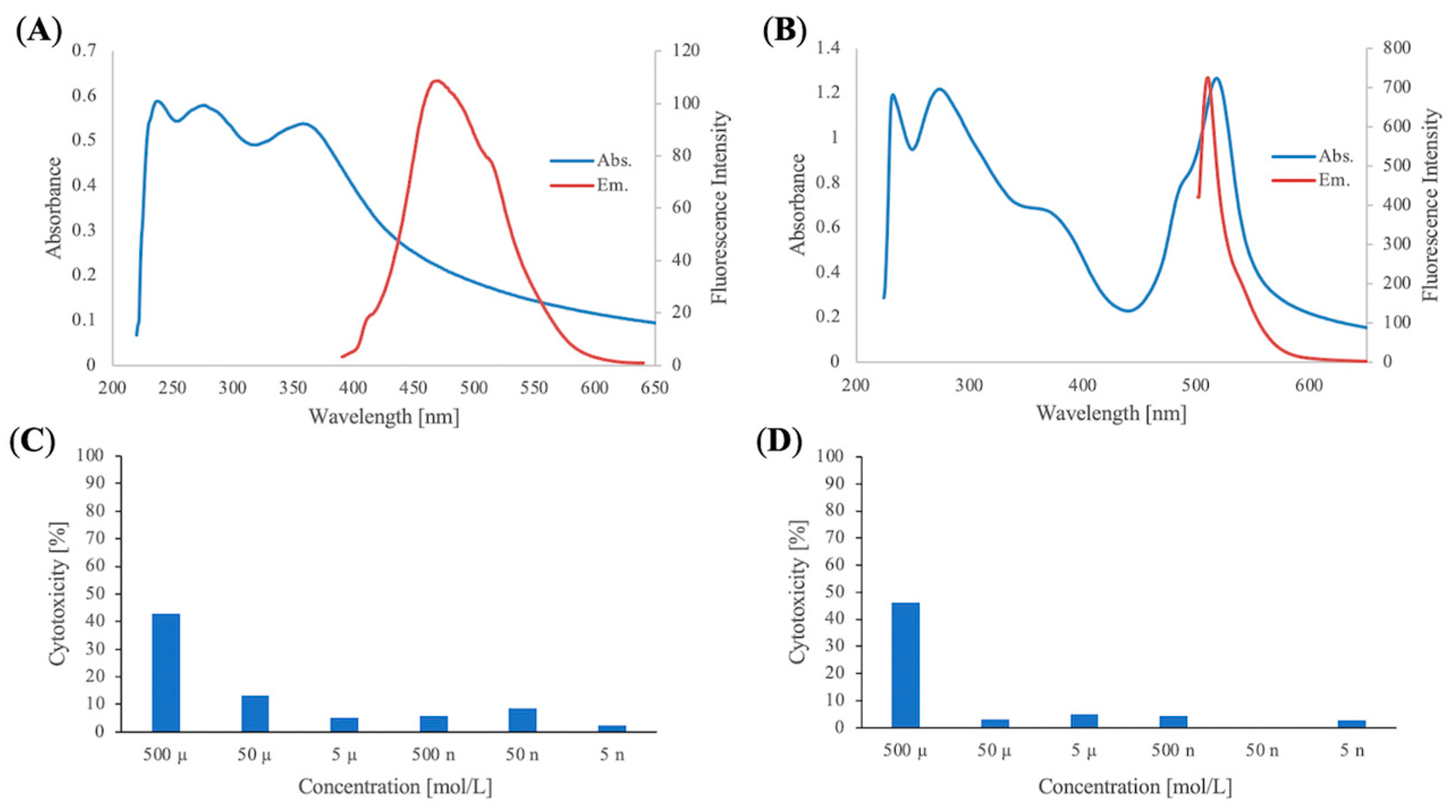
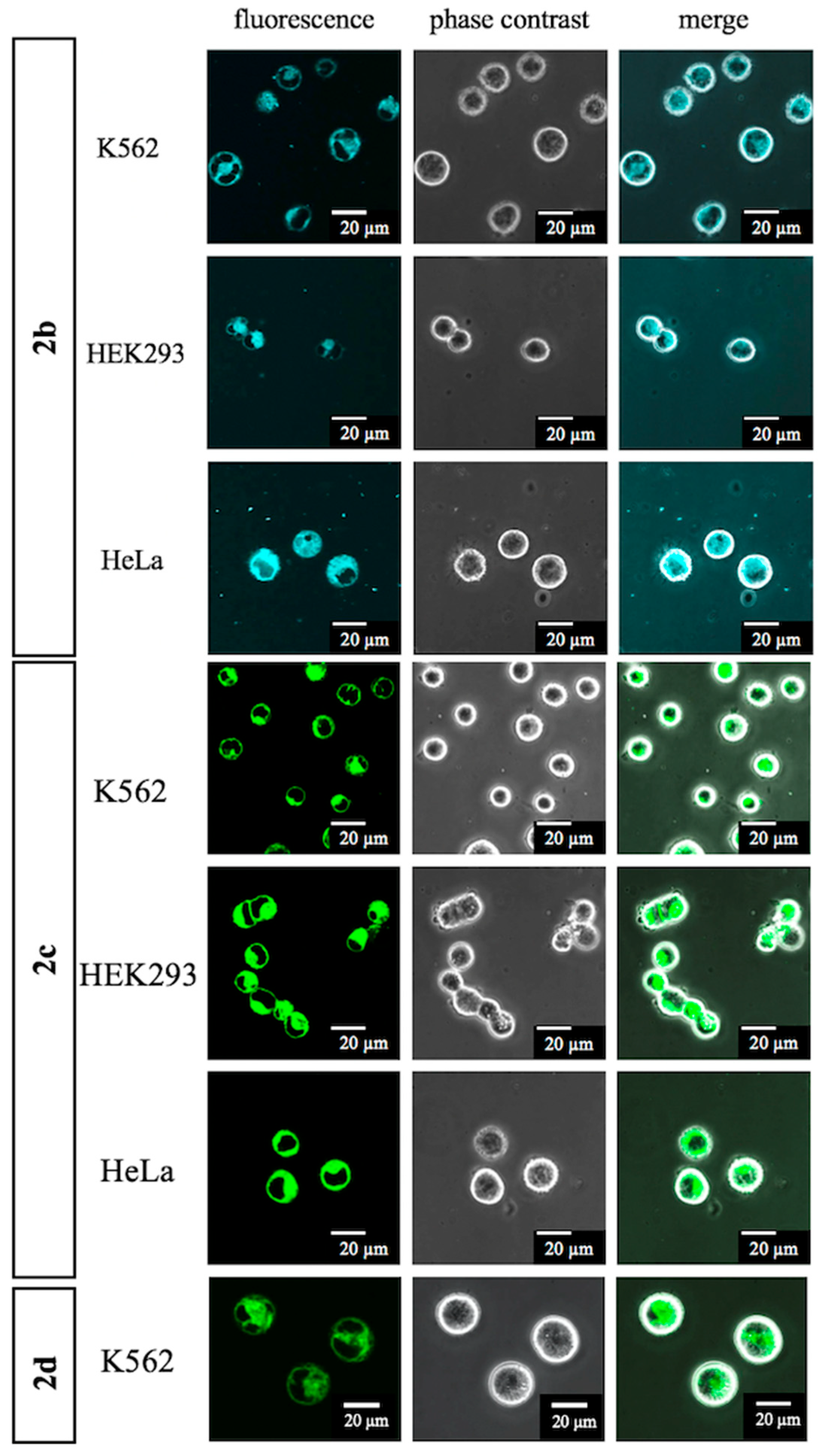
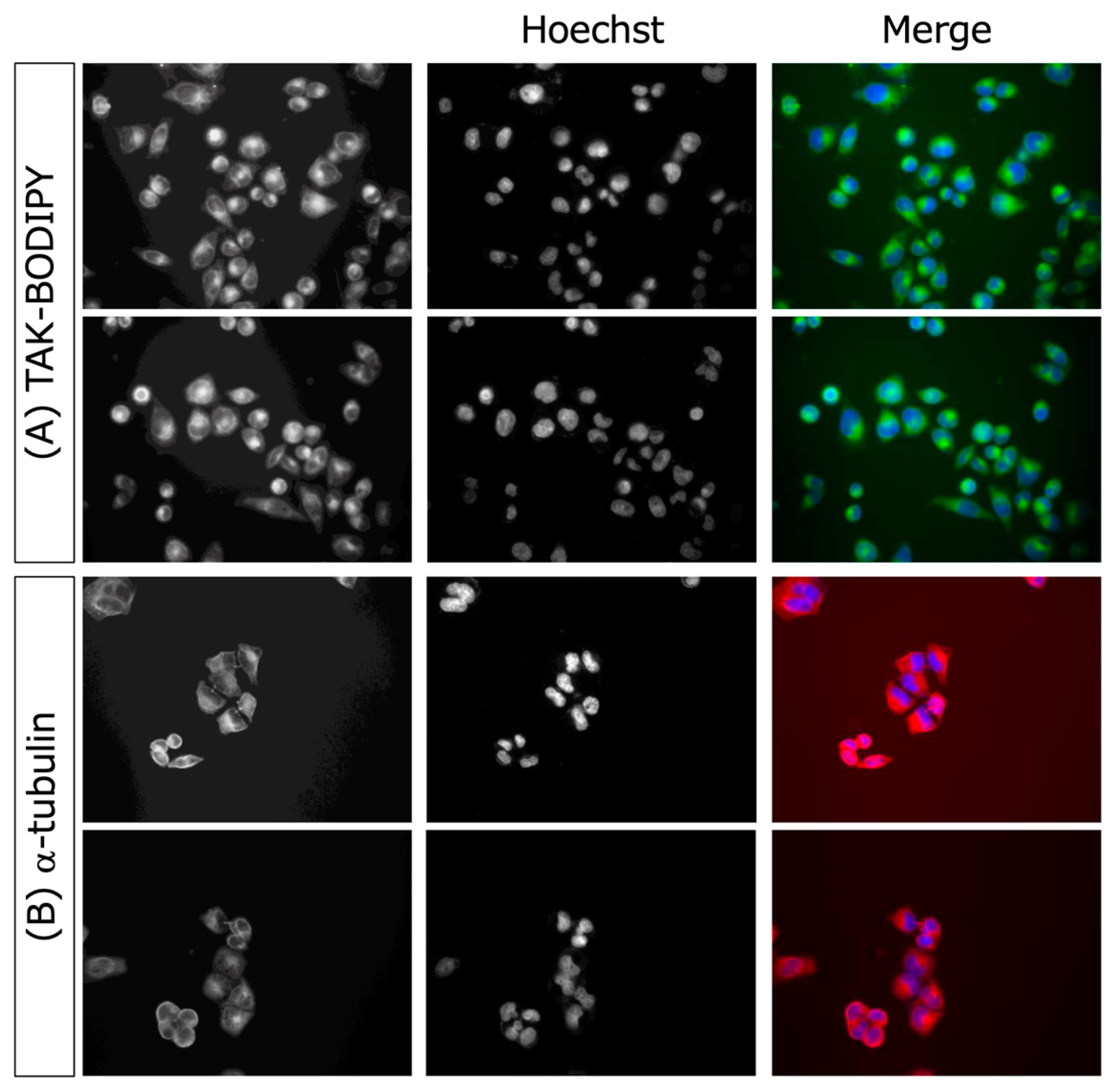
3. Experimental
3.1. General
All solvents were of reagent grade. THF was distilled from sodium and benzophenone ketyl. CH2Cl2 was distilled from CaH2. All commercial reagents were of the highest purity available. Analytical TLC was performed on silica gel (60 F-254, Plates 0.25 mm). Column chromatography was carried out on silica gel 60 N (Kanto Chemical Co., Tokyo, Japan, 40–100 µm). 1H (600, 500 or 400 MHz) and 13C (150, 125 or 100 MHz) NMR spectra were recorded on a JEOL JNM-ECX 600, JNM-ECX 500, or JNM-ECX 400 (JEOL, Ltd., Tokyo, Japan). Chemical shifts are expressed in ppm relative to TMS (0 ppm), CHCl3 (7.26 ppm for 1H and 77.0 ppm for 13C), or DMSO-d6 (2.50 ppm for 1H and 39.5 ppm for 13C). IR spectra were obtained on a JASCO FT/IR-460 Plus spectrometer (Tokyo, Japan). High-resolution mass spectra (HRMS) were obtained on a JEOL The AccuTOF JMS-T100LC (ESI). Melting points were measured on an AS ONE ATM-02 (Osaka, Japan). Optical rotations were measured on a JASCO DIP-371. Ultraviolet and visible absorption spectra were obtained on a HITACHI U-1900 or U-2900 (Chiyoda, Japan). Fluorescence spectra were recorded on a JASCO FP-8200 or HITACHI F-2700. Absorbances were measured on a CORONA ELECTRIC MTP-310 Lab (Hitachinaka, Japan).
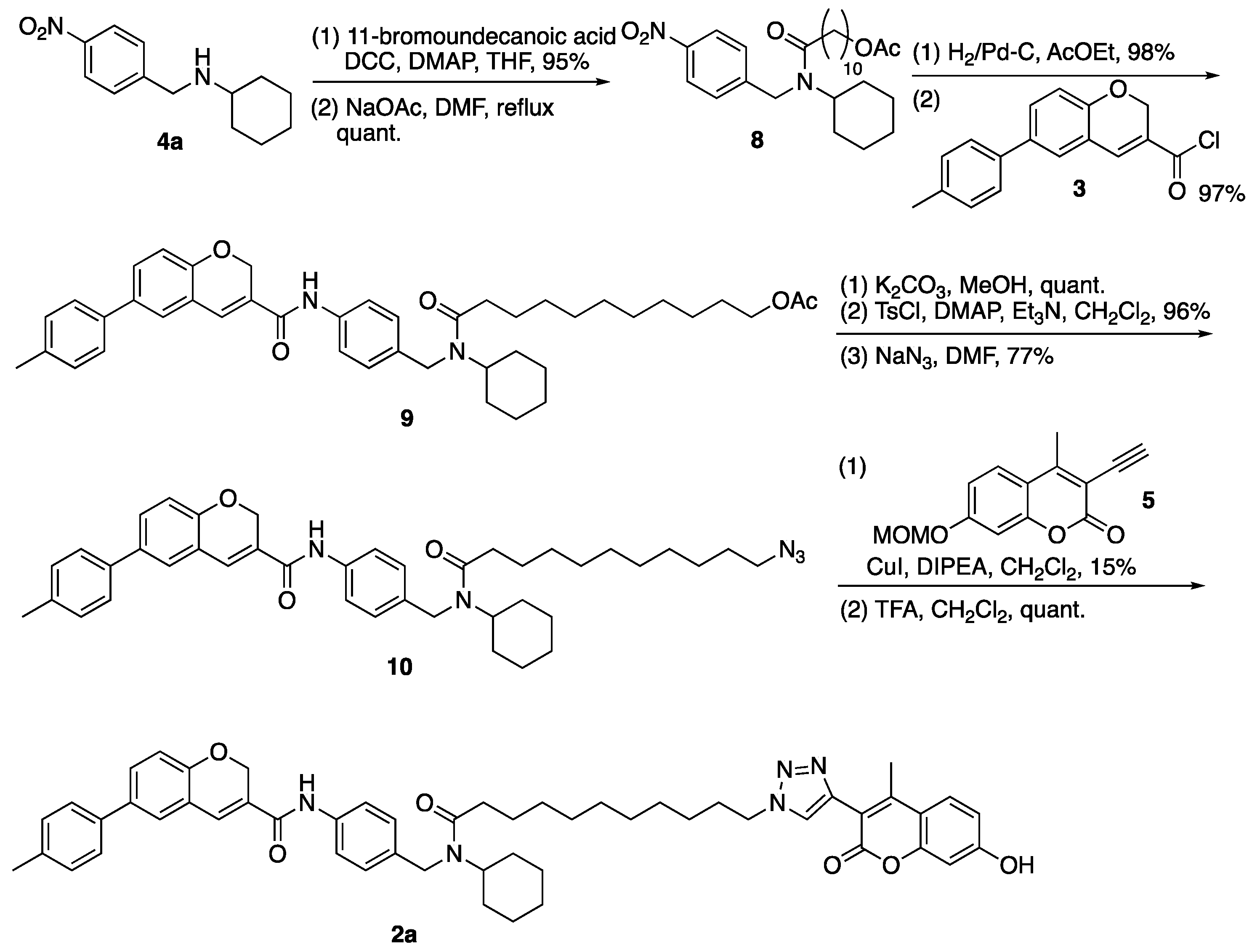
3.2. N-Cyclohexyl-N-(4-nitrobenzyl)-11-undecanamido Acetate (8)
(1) To a solution of 11-bromoundecanoic acid (0.85 g, 3.21 mmol) in THF were added DCC (0.66 g, 3.20 mmol), DMAP (0.04 g, 0.33 mmol), and 4a (0.50 g, 2.13 mmol). The reaction mixture was stirred overnight at room temperature, filtered through a celite pad, to which was added AcOEt and H2O, and separated. The