
Gaussian Process Regression for Mapping Free EnergyLandscape of Mg2+-Cl− Ion Pairing in Aqueous Solution: Molecular Insights and Computational Efficiency


Abstract
1. Introduction
2. Results and Discussion
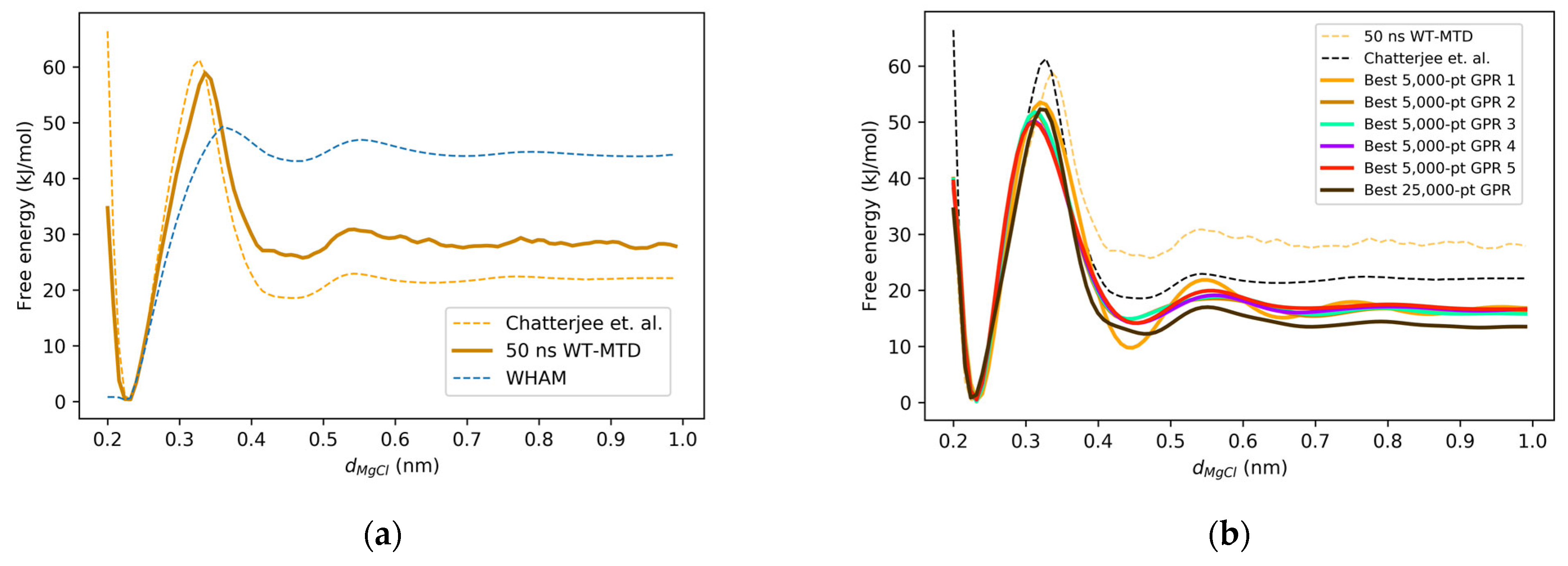
2.1. Mg2+-Cl− One-Dimensional Free Energy Surface
2.2. GPR Reconstruction of the One-Dimensional Free Energy Surface
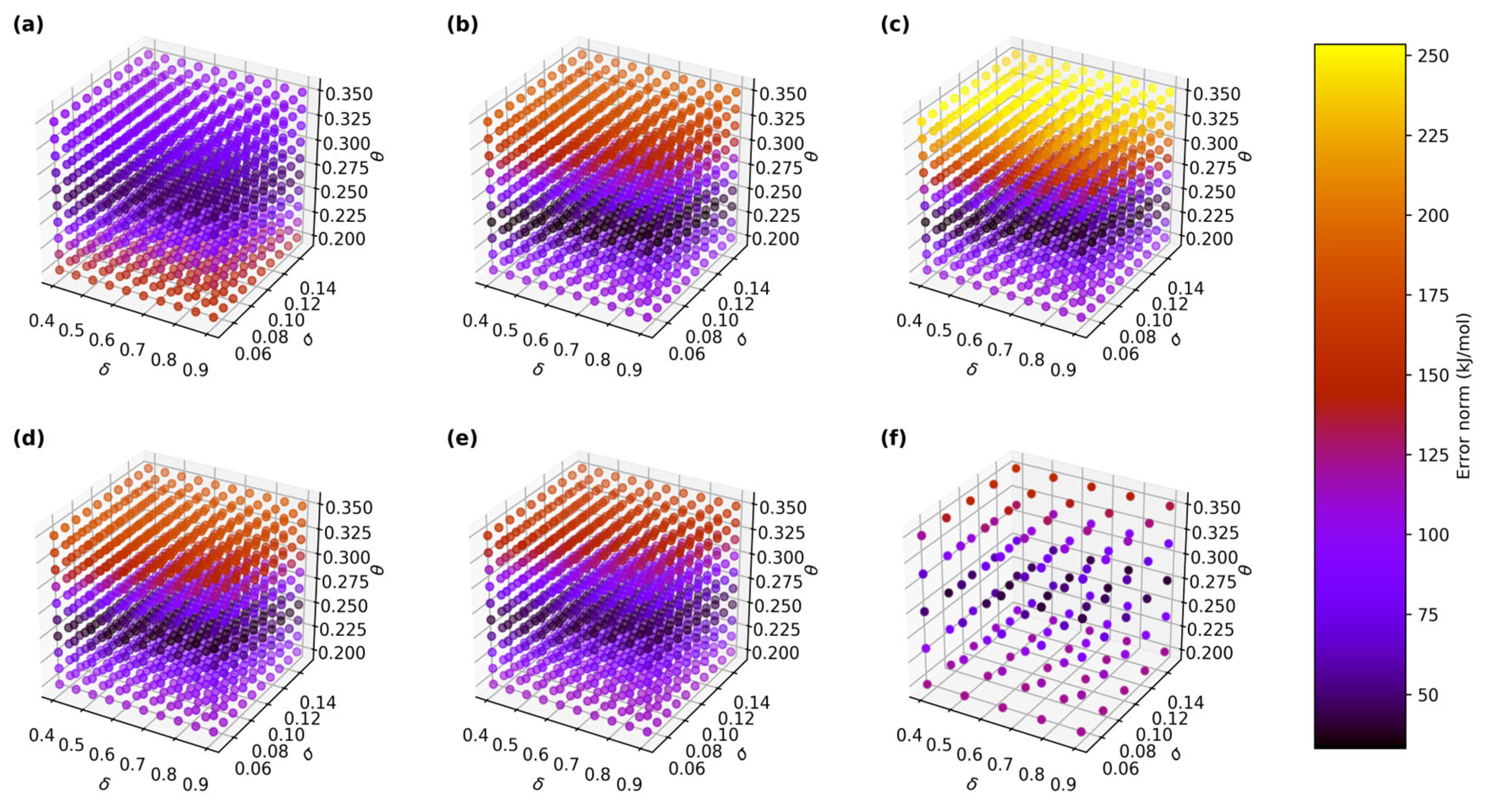
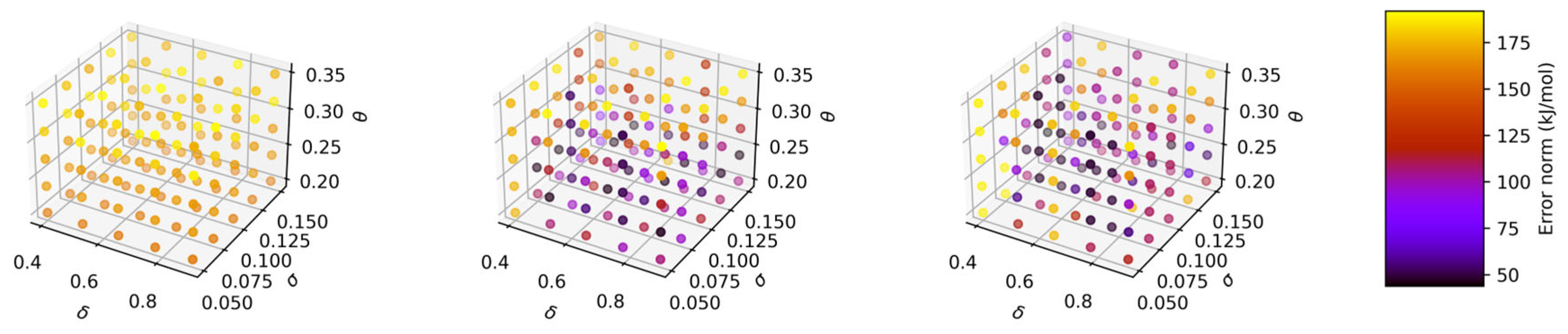
2.3. Addressing High GPR Computational Cost in Large Data Regime
2.4. Extending GPR Beyond One-Dimensional Free Energy Reconstruction
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torrie, G.M.; Valleau, J.P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- E, W.; Ren, W.; Vanden-Eijnden, E. String Method for the Study of Rare Events. Phys. Rev. B 2002, 66, 052301. [Google Scholar] [CrossRef]
- E, W.; Ren, W.; Vanden-Eijnden, E. Finite Temperature String Method for the Study of Rare Events. J. Phys. Chem. B 2005, 109, 6688–6693. [Google Scholar] [CrossRef]
- E, W.; Ren, W.; Vanden-Eijnden, E. Simplified and Improved String Method for Computing the Minimum Energy Paths in Barrier-Crossing Events. J. Chem. Phys. 2007, 126, 164103. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella Sampling. WIREs Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Grossfield, A. WHAM: The Weighted Histogram Analysis Method, version 2.1.0. Available online: http://membrane.urmc.rochester.edu/wordpress/?page_id=126 (accessed on 20 May 2025).