aqueous layer was extracted with AcOEt and the combined organic layer was washed successively with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 5:1) to give amide (4c) (0.94 g, 2.01 mmol, 95%) as a brown oil. 1H-NMR (CDCl3, 600 MHz) δ 8.10 (m, 2H), 7.33 (m, 2H), 4.55 (m, 2H), 4.50 (m, 1H), 3.35–3.31 (m, 2H), 2.42–1.73 (m, 6H), 1.67–1.16 (m, 21H), 1.00 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 173.33, 173.29, 147.6, 146.9, 146.7, 146.4, 127.3, 126.5, 123.7, 123.3, 57.3, 53.4, 46.2, 44.3, 33.8, 33.7, 33.4, 32.6, 31.9, 30.7, 30.5, 29.22, 29.16, 29.1, 29.0, 28.49, 28.45, 27.90, 27.87, 25.7, 25.5, 25.23, 25.20, 25.1, 24.9. IR (film, cm−1) νmax 2927, 2853, 1644, 1601, 1521, 1408, 1343, 1109, 1013, 858, 736. ESI-HRMS m/z 481.2036 [M + H]+ (calcd. for C24H38BrN2O3, 481.2066).
(2) To a solution of amide (4c) (1.50 g, 3.12 mmol) in DMF was added NaOAc (1.23 g, 15.0 mmol). The reaction mixture was refluxed for 4 h, diluted with AcOEt and H2O. The aqueous layer was extracted with AcOEt and the combined organic layer was washed successively with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 3:1) to give 8 (1.52 g, quant.) as a brown oil. 1H-NMR (CDCl3, 600 MHz) δ 8.11 (m, 2H), 7.33 (m, 2H), 4.56–4.54 (m, 2H), 4.60 (m, 1H), 3.98 (m, 2H), 2.43–2.08 (m, 2H), 1.98 (m, 3H), 1.76–1.51 (m, 9H), 1.36–1.13 (m, 16H), 0.98 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 173.41, 173.37, 161.0, 147.7, 147.0, 146.7, 146.5, 127.4, 126.5, 123.7, 123.3, 64.4, 57.4, 53.4, 46.2, 44.4, 33.8, 33.4, 32.0, 30.6, 29.30, 29.27, 29.24, 29.22, 29.14, 29.11, 29.04, 28.99, 28.4, 25.7, 25.5, 25.3, 25.2, 25.1, 25.0. IR (film, cm−1) νmax 2928, 2854, 1736, 1646, 1521, 1343, 1238, 1109, 1033, 737. ESI-HRMS m/z 461.2986 [M + H]+ (calcd. for C26H41N2O5 461.3016).
3.3. N-Cyclohexyl-N-(4-{N-[6-(4-methylphenyl)-2H-1-benzopyran-3-carbonyl]amino}benzyl)-11-undecanamido Acetate (9)
(1) To a solution of 8 (0.62 g, 1.35 mmol) in AcOEt was added 5% Pt/C (2 mg). The reaction mixture was vigorously stirred at room temperature under a hydrogen atmosphere for 4 h and filtered through a celite pad, which was washed with AcOEt. The filtrate was concentrated in vacuo. The residue was purified by column chromatography (AcOEt) to give amine (8a) (0.57 g, 1.32 mmol, 98%) as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 6.94 (m, 2H), 6.56 (m, 2H), 4.49 (m, 1H), 4.36 (m, 2H), 3.99 (m, 2H), 3.70 (s, 2H), 2.38–2.16 (m, 2H), 1.99 (s, 3H), 1.73–1.53 (m, 9H), 1.41–1.18 (m, 16H), 0.98 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ: 173.6, 173.0, 161.0, 145.3, 144.8, 129.8, 128.12, 128.05, 126.6, 114.9, 114.8, 64.4, 60.1, 57.4, 53.5, 46.2, 43.8, 33.8, 33.6, 32.0, 30.5, 29.3, 29.2, 29.1, 29.01, 28.98, 28.4, 25.9, 25.6, 25.5, 25.4, 25.3, 25.1. IR (film, cm−1) νmax 3351, 2927, 2853, 1735, 1629, 1518, 1419, 1363, 1239, 1176, 1035, 816. ESI-HRMS m/z 431.3273 [M + H]+ (calcd. for C26H43N2O3 431.3274).
(2) To a solution of carboxylic acid (S5) (0.10 g, 0.38 mmol) in CHCl3 were added SOCl2 (0.10 mL, 1.38 mmol) and a DMF (1 drop) on ice. The reaction mixture was stirred at room temperature under nitrogen atmosphere for 2 h and concentrated in vacuo to give 3. To a solution of 3 in THF was dropwise added a solution of amine (8a) (0.20 g, 0.46 mmol) and TEA (0.14 mL, 1.00 mmol) in THF on ice. The reaction mixture was stirred overnight at room temperature under nitrogen atmosphere, then diluted with AcOEt and H2O. The aqueous layer was extracted with AcOEt and the combined organic layer was washed successively with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 2:1) to give 9 (0.25 g, 0.37 mmol, 97%) as a yellow oil. 1H-NMR (CDCl3, 500 MHz) δ 8.87 (m, 1H), 7.54 (m, 2H), 7.41 (m, 4H), 7.26 (m, 1H), 7.22 (d, J = 8.5 Hz, 2H), 7.08 (m, 2H), 6.91 (m, 1H), 5.11 (s, 2H), 4.49 (m, 2H), 4.54–3.61 (m, 1H), 4.08 (m, 2H), 2.45–2.17 (m, 2H), 2.38 (s, 3H), 2.04 (m, 3H), 1.78–1.55 (m, 9H), 1.43–1.19 (m, 16H), 1.01 (m, 1H). 13C-NMR (CDCl3, 125 MHz) δ: 174.2, 173.5, 163.6, 163.5, 154.1, 137.24, 137.20, 136.73, 136.69, 136.3, 135.6, 134.8, 134.7, 134.2, 129.8, 129.7, 129.4, 128.1, 128.0, 127.3, 127.2, 126.58, 126.55, 126.3, 126.1, 121.3, 121.2, 120.6, 120.3, 116.31, 116.26, 65.00, 64.96, 64.6, 57.8, 53.7, 46.2, 44.2, 34.3, 34.1, 33.8, 32.1, 30.7, 29.5, 29.33, 29.28, 29.14, 29.09, 28.49, 28.46, 25.9, 25.8, 25.7, 25.6, 25.3, 25.1, 24.9. IR (film, cm−1) νmax 2927, 2854, 1735, 1600, 1531, 1488, 1411, 1319, 1247, 1020, 812. ESI-HRMS m/z: 679.4127 [M + H]+ (calcd. for C43H55N2O5 679.4111).
3.4. N-[4-({[N-(11-Azido-1-oxoundecanyl)-N-cyclohexyl]amino}methyl)phenyl]-6-(4-methylphenyl)-2H-1-benzopyran-3-carboxamide (10)
(1) To a solution of 9 (0.25 g, 0.37 mmol) in MeOH was added K2CO3 (0.14 g, 1.01 mmol). The reaction mixture was stirred at room temperature for 2 h, diluted with AcOEt, and saturated NH4Cl aq. The aqueous layer was extracted with AcOEt and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 1:1) to give alcohol (9a) (0.26 g, quant.) as a brown oil. 1H-NMR (CDCl3, 600 MHz) δ 9.15 (m, 1H), 7.50 (m, 2H), 7.40 (m, 4H), 7.26 (m, 1H), 7.20 (d, J = 7.8 Hz, 2H), 7.07 (m, 2H), 6.89 (m, 1H), 5.09 (s, 2H), 4.47 (m, 2H), 4.62 (m, 1H), 3.59 (m, 2H), 2.43–2.16 (m, 3H), 2.36 (s, 3H), 1.76–1.49 (m, 9H), 1.40–1.16 (m, 16H), 1.00 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ: 174.2, 173.6, 163.7, 163.6, 154.0, 137.2, 137.15, 137.10, 136.60, 136.57, 136.4, 135.4, 134.7, 134.6, 134.0, 129.63, 129.56, 129.4, 128.2, 128.1, 127.10, 127.06, 126.50, 126.48, 126.3, 126.2, 126.0, 121.3, 121.2, 120.5, 120.4, 116.2, 116.1, 64.92, 64.89, 62.6, 57.8, 53.7, 46.2, 44.2, 33.9, 33.7, 32.60, 32.55, 32.0, 30.6, 29.29, 29.27, 29.25, 29.21, 29.17, 29.16, 29.13, 29.10, 25.8, 25.7, 25.61, 25.56, 25.4, 25.3, 25.0. IR (film, cm−1) νmax 3298, 2928, 2854, 1653, 1620, 1533, 1487, 1412, 1320, 1253, 1136, 1020, 918, 812, 755, 665. ESI-HRMS m/z 659.3833 [M + Na]+ (calcd. for C41H52N2NaO4 659.3825).
(2) To a solution of alcohol (9a) (1.42 g, 2.23 mmol) in CH2Cl2 were added TEA (2.80 mL, 20.1 mmol), TsCl (1.90 g, 9.97 mmol), and DMAP (0.12 g, 0.98 mmol). The reaction mixture was stirred at room temperature for 1.3 h, diluted with CH2Cl2, and saturated with NH4Cl aq. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 2:1–1:1) to give tosylate (9b) (1.70 g, 2.15 mmol, 96%) as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 8.62–6.88 (m, 17H), 5.12 (m, 2H), 4.47 (m, 2H), 4.52 (m, 1H), 3.99 (m, 2H), 2.44–2.16 (m, 8H), 1.77–1.58 (m, 9H), 1.40–1.15 (m, 16H), 1.00 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ: 174.1, 173.5, 163.6, 163.4, 154.1, 144.6, 143.2, 139.3, 137.19, 137.15, 137.0, 136.73, 136.69, 136.3, 135.7, 134.8, 134.7, 134.4, 133.1, 132.99, 132.96, 129.7, 129.5, 129.4, 128.0, 127.8, 127.4, 126.8, 126.6, 126.33, 126.28, 126.2, 120.3, 116.3, 116.2, 70.7, 64.9, 57.8, 53.7, 46.3, 44.2, 33.9, 33.7, 32.1, 30.6, 29.4, 29.3, 29.2, 29.11, 29.08, 28.8, 28.7, 25.9, 25.7, 25.4, 25.2, 25.1, 21.5, 21.4, 21.0. IR (film, cm−1) νmax 2927, 2854, 1599, 1530, 1488, 1412, 1356, 1249, 1175, 1097, 926, 812, 755, 663, 554.
(3) To a solution of tosylate (9b) (70 mg, 89 µmol) in DMF was added NaN3 (65 mg, 1.0 mmol). The reaction mixture was stirred at room temperature for 10 h, diluted with AcOEt and H2O. The aqueous layer was extracted with AcOEt and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 1:1) to give 10 (45 mg, 67.5 mmol, 77%) as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 9.07 (m, 1H), 7.87–6.88 (m, 12H), 5.10 (m, 2H), 4.48 (m, 2H), 4.53 (m, 1H), 3.22 (m, 2H), 2.45–2.18 (m, 5H), 1.77–1.52 (m, 9H), 1.39–1.19 (m, 16H), 1.02 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ: 174.2, 173.5, 163.6, 163.5, 154.0, 137.3, 137.12, 137.08, 136.60, 136.57, 136.4, 135.4, 134.62, 134.59, 134.0, 129.7, 129.6, 129.5, 129.4, 128.1, 127.9, 127.5, 127.32, 127.27, 127.1, 126.7, 126.5, 126.4, 126.3, 126.2, 126.0, 121.24, 121.20, 120.7, 120.5, 120.35, 120.28, 116.20, 116.15, 64.93, 64.91, 57.7, 53.7, 51.3, 46.2, 44.1, 34.0, 33.7, 32.0, 30.6, 29.4, 29.23, 29.19, 28.94, 28.92, 28.6, 26.53, 26.51, 25.8, 25.7, 25.6, 25.5, 25.4, 25.3, 25.0, 20.9. IR (film, cm−1) νmax 2928, 2854, 2094, 1718, 1702, 1619, 1533, 1516, 1487, 1467, 1451, 1411, 1319, 1287, 1252, 1190, 1137, 1066, 1019, 1002, 917, 812, 755, 664.