- Thiede, E.H.; Van Koten, B.; Weare, J.; Dinner, A.R. Eigenvector Method for Umbrella Sampling Enables Error Analysis. J. Chem. Phys. 2016, 145, 84115. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping Free-Energy Minima. Proc. Nat. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 20603. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bonomi, M.; Parrinello, M. Linking Well-Tempered Metadynamics Simulations with Experiments. Biophys. J. 2010, 98, L44–L46. [Google Scholar] [CrossRef] [PubMed]
- Valsson, O.; Parrinello, M. Well-Tempered Variational Approach to Enhanced Sampling. J. Chem. Theory Comput. 2015, 11, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Maragliano, L.; Vanden-Eijnden, E. A Temperature Accelerated Method for Sampling Free Energy and Determining Reaction Pathways in Rare Events Simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar] [CrossRef]
- Darve, E.; Pohorille, A. Calculating Free Energies Using Average Force. J. Chem. Phys. 2001, 115, 9169–9183. [Google Scholar] [CrossRef]
- Maragliano, L.; Vanden-Eijnden, E. Single-Sweep Methods for Free Energy Calculations. J. Chem. Phys. 2008, 128, 184110. [Google Scholar] [CrossRef]
- Mones, L.; Bernstein, N.; Csányi, G. Exploration, Sampling, And Reconstruction of Free Energy Surfaces with Gaussian Process Regression. J. Chem. Theory Comput. 2016, 12, 5100–5110. [Google Scholar] [CrossRef]
- Leong, K.W.; Pan, W.; Yi, X.; Luo, S.; Zhao, X.; Zhang, Y.; Wang, Y.; Mao, J.; Chen, Y.; Xuan, J.; et al. Next-Generation Magnesium-Ion Batteries: The Quasi-Solid-State Approach to Multivalent Metal Ion Storage. Sci. Adv. 2023, 9, eadh1181. [Google Scholar] [CrossRef]
- León-Pimentel, C.I.; Saint-Martin, H.; Ramirez-Solis, A. Mg(II) and Ca(II) Microsolvation by Ammonia: BornOppenheimer Molecular Dynamics Studies. J. Phys. Chem. A 2021, 125, 4565–4577. [Google Scholar] [CrossRef]
- van der Vegt, N.F.A.; Haldrup, K.; Roke, S.; Zheng, J.; Lund, M.; Bakker, H.J. Water-Mediated Ion Pairing: Occurrence and Relevance. Chem. Rev. 2016, 116, 7626–7641. [Google Scholar] [CrossRef]
- Richens, D.T. Ligand Substitution Reactions at Inorganic Centers. Chem. Rev. 2005, 105, 1961–2002. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Boero, M.; Terakura, K. Hydration Properties of Magnesium and Calcium Ions from Constrained First Principles Molecular Dynamics. J. Chem. Phys. 2007, 127, 74503. [Google Scholar] [CrossRef] [PubMed]
- Ohtaki, H.; Radnai, T. Structure and Dynamics of Hydrated Ions. Chem. Rev. 1993, 93, 1157–1204. [Google Scholar] [CrossRef]
- Martínez, J.M.; Pappalardo, R.R.; Sánchez Marcos, E. First-Principles Ion−Water Interaction Potentials for Highly Charged Monatomic Cations. Computer Simulations of Al3+, Mg2+, and Be2+ in Water. J. Am. Chem. Soc. 1999, 121, 3175–3184. [Google Scholar] [CrossRef]
- Chatterjee, A.; Dixit, M.K.; Tembe, B.L. Solvation Structures and Dynamics of the Magnesium Chloride (Mg2+-Cl−) Ion Pair in Water-Ethanol Mixtures. J. Phys. Chem. A 2013, 117, 8703–8709. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Schwierz, N. Kinetic Pathways of Water Exchange in the First Hydration Shell of Magnesium. J. Chem. Phys. 2020, 152, 224106. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1 and χ 2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| WHAM | WT-MTD | Chatterjee et al. | Experimental | |