3.5. N-(4-{[(N-{11-[4-(7-Hydroxy-4-methyl-2-oxo-2H-1-benzopyran-3-)-1,2,3-triazolyl]-1-oxoundecanyl}-N-cyclohexyl)amino]methyl}phenyl)-6-(4-methylphenyl)-2H-1-benzopyran-3-carboxamide (2a)
(1) To a solution of 10 (250 mg, 0.38 mmol) in CH2Cl2 were added 5 (160 mg, 0.66 mmol), CuI (76.0 mg, 0.40 mmol), and DIPEA (175 µL, 1.00 mmol). The reaction mixture was stirred at room temperature for 3 h, diluted with CH2Cl2, and saturated with NH4Cl aq. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 1:1) to give triazole (10a) (51 mg, 56.7 µmol, 15%) as a brown oil. 1H-NMR (CDCl3, 600 MHz) δ 8.60 (m, 1H), 8.13 (m, 1H), 7.66–6.83 (m, 15H), 5.24–5.07 (m, 4H), 4.49 (m, 2H), 4.54 (m, 1H), 4.38 (m, 2H), 3.47 (m, 3H), 2.36 (s, 3H), 2.16 (s, 3H), 1.93 (m, 2H), 1.77–1.56 (m, 9H), 1.42–1.21 (m, 16H), 1.02 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 174.1, 173.5, 163.7, 163.5, 160.6, 160.5, 159.9, 159.6, 159.5, 154.5, 154.1, 153.4, 153.24, 153.17, 146.0, 141.1, 137.14, 137.05, 136.7, 134.7, 129.8, 129.7, 129.4, 128.0, 127.8, 127.4, 127.3, 126.9, 126.55, 126.52, 126.3, 126.2, 125.6, 121.1, 120.4, 116.2, 114.3, 113.6, 113.5, 103.6, 103.3, 94.3, 82.2, 65.0, 57.7, 56.3, 53.7, 51.1, 50.4, 46.3, 44.2, 33.8, 33.7, 32.1, 30.7, 30.1, 30.0, 29.6, 29.3, 29.24, 29.16, 29.1, 29.0, 28.94, 28.87, 28.8, 28.7, 28.6, 26.4, 26.3, 26.2, 25.9, 25.7, 25.6, 25.4, 25.3, 25.24, 25.17, 21.1, 21.0, 17.0, 16.6. IR (film, cm−1) νmax 2928, 2859, 1609, 1514, 996, 754. ESI-MS m/z 906.4 [M + H]+ (calcd. for C55H64N5O7; 906.5).
(2) A solution of triazole (10a) (12.8 mg, 14.2 µmol) in CH2Cl2 was dropwise added to 50% TFA/CH2Cl2. The reaction mixture was stirred at room temperature for 1.5 h and concentrated in vacuo. The residue was diluted with CHCl3 and H2O. The aqueous layer was extracted with CHCl3, and the combined organic layer was washed with 1M NaOH, dried over MgSO4, and concentrated in vacuo. 2a (13 mg, quant.) was obtained as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 9.65 (m, 1H), 9.00 (m, 1H), 8.03 (m, 1H), 7.90–6.61 (m, 15H), 5.09 (m, 2H), 4.57 (m, 3H), 4.25 (m, 2H), 2.67–2.18 (m, 8H), 1.88–1.58 (m, 9H), 1.49–1.17 (m, 16H), 1.05 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 174.3, 163.8, 154.7, 154.2, 147.2, 143.7, 136.7, 136.1, 135.1, 129.9, 129.5, 128.2, 127.6, 126.9, 126.64, 126.59, 126.5, 126.4, 125.4, 121.9, 121.5, 120.6, 116.3, 113.9, 103.2, 65.1, 58.0, 51.1, 50.3, 46.5, 45.0, 33.7, 32.0, 30.7, 30.0, 29.6, 29.3, 29.1, 28.9, 28.84, 28.78, 28.7, 28.6, 26.1, 25.9, 25.8, 25.7, 25.5, 25.1, 21.1, 16.6. IR (film, cm−1) νmax 2923, 2854, 1713, 1601, 1512, 1487, 1318, 1214, 813, 750. ESI-HRMS m/z 862.4517 [M+H]+ (calcd. for C53H60N5O6 862.4544).
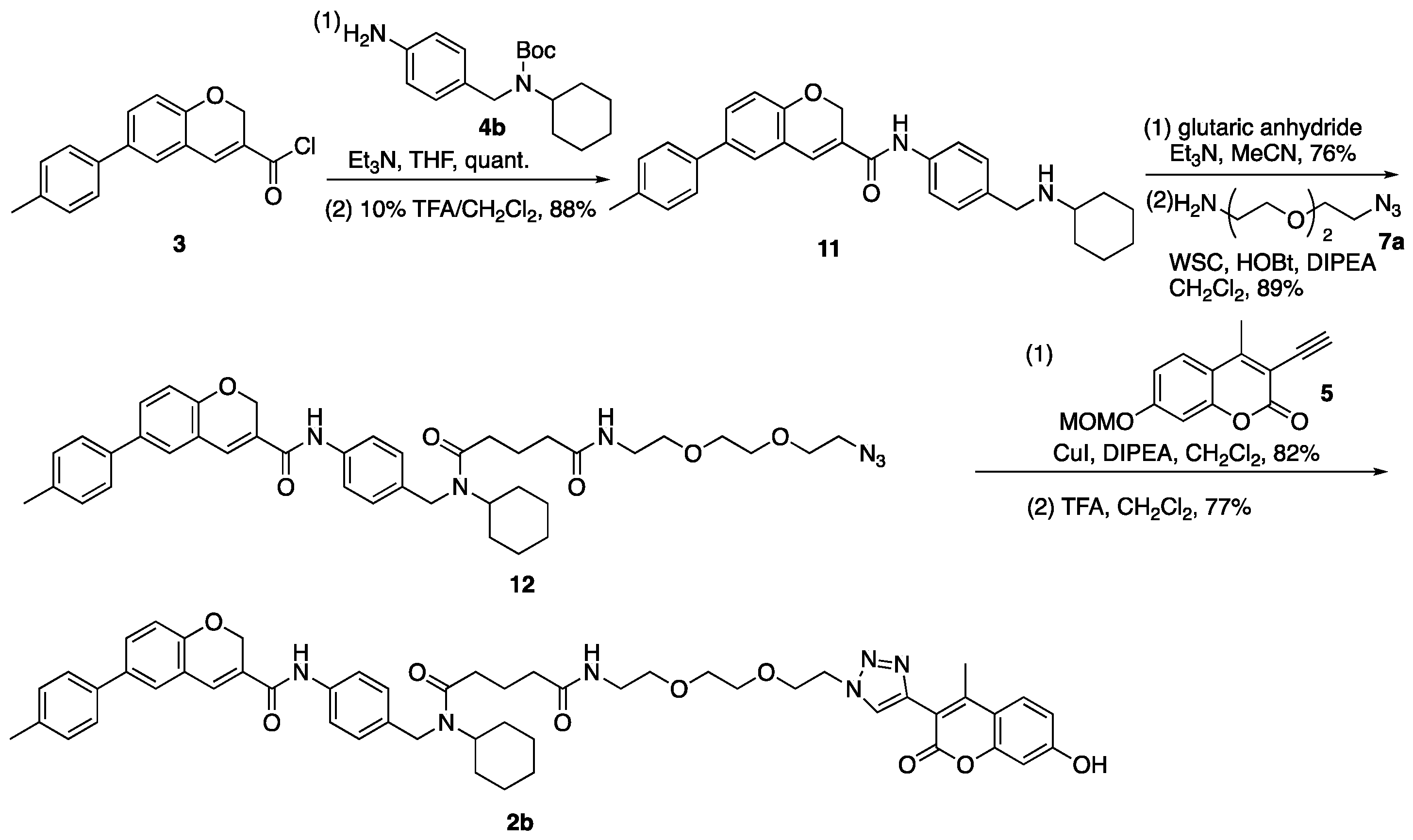
3.6. N-{4-[(Cyclohexylamino)methyl]phenyl}-6-(4-methylphenyl)-2H-chromene-3-carboxamide (11)
(1) To a solution of carboxylic acid (S5) (300 mg, 1.13 mmol) in CHCl3 (2.50 mL) was added SOCl2 (0.66 mL, 9.10 mmol) on ice. The reaction mixture was stirred at room temperature for 10 min, then DMF (71 µL, 0.92 mmol) was added to the mixture. The mixture was stirred at room temperature for 2 h and concentrated in vacuo to give 3. To a solution of 3 in THF (1.30 mL) were dropwise added a solution of 4b (446 mg, 1.47 mmol) and Et3N (613 µL, 4.40 mmol) in THF (1.30 mL) on ice. The reaction mixture was stirred at room temperature for 19 h, concentrated in vacuo, and diluted with CHCl3 and NH4Cl aq. The aqueous layer was extracted with CHCl3, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexane/AcOEt = 6:1–4:1) to give carbamate (3a) (625 mg, 1.13 mmol, quant.) as a pale-yellow powder. 1H-NMR (CDCl3, 600 MHz) δ 8.31 (m, 1H), 7.52 (m, 2H), 7.44 (dd, J = 8.4, 2.4 Hz, 1H), 7.40 (d, J = 7.8 Hz, 2H), 7.31 (s, 1H), 7.27 (d, J = 1.8 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 7.2 Hz, 2H), 6.92 (d, J = 8.4 Hz, 1H), 5.11 (s, 2H), 4.38–3.64 (m, 3H), 2.38 (s, 3H), 1.67–1.18 (m, 18H), 0.99 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 163.4, 155.8, 154.1, 137.2, 136.7, 136.5, 136.2, 134.8, 129.8, 129.5, 127.8, 127.3, 126.9, 126.6, 126.3, 121.1, 120.0, 116.3, 79.7, 64.9, 55.3, 45.8, 31.1, 28.4, 25.8, 25.4, 21.0. IR (KBr, cm−1) νmax 3286, 3195, 3126, 3058, 2971, 2935, 2852, 1687, 1644, 1600, 1534, 1515, 1486, 1447, 1404, 1365, 1326, 1279, 1247, 1164, 1148, 1100, 1022, 1006, 962, 944, 907, 880, 862, 830, 810, 780, 510. ESI-HRMS m/z 575.2879 [M + Na]+ (calcd. for C35H40N2NaO4 575.2886. m.p. 188–189 °C.
(2) To a solution of carbamate (3a) (2.03 g, 3.67 mmol) in CH2Cl2 (18.0 mL) was dropwise added TFA (1.00 mL) on ice. The reaction mixture was stirred at room temperature for 30 min, then TFA (1.00 mL) was added to the mixture on ice. The mixture was stirred at room temperature for 1.5 h, then CH2Cl2 (18.0 mL) and TFA (2.0 mL) were added to the mixture on ice. The mixture was stirred at room temperature for 2 h, diluted with CH2Cl2 and 1 M NaHCO3 aq. on ice. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 95:5–70:30) to give 11 (1.47 g, 3.25 mmol, 88%) as a yellow oil. 1H-NMR (CDCl3, 500 MHz) δ 8.05 (s, 1H), 7.51 (d, J = 8.5 Hz, 2H), 7.42 (dd, J = 8.5, 2.0 Hz, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 2.5 Hz, 1H), 7.21 (d, J = 8.0 Hz, 2H), 7.10 (s, 1H), 6.89 (d, J = 8.5 Hz, 1H), 5.05 (s, 2H), 3.76 (s, 2H), 2.51–2.45 (m, 1H), 2.38 (s, 3H), 2.05 (s, 1H), 1.91 (m, 2H), 1.73 (m, 2H), 1.62 (m, 1H), 1.19 (m, 5H). 13C-NMR (CDCl3, 150 MHz) δ 163.4, 154.0, 137.1, 136.8, 136.7, 136.3, 134.9, 129.9, 129.5, 128.8, 127.7, 127.3, 126.6, 126.4, 120.9, 120.3, 116.3, 64.9, 56.1, 50.2, 33.2, 26.0, 24.9, 21.0. IR (film, cm−1) νmax 3299, 3189, 3119, 3023, 2926, 2852, 1651, 1600, 1518, 1487, 1450, 1412, 1320, 1254, 1215, 1186, 1135, 1111, 1020, 1006, 911, 891, 811, 756, 665. ESI-HRMS m/z: 453.2571 [M + H]+ (calcd. for C30H33N2O2 453.2542).
3.7. N-{2-[2-(2-Azidoethoxy)ethoxy]ethyl}-N′-cyclohexyl-N′-({4-[6-(4-methylphenyl)-2H-chromene-3-amido]phenyl}methyl)pentanediamide (12)
(1) To a solution of 11 (400 mg, 0.88 mmol) in MeCN (3.00 mL) were added Et3N (616 µL, 4.42 mmol) and glutaric anhydride (303 mg, 2.66 mmol). The reaction mixture was stirred at room temperature for 80 min, then glutaric anhydride (202 mg, 1.77 mmol) was added to the mixture. The mixture was stirred at room temperature for 70 min, concentrated in vacuo, and diluted with AcOEt and 1 M HCl. The aqueous layer was extracted with AcOEt and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 95:5) to give carboxylic acid (11a) (382 mg, 0.67 mmol, 76%) as a yellow oil. 1H-NMR (CDCl3, 500 MHz) δ 10.23 (s, 1H), 8.90 (m, 1H), 7.51 (m, 2H), 7.39 (m, 3H), 7.30 (m, 1H), 7.24 (m, 1H), 7.19 (d, J = 7.0 Hz, 2H), 7.05 (m, 2H), 6.88 (d, J = 8.5 Hz, 1H), 5.05 (s, 2H), 4.48 (m, 3H), 2.45 (m, 2H), 2.36 (s, 3H), 2.27 (m, 2H), 1.93 (m, 2H), 1.63 (m, 5H), 1.38–1.15 (m, 4H), 0.97 (m, 1H). 13C-NMR (CDCl3, 125 MHz) δ 177.3, 177.2, 173.6, 173.0, 163.9, 163.7, 154.0, 137.1, 137.0, 136.9, 136.7, 136.6, 136.3, 135.1, 134.7, 134.0, 129.72, 129.65, 129.4, 128.4, 128.2, 127.1, 127.0, 126.9, 126.5, 126.3, 126.1, 121.2, 121.1, 120.9, 120.6, 116.22, 116.18, 64.9, 64.8, 57.6, 53.9, 46.2, 44.4, 33.1, 32.9, 32.7, 32.5, 31.8, 30.5, 25.7, 25.5, 25.2, 25.0, 20.9, 20.6, 20.4. IR (film, cm−1) νmax 3296, 3191, 3119, 3017, 2932, 2856, 1709, 1651, 1600, 1530, 1514, 1487, 1469, 1450, 1412, 1320, 1286, 1251, 1216, 1194, 1138, 1019, 1003, 914, 812, 755, 666. ESI-HRMS m/z 589.2693 [M + Na]+ (calcd. for C35H38N2NaO5 589.2678).
(2) To a solution of carboxylic acid (11a) (176 mg, 0.31 mmol) in CH2Cl2 (0.50 mL) were added DIPEA (307 µL, 1.76 mmol), WSC•HCl (135 mg, 0.70 mmol), HOBt•H2O (108 mg, 0.71 mmol), and a solution of 7a (68 mg, 0.39 mmol) in CH2Cl2 (0.20 mL). The reaction mixture was stirred at room temperature for 13 h, diluted with CH2Cl2 and H2O. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 100:0–97:3) to give 12 (200 mg, 0.28 mmol, 89%) as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 9.15 (m, 1H), 7.53 (m, 2H), 7.35 (m, 3H), 7.29 (m, 1H), 7.20 (s, 1H), 7.15 (d, J = 7.8 Hz, 2H), 7.03 (m, 2H), 6.84 (m, 1H), 6.41 (m, 1H), 5.02 (s, 2H), 4.44–3.29 (m, 15H), 2.45–2.08 (m, 4H), 2.31 (s, 3H), 1.88 (m, 2H), 1.69–1.50 (m, 5H), 1.24 (m, 4H), 0.95 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ: 172.9, 172.5, 172.4, 163.5, 163.4, 153.79, 153.77, 137.0, 136.84, 136.82, 136.5, 136.42, 136.37, 135.1, 134.5, 134.4, 133.9, 129.4, 129.3, 129.2, 128.0, 127.8, 127.0, 126.93, 126.91, 126.3, 126.0, 125.9, 121.08, 121.05, 120.4, 120.2, 116.01, 115.98, 70.1, 69.83, 69.81, 69.7, 69.6, 69.45, 69.42, 64.73, 64.72, 57.2, 53.6, 50.2, 46.0, 44.0, 38.85, 38.82, 35.22, 35.16, 32.8, 32.4, 31.8, 30.3, 25.5, 25.4, 25.1, 24.9, 21.33, 21.27, 20.7. IR (film, cm−1) νmax 3423, 3307, 3118, 3056, 3006, 2931, 2858, 2107, 1731, 1651, 1531, 1488, 1467, 1453, 1412, 1320, 1286, 1253, 1185, 1136, 1020, 997, 920, 899, 813, 755, 665, 643, 613, 572, 558, 513. ESI-HRMS m/z 745.3668 [M + Na]+ (calcd. for C41H50N6NaO6 745.3690).