|---|---|---|---|---|
| CIP Location (dMg-Cl nm) | 0.23 | 0.23 | 0.23 | 0.20–0.22 |
| SSIP Location (dMg-Cl nm) | 0.47 | 0.47 | 0.47 | 0.41–0.43 |
| Barrier Location (dMg-Cl nm) | 0.36 | 0.34 | 0.33 | n/a |
| Training Data/GPR Models | δ | σ | θ | Error Norm (kJ/mol) | Error per Data Point (kJ/mol) |
|---|---|---|---|---|---|
| 5000 points/regular (Batch 1) | 0.40 | 0.11 | 0.25 | 44.35 | 0.44 |
| 5000 points/regular (Batch 1) | 0.57 | 0.15 | 0.25 | 44.35 | 0.44 |
| 5000 points/regular (Batch 2) | 0.40 | 0.14 | 0.22 | 33.34 | 0.33 |
| 5000 points/regular (Batch 2) | 0.62 | 0.13 | 0.23 | 33.82 | 0.34 |
| 5000 points/regular (Batch 2) | 0.73 | 0.15 | 0.23 | 33.80 | 0.34 |
| 5000 points/regular (Batch 3) | 0.40 | 0.14 | 0.22 | 35.09 | 0.35 |
| 5000 points/regular (Batch 3) | 0.62 | 0.13 | 0.23 | 35.40 | 0.35 |
| 5000 points/regular (Batch 3) | 0.79 | 0.15 | 0.23 | 35.24 | 0.35 |
| 5000 points/regular (Batch 4) | 0.46 | 0.12 | 0.23 | 36.17 | 0.36 |
| 5000 points/regular (Batch 4) | 0.51 | 0.13 | 0.23 | 36.03 | 0.36 |
| 5000 points/regular (Batch 4) | 0.84 | 0.13 | 0.25 | 36.68 | 0.37 |
| 5000 points/regular (Batch 5) | 0.40 | 0.13 | 0.23 | 39.90 | 0.40 |
| 5000 points/regular (Batch 5) | 0.51 | 0.15 | 0.23 | 40.54 | 0.41 |
| 5000 points/regular (Batch 5) | 0.79 | 0.15 | 0.25 | 40.65 | 0.41 |
| 25,000 points/regular | 0.40 | 0.08 | 0.28 | 40.15 | 0.40 |
| 25,000 points/regular | 0.65 | 0.13 | 0.28 | 40.34 | 0.40 |
| 25,000 points/regular | 0.90 | 0.15 | 0.28 | 40.73 | 0.41 |
| 25,000 points/sparse grid size = 10 | 0.40 | 0.15 | 0.20 | 158.13 | 1.58 |
| 25,000 points/sparse grid size = 25 | 0.40 | 0.10 | 0.24 | 45.36 | 0.45 |
| 25,000 points/sparse grid size = 25 | 0.53 | 0.15 | 0.24 | 45.13 | 0.45 |
| 25,000 points/sparse grid size = 25 | 0.90 | 0.05 | 0.24 | 46.85 | 0.47 |
| 25,000 points/sparse grid size = 50 | 0.40 | 0.13 | 0.24 | 43.85 | 0.44 |
| 25,000 points/sparse grid size = 50 | 0.53 | 0.10 | 0.28 | 47.50 | 0.48 |
| 25,000 points/sparse grid size = 50 | 0.78 | 0.15 | 0.24 | 47.20 | 0.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pornpatcharapong, W. Gaussian Process Regression for Mapping Free EnergyLandscape of Mg2+-Cl− Ion Pairing in Aqueous Solution: Molecular Insights and Computational Efficiency. Molecules 2025, 30, 2595. https://doi.org/10.3390/molecules30122595
Pornpatcharapong W. Gaussian Process Regression for Mapping Free EnergyLandscape of Mg2+-Cl− Ion Pairing in Aqueous Solution: Molecular Insights and Computational Efficiency. Molecules. 2025; 30(12):2595. https://doi.org/10.3390/molecules30122595
Chicago/Turabian StylePornpatcharapong, Wasut. 2025. "Gaussian Process Regression for Mapping Free EnergyLandscape of Mg2+-Cl− Ion Pairing in Aqueous Solution: Molecular Insights and Computational Efficiency" Molecules 30, no. 12: 2595. https://doi.org/10.3390/molecules30122595
APA StylePornpatcharapong, W. (2025). Gaussian Process Regression for Mapping Free EnergyLandscape of Mg2+-Cl− Ion Pairing in Aqueous Solution: Molecular Insights and Computational Efficiency. Molecules, 30(12), 2595. https://doi.org/10.3390/molecules30122595