3.8. N′-Cyclohexyl-N-[2-(2-{2-[4-(7-hydroxy-4-methyl-2-oxo-2H-chromen-3-yl)-1H-1,2,3-triazol-1-yl]ethoxy}ethoxy)ethyl]-N′-({4-[6-(4-methylphenyl)-2H-chromene-3-amido]phenyl}methyl)pentanediamide (2b)
(1) To a solution of 12 (200 mg, 0.28 mmol) and 5 (72 mg, 0.29 mmol) in toluene (600 µL) were added CuI (79 mg, 0.41 mmol) and DIPEA (120 µL, 0.69 mmol). The reaction mixture was stirred at room temperature for 60 h, diluted with CH2Cl2 and NH4Cl aq. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 100:0–98:2) to give triazole (12a) (220 mg, 0.23 mmol, 82%) as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 9.26 (m, 1H), 8.18 (m, 1H), 7.49 (m, 3H), 7.25 (m, 4H), 7.11 (s, 1H), 7.03 (d, J = 6.0 Hz, 2H), 6.96 (m, 2H), 6.85 (d, J = 9.0 Hz, 1H), 6.75 (m, 3H), 5.07 (m, 2H), 4.95 (s, 2H), 4.43–3.54 (m, 5H), 3.75 (m, 2H), 3.46–3.23 (m, 11H), 2.56 (m, 3H), 2.43–2.09 (m, 4H), 2.21 (s, 3H), 1.87 (m, 2H), 1.61–1.42 (m, 5H), 1.28–1.10 (m, 4H), 0.90 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 172.7, 172.5, 172.2, 163.3, 163.2, 160.2, 160.1, 159.5, 153.5, 152.9, 149.53, 149.50, 140.5, 136.9, 136.51, 136.49, 136.3, 136.14, 136.11, 135.0, 134.1, 134.0, 133.7, 129.0, 127.8, 127.6, 126.8, 126.7, 126.3, 126.1, 126.0, 125.7, 125.6, 120.9, 120.8, 120.1, 119.9, 115.72, 115.69, 114.5, 113.2, 113.00, 112.98, 102.61, 102.59, 93.8, 69.9, 69.7, 69.6, 69.4, 68.7, 64.5, 57.0, 55.8, 53.3, 49.6, 45.8, 43.8, 38.8, 38.7, 35.0, 32.7, 32.3, 31.6, 30.2, 25.34, 25.28, 24.9, 24.7, 21.25, 21.22, 20.5, 16.4. IR (film, cm−1) νmax 3320, 3190, 3123, 3056, 3006, 2932, 2858, 1709, 1659, 1616, 1578, 1530, 1516, 1488, 1467, 1453, 1412, 1385, 1337, 1320, 1279, 1254, 1221, 1143, 1073, 1019, 998, 970, 924, 814, 754, 711, 692, 665, 612, 512. ESI-HRMS m/z 989.4419 [M + Na]+ (calcd. for C55H62N6NaO10 989.4425).
(2) Triazole (12a) (270 mg, 0.28 mmol) was added to 50% TFA/CH2Cl2 (2.00 mL) on ice. The reaction mixture was stirred at room temperature for 30 min, diluted with CH2Cl2 and NaHCO3 aq. on ice. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 100:0–97:3) to give 2b (198 mg, 0.21 mmol, 77%) as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 10.24 (m, 1H), 9.06 (m, 1H), 8.22 (m, 1H), 7.50 (m, 2H), 7.43 (m, 1H), 7.34 (m, 4H), 7.21 (m, 1H), 7.14 (d, J = 8.4 Hz, 2H), 7.02 (m, 3H), 6.80 (m, 2H), 6.67 (s, 1H), 5.04 (m, 2H), 4.47–3.63 (m, 5H), 3.80 (m, 2H), 3.52–3.33 (m, 8H), 2.58 (s, 3H), 2.52–2.20 (m, 4H), 2.31 (s, 3H), 1.96 (m, 2H), 1.60 (m, 5H), 1.36–1.13 (m, 4H), 0.90 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ: 173.4, 173.3, 173.2, 173.0, 163.70, 163.68, 161.21, 161.18, 161.15, 161.1, 153.9, 153.6, 150.9, 141.0, 136.9, 136.6, 136.2, 135.0, 134.6, 134.5, 134.0, 129.6, 129.5, 129.4, 128.3, 128.1, 127.0, 126.8, 126.7, 126.6, 126.5, 126.44, 126.38, 126.2, 126.0, 121.2, 121.1, 120.8, 120.6, 116.1, 113.7, 113.0, 111.8, 102.51, 102.45, 70.30, 70.25, 70.0, 69.9, 69.7, 69.0, 64.9, 64.8, 57.5, 53.9, 50.0, 46.2, 44.5, 39.3, 35.39, 35.35, 33.1, 32.6, 31.8, 30.5, 25.6, 25.5, 25.2, 25.0, 21.62, 21.58, 16.7. IR (film, cm−1) νmax 3306, 3011, 2932, 2858, 1698, 1650, 1614, 1601, 1583, 1530, 1512, 1487, 1453, 1412, 1384, 1320, 1287, 1251, 1216, 1189, 1131, 1077, 1022, 982, 932, 894, 853, 814, 755, 687, 666, 612, 511. ESI-HRMS m/z: 945.4151 [M + Na]+ (calcd. for C53H58N6NaO9 945.4163).
3.9. Tert-Butyl N-{2-[2-(2-{4-[Cyclohexyl({4-[6-(4-methylphenyl)-2H-chromene-3-amido]phenyl}methyl)carbamoyl]butanamido}ethoxy)ethoxy]ethyl}carbamate (13)
(1) To a solution of 11 (400 mg, 0.88 mmol) in MeCN (3.00 mL) were added TEA (616 µL, 4.42 mmol) and glutaric anhydride (303 mg, 2.66 mmol). The reaction mixture was stirred at room temperature for 80 min, then glutaric anhydride (202 mg, 1.77 mmol) was added to the mixture. The mixture was stirred at room temperature for 70 min, concentrated in vacuo, and diluted with AcOEt and 1M HCl. The aqueous layer was extracted with AcOEt and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 95:5) to give carboxylic acid (11a) (382 mg, 0.67 mmol, 76%) as a yellow oil. 1H-NMR (CDCl3, 500 MHz) δ 10.23 (s, 1H), 9.89 (m, 1H), 7.51 (m, 2H), 7.40 (m, 3H), 7.30 (m, 1H), 7.24 (m, 1H), 7.19 (d, J = 7.0 Hz, 2H), 7.06 (m, 2H), 6.88 (d, J = 8.5 Hz, 1H), 5.05 (s, 2H), 4.41 (m, 3H), 2.45 (m, 2H), 2.36 (s, 3H), 2.28 (m, 2H), 1.97 (m, 2H), 1.63 (m, 5H), 1.38–1.15 (m, 4H), 0.96 (m, 1H). 13C-NMR (CDCl3, 125 MHz) δ 177.3, 177.2, 173.6, 173.0, 163.9, 163.7, 154.0, 137.1, 137.0, 136.9, 136.7, 136.6, 136.3, 135.1, 134.7, 134.0, 129.72, 129.65, 129.4, 128.4, 128.2, 127.1, 127.0, 126.9, 126.5, 126.3, 126.1, 121.17 121.14, 120.9, 120.6, 116.22, 116.18, 64.85, 64.82, 57.6, 53.9, 46.2, 44.4, 33.1, 32.9, 32.7, 32.5, 31.8, 30.5, 25.7, 25.5, 25.2, 25.0, 20.9, 20.6, 20.4. IR (film, cm−1) νmax 3296, 3191, 3119, 3017, 2932, 2856, 1709, 1651, 1600, 1530, 1514, 1487, 1469, 1450, 1412, 1320, 1286, 1251, 1216, 1194, 1138, 1019, 1003, 914, 812, 755, 666. ESI-HRMS m/z: 589.2693 [M + Na]+ (calcd. for C35H38N2NaO5 589.2678).
(2) To a solution of carboxylic acid (11a) (190 mg, 0.34 mmol) in CH2Cl2 (0.50 mL) were added DIPEA (292 µL, 1.68 mmol), WSC•HCl (129 mg, 0.67 mmol), HOBt•H2O (103 mg, 0.67 mmol), and a solution of 7b (108 mg, 0.43 mmol) in CH2Cl2 (0.20 mL). The reaction mixture was stirred at room temperature for 14 h, diluted with CH2Cl2 and NaCl aq. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 100:0–98:2) to give 13 (241 mg, 0.30 mmol, 90%) as a yellow oil. 1H-NMR (CDCl3, 600 MHz) δ 8.79 (m, 1H), 7.53 (m, 2H), 7.40 (m, 3H), 7.29 (m, 1H), 7.27 (s, 1H), 7.20 (d, J = 7.8 Hz, 2H), 7.09 (m, 2H), 6.89 (m, 1H), 6.47 (m, 1H), 5.18 (s, 1H), 5.07 (s, 2H), 4.49–3.62 (m, 3H), 3.55–3.23 (m, 12H), 2.50–2.13 (m, 4H), 2.36 (s, 3H), 2.01–1.86 (m, 2H), 1.74–1.55 (m, 5H), 1.42–1.19 (m, 13H), 0.99 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 173.2, 172.71, 172.65, 163.6, 163.5, 156.0, 154.0, 137.2, 137.1, 137.0, 136.74, 136.70, 136.4, 135.5, 134.80, 134.76, 134.2, 129.8, 129.7, 129.4, 128.0, 127.8, 127.3, 127.2, 126.5, 126.3, 126.1, 121.21, 121.17, 120.5, 120.3, 116.3, 116.2, 79.2, 70.1, 70.0, 69.72, 69.67, 65.0, 64.9, 57.5, 53.8, 46.2, 44.3, 40.2, 39.1, 39.0, 35.6, 35.5, 32.9, 32.5, 32.0, 30.6, 28.3, 25.8, 25.7, 25.3, 25.1, 21.6, 21.5, 21.0. IR (film, cm−1) νmax 3319, 3002, 2975, 2932, 2859, 1695, 1652, 1532, 1488, 1453, 1412, 1390, 1365, 1320, 1283, 1252, 1173, 1136, 1109, 1020, 1006, 921, 858, 813, 755, 666, 612, 511. ESI-HRMS m/z 819.4286 [M + Na]+ (calcd. for C46H60N4NaO8 819.4309).
3.10. N′-Cyclohexyl-N′-({4-[6-(4-methylphenyl)-2H-chromene-3-amido] phenyl}methyl)carbamoyl]butanamido-N′-(ethoxy)ethoxy]ethyl}carbamoyl)ethyl]-2,2-difluoro-4,6-dimethyl-1λ⁵,3-diaza-2-boratricyclo [7.3.0.03,7]dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (2c)
(1) To a solution of 13 (1.25 g, 1.57 mmol) in CH2Cl2 (27.0 mL) was added TFA (3.00 mL) on ice. The reaction mixture was stirred at room temperature for 2 h, diluted with CH2Cl2 and an excess amount of NaHCO3 aq. on ice. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo to give crude amine (13a).
(2) To a solution of 6 (37 mg, 0.13 mmol) in CH2Cl2 (200 µL) were added DIPEA (110 µL, 0.63 mmol), WSC•HCl (49 mg, 0.26 mmol), HOBt•H2O (39 mg, 0.25 mmol), and a solution of the residue (13a) (90 mg) in CH2Cl2 (500 µL). The reaction mixture was stirred at room temperature for 17 h, diluted with CH2Cl2 and H2O. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 100:0–97:3) to give 2c (85 mg, 87.5 µmol, 68%, 2 steps) as a red oil. 1H-NMR (CDCl3, 600 MHz) δ 8.93–8.73 (m, 1H), 7.59–7.47 (m, 2H), 7.38 (m, 3H), 7.26 (m, 1H), 7.22 (m, 1H), 7.19 (d, J = 7.2 Hz, 2H), 7.07 (m, 2H), 6.99 (m, 1H), 6.87 (m, 1H), 6.76 (m, 1H), 6.47 (m, 2H), 6.16 (m, 1H), 6.02 (m, 1H), 5.05 (m, 2H), 4.47–3.61 (m, 3H), 3.41 (m, 12H), 3.23 (m, 2H), 2.60–2.10 (m, 6H), 2.47 (m, 3H), 2.34 (s, 3H), 2.16 (m, 3H), 1.99–1.84 (m, 2H), 1.73–1.54 (m, 5H), 1.38–1.17 (m, 4H), 0.98 (m, 1H) ppm. 13C-NMR (CDCl3, 150 MHz) δ 173.5, 173.1, 172.67, 172.65, 172.5, 172.2, 171.9, 171.8, 171.7, 163.5, 163.4, 159.9, 159.8, 157.5, 157.3, 153.99, 153.96, 143.74, 143.67, 137.10, 137.07, 137.0, 136.7, 136.6, 136.4, 135.3, 134.91, 134.88, 134.7, 134.6, 134.2, 133.2, 129.7, 129.6, 129.4, 128.13, 128.10, 128.0, 127.8, 127.2, 127.11, 127.07, 126.5, 126.3, 126.1, 123.64, 123.60, 121.2, 120.5, 120.3, 117.00, 116.96, 116.20, 116.15, 70.03, 69.98, 69.60, 69.56, 64.92, 64.90, 57.4, 53.7, 46.1, 44.2, 39.1, 39.01, 38.95, 35.53, 35.48, 35.46, 35.2, 32.93, 32.89, 32.4, 31.9, 30.6, 25.7, 25.6, 25.3, 25.0, 24.6, 24.5, 21.5, 21.4, 20.9, 14.8, 11.1. IR (film, cm−1) νmax 3413, 3307, 3056, 3006, 2931, 2858, 1721, 1651, 1605, 1530, 1488, 1443, 1412, 1373, 1321, 1252, 1216, 1194, 1175, 1136, 1087, 1064, 1019, 1000, 975, 910, 813, 754, 666, 612, 590, 512. ESI-HRMS m/z 993.4866 [M + Na]+ (calcd. for C55H65BF2N6NaO7 993.4874).
3.11. 12-(2-{[(1S)-3-Carboxy-1-({2-[2-(2-{4-[cyclohexyl({4-[6-(4-methylphenyl)-2H-chromene-3-amido]phenyl}methyl)carbamoyl]butanamido}ethoxy)ethoxy]ethyl} carbamoyl)propyl]-carbamoyl}ethyl)-2,2-difluoro-4,6-dimethyl-1λ⁵,3-diaza-2-boratricyclo [7.3.0.03,⁷]dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (2d)
(1) To a solution of 13 (1.25 g, 1.57 mmol) in CH2Cl2 (27.0 mL) was added TFA (3.0 mL) on ice. The reaction mixture was stirred at room temperature for 2 h, diluted with CH2Cl2 and 1 M NaHCO3 aq. on ice. The aqueous layer was extracted with CH2Cl2, and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo to give activated aster (6b).
(2) To a solution of 6a (5.0 mg, 10.5 µmol) in DMF (200 µL) were added DIPEA (9 µL, 51.7 µmol), WSC•HCl (4.0 mg, 20.9 µmol), HOBt•H2O (3.2 mg, 20.9 µmol), and a solution of the residue (6b) (8.0 mg) in DMF (200 µL). The reaction mixture was stirred at room temperature for 80 min, then a solution of the residue (6b) (5.0 mg) in DMF (100 µL) was added to the mixture. The mixture was stirred at room temperature for 40 min, diluted with AcOEt and NH4Cl aq. The aqueous layer was extracted with AcOEt and the combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 100:0–96:4) to give ester (14) (7.0 mg, 6.05 µmol, 58%, 2 steps) as a red oil. 1H-NMR (CDCl3, 600 MHz) δ 8.60 (m, 1H), 7.54 (m, 2H), 7.40 (m, 3H), 7.23 (m, 4H), 7.10 (m, 3H), 6.99 (m, 1H), 6.83 (m, 1H), 6.88 (m, 1H), 6.81 (m, 2H), 6.17 (m, 1H), 6.02 (m, 1H), 5.06 (m, 2H), 4.49–3.64 (m, 4H), 3.60–3.29 (m, 12H), 3.21 (m, 2H), 2.60 (m, 3H), 2.48 (m, 3H), 2.37 (s, 3H), 2.31–2.15 (m, 8H), 2.04–1.77 (m, 4H), 1.75–1.56 (m, 5H), 1.40–1.19 (m, 13H), 1.00 (m, 1H). 13C-NMR (CDCl3, 150 MHz) δ 173.2, 172.95, 172.91, 172.8, 172.7, 172.5, 171.91, 171.86, 171.33, 171.27, 163.5, 163.4, 160.3, 160.2, 156.9, 156.8, 154.03, 154.01, 144.0, 143.9, 137.13, 137.09, 137.0, 136.75, 136.70, 136.4, 135.3, 135.1, 135.0, 134.75, 134.68, 134.3, 133.2, 129.7, 129.6, 129.5, 128.04, 127.99, 127.8, 127.23, 127.18, 127.0, 126.6, 126.3, 126.2, 123.71, 123.68, 121.20, 121.16, 120.5, 120.4, 120.3, 116.9, 116.3, 116.2, 80.7, 80.6, 70.14, 70.12, 70.06, 69.73, 69.70, 69.4, 64.9, 57.5, 53.7, 52.5, 46.2, 44.4, 39.2, 39.12, 39.07, 35.63, 35.58, 35.2, 35.1, 33.0, 32.5, 31.9, 31.5, 30.6, 28.0, 27.4, 25.8, 25.7, 25.4, 25.1, 24.4, 21.7, 21.5, 21.0, 14.8, 11.2. IR (film, cm−1) νmax 3300, 3006, 2975, 2932, 2858, 1724, 1651, 1606, 1530, 1488, 1448, 1412, 1367, 1320, 1252, 1216, 1174, 1136, 1087, 1064, 1019, 1000, 974, 910, 813, 754, 666, 512. ESI-HRMS m/z 1178.5929 [M + Na]+ (calcd. for C64H80BF2N7NaO10 1178.5925). [α]D28 −60.0 (c 0.1, CHCl3).
(3) 4 N HCl/AcOEt (0.30 mL) was added to ester (14) (3.0 mg, 2.59 µmol) on ice and the reaction mixture was stirred at room temperature for 2 h and concentrated in vacuo. The residue was purified by column chromatography (CHCl3/MeOH = 95:5) to give 2d (2.1 mg, 1.91 µmol, 74%) as a red oil. 1H-NMR (CDCl3, 600 MHz) δ 8.68 (m, 1H), 7.54 (m, 2H), 7.40 (m, 3H), 7.23 (m, 5H), 7.06 (m, 4H), 6.87 (m, 2H), 6.76 (m, 1H), 6.15 (m, 1H), 6.03 (m, 1H), 5.06 (m, 2H), 4.57–3.65 (m, 4H), 3.55–3.27 (m, 12H), 3.21 (t, J = 7.2 Hz, 2H), 2.66–2.50 (m, 3H), 2.48 (m, 3H), 2.40–2.12 (m, 5H), 2.37 (s, 3H), 2.16 (m, 3H), 2.05–1.56 (m, 9H), 1.44–1.19 (m, 4H), 1.00 (m, 1H). 13C- NMR (CDCl3, 150 MHz) δ 175.24, 175.16, 174.0, 173.44, 173.36, 173.2, 172.2, 172.1, 171.40, 171.36, 163.55, 163.51, 160.29, 160.25, 156.9, 156.8, 156.5, 154.0, 143.9, 137.2, 137.1, 137.0, 136.8, 136.7, 136.5, 135.1, 134.9, 134.8, 134.7, 134.0, 133.2, 129.8, 129.7, 129.49, 129.47, 128.2, 128.1, 128.0, 127.9, 127.21, 127.18, 127.0, 126.7, 126.4, 126.3, 123.8, 121.25, 121.18, 120.6, 120.5, 120.4, 116.9, 116.3, 116.2, 70.2, 70.13, 70.09, 69.9, 69.8, 69.3, 64.97, 64.95, 57.7, 54.0, 52.21, 52.17, 46.3, 44.6, 39.3, 39.19, 39.15, 35.5, 35.4, 35.1, 33.1, 32.4, 31.9, 31.8, 30.6, 30.0, 29.8, 29.6, 27.9, 27.8, 25.74, 25.66, 25.3, 25.1, 24.44, 24.39, 21.7, 21.6, 21.0, 14.9, 11.2. IR (film, cm−1) νmax 3298, 3006, 2932, 2858, 1720, 1650, 1606, 1530, 1488, 1445, 1412, 1376, 1321, 1252, 1216, 1175, 1137, 1087, 1063, 1017, 999, 974, 910, 813, 754, 665, 512. ESI-HRMS m/z 1122.5288 [M + Na]+ (calcd. for C60H72BF2N7NaO10 1122.5299). [α]D28 −112.0 (c 0.1, CHCl3).
3.12. 12-[2-({2-[2-(2-Azidoethoxy)ethoxy]ethyl}carbamoyl)ethyl]-2,2-difluoro-4,6-dimethyl-1λ⁵,3-diaza-2-boratricyclo [7.3.0.03,⁷]dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (15)
To a solution of BODIPY FL propionic acid (6) (80 mg, 0.27 mmol) in DMF (1.0 mL) was added DIPEA (238 µL, 1.37 mmol), WSC•HCl (105 mg, 0.55 mmol), HOBt•H2O (84 mg, 0.55 mmol), and 1-(2-aminoethoxy)-2-(2-azidoethoxy)ethane (7a) (95 mg, 0.55 mmol) at room temperature. After 3 h, the mixture was added to AcOEt and H2O. The organic layer was separated, washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified with silica gel column chromatography (CHCl3/MeOH = 97:3) to give 12-[2-({2-[2-(2-azidoethoxy)ethoxy]ethyl}carbamoyl)ethyl]-2,2-difluoro-4,6-dimethyl-1λ5,3-diaza-2-boratricyclo [7.3.0.03,7]dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (15) (70 mg, 0.16 mmol, 57%) as a red oil. 1H-NMR (CDCl3, 600 MHz) δ 7.06 (s, 1H), 6.86 (d, J = 4.2 Hz, 1H), 6.27 (d, J = 3.6 Hz, 1H), 6.09 (m, 2H), 3.63 (t, J = 4.8 Hz, 2H), 3.60–3.56 (m, 4H), 3.50 (t, J = 4.8 Hz, 2H), 3.43–3.40 (m, 2H), 3.34 (t, J = 4.8 Hz, 2H), 3.26 (t, J = 7.5 Hz, 2H), 2.61 (t, J = 7.5 Hz, 2H), 2.54 (s, 3H), 2.23 (s, 3H). 13C-NMR (CDCl3, 150 MHz) δ: 171.6, 160.0, 157.6, 143.7, 135.0, 133.3, 128.2, 123.7, 120.3, 117.4, 70.4, 70.2, 70.0, 69.8, 50.5, 39.1, 35.8, 24.7, 14.8, 11.2. IR (film, cm−1) νmax 3418, 3311, 3096, 3070, 2924, 2870, 2108, 1656, 1606, 1529, 1488, 1442, 1345, 1305, 1252, 1174, 1136, 1185, 973, 933, 817, 750, 666. ESI-HRMS m/z 471.2132 [M + Na]+ (calcd. for C20H27BF2N6NaO3 471.2103).
3.13. 12-{2-[(2-{2-[2-(4-Carboxybutanamido)ethoxy]ethoxy}ethyl)carbamoyl]ethyl}-2,2-difluoro-4,6-dimethyl-1λ5,3-diaza-2-boratricyclo [7.3.0.03,7]dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (16)
To a solution of 12-[2-({2-[2-(2-Azidoethoxy)ethoxy]ethyl}carbamoyl)ethyl]-2,2-difluoro-4,6-dimethyl-1λ5,3-diaza-2-boratricyclo [7.3.0.03,7]dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (15) (70 mg, 0.16 mmol) in MeOH (520 µL) was added glutaric anhydride (53 mg, 0.46 mmol) and 10% Pd/C (4.0 mg). The mixture was vigorously stirred at room temperature in H2 atmosphere. After 1 h, to the mixture was added glutaric anhydride (36 mg, 0.32 mmol) and Et3N (150 µL, 1.08 mmol) and stirred at room temperature. After 10 min, the mixture was filtrated with a celite pad and concentrated in vacuo. The residue was added to H2O, 1M HCl (adjusted to pH 2), and AcOEt. The organic layer was separated and washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified with silica gel column chromatography (CHCl3/MeOH = 90:10) to give 12-{2-[(2-{2-[2-(4-Carboxybutanamido)ethoxy]ethoxy}ethyl)carbamoyl]ethyl}-2,2-difluoro-4,6-dimethyl-1λ5,3-diaza-2-boratricyclo [7.3.0.03,7]dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (16) (63 mg, 0.12 mmol, 75%) as a red oil. 1H-NMR (CDCl3, 600 MHz) δ: 7.07 (s, 1H), 6.86 (d, J = 4.2 Hz, 1H), 6.59–6.54 (m, 2H), 6.27 (d, J = 3.6 Hz, 1H), 6.10 (s, 1H), 3.54 (m, 4H), 3.51 (t, J = 5.4 Hz, 2H), 3.49 (t, J = 5.4 Hz, 2H), 3.40 (m, 4H), 3.25 (t, J = 7.5 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H), 2.52 (s, 3H), 2.35 (t, J = 6.6 Hz, 2H), 2.25 (t, J = 7.2 Hz, 2H), 2.23 (s, 3H), 1.90 (quin, J = 7.2 Hz, 2H). 13C-NMR (CDCl3, 150 MHz) δ 175.9, 172.9, 172.4, 160.1, 157.4, 143.9, 135.0, 133.3, 128.2, 123.8, 120.4, 117.3, 70.1, 70.0, 69.8, 69.7, 39.2, 39.1, 35.6, 35.0, 32.7, 24.7, 20.7, 14.9, 11.2. IR (film, cm−1) νmax 3306, 3094, 3307, 2932, 2871, 1725, 1649, 1607, 1530, 1488, 1442, 1347, 1253, 1175, 1137, 1086, 973, 899, 812, 752, 666. ESI-HRMS m/z 559.2509 [M + Na]+ (calcd. for C25H35BF2N4NaO6 559.2515).
3.14. 12-(2-{[2-(2-{2-[4-(Diethylcarbamoyl)butanamido]ethoxy}ethoxy)ethyl]-carbamoyl}ethyl)-2,2-difluoro-4,6-dimethyl-1λ5,3-diaza-2-boratricyclo [7.3.0.03,7]-dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (17)
To a solution of 12-{2-[(2-{2-[2-(4-Carboxybutanamido)ethoxy]ethoxy}ethyl)-carbamoyl]ethyl}-2,2-difluoro-4,6-dimethyl-1λ5,3-diaza-2-boratricyclo [7.3.0.03,7]-dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (16) (10.0 mg, 18.6 µmol) in MeCN (300 µL) was added DIPEA (16 µL, 91.9 µmol), WSC•HCl (7.0 mg, 36.5 µmol), HOBt•H2O (6.0 mg, 39.2 µmol), and Et2NH (4 µL, 38.7 µmol) and stirred at room temperature. After 1 h, the mixture was added AcOEt and H2O and the organic layer was separated. The organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified with silica gel column chromatography (CHCl3/MeOH = 95:5) to give 12-(2-{[2-(2-{2-[4-(Diethylcarbamoyl)butanamido]ethoxy}ethoxy)ethyl]-carbamoyl}ethyl)-2,2-difluoro-4,6-dimethyl-1λ5,3-diaza-2-boratricyclo [7.3.0.03,7]-dodeca-1(12),4,6,8,10-pentaen-1-ylium-2-uide (17) (6.0 mg, 10.1 µmol, 54%) as a red oil. 1H-NMR (CDCl3, 600 MHz) δ 7.08 (s, 1H), 6.87 (d, J = 3.6 Hz, 1H), 6.41 (t, J = 4.8 Hz, 1H), 6.38 (t, J = 4.8 Hz, 1H), 6.29 (d, J = 3.6 Hz, 1H), 6.11 (s, 1H), 3.56 (s, 4H), 3.52 (t, J = 5.4 Hz, 4H), 3.44–3.40 (m, 4H), 3.34 (q, J = 7.2 Hz, 2H), 3.28 (t, J = 7.8 Hz, 2H), 3.28 (q, J = 7.2 Hz, 2H), 2.63 (t, J = 7.2 Hz, 2H), 2.55 (s, 3H), 2.35 (t, J = 7.2 Hz, 2H), 2.25 (s, 3H), 2.24 (t, J = 7.2 Hz, 2H), 1.93 (quin, J = 7.2 Hz, 2H), 1.14 (t, J = 7.2 Hz, 3H), 1.08 (t, J = 7.2 Hz, 3H). 13C-NMR (CDCl3, 150 MHz) δ 172.8, 171.8, 171.6, 160.0, 157.8, 143.7, 135.0, 133.3, 128.3, 123.7, 120.3, 117.4, 70.20, 70.17, 69.9, 69.8, 41.9, 40.1, 39.2, 39.1, 35.8, 35.7, 31.9, 24.8, 21.5, 14.9, 14.3, 13.1, 11.3. IR (film, cm−1) νmax 3419, 3299, 3082, 2971, 2931, 2874, 1647, 1606, 1529, 1488, 1439, 1374, 1253, 1175, 1136, 1086, 973, 750, 666. ESI-HRMS m/z 614.3308 [M + Na]+ (calcd. for C29H44BF2N5NaO5 614.3301).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}