
Chelerythrine Chloride Alleviated Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting Glycolytic Pathway Through Targeting Glyceraldehyde-3-Phosphate Dehydrogenase

 , ,
, ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
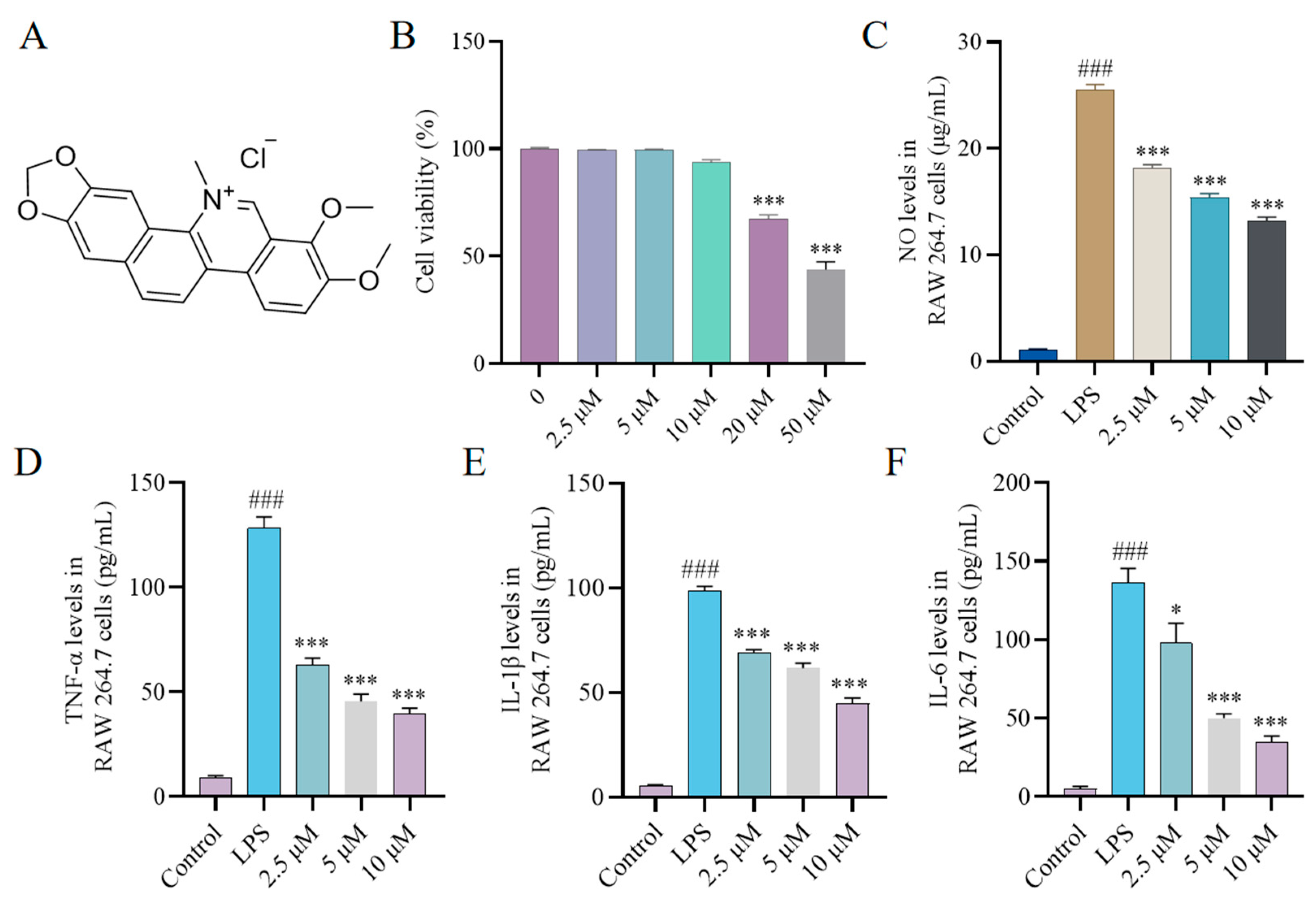
2.1. Effect of CH on Cytokine Production In Vitro
2.2. Effect of CH on Nitric Oxide Synthase (iNOS) and Cyclooxygenase-2 (COX-2) Expression In Vitro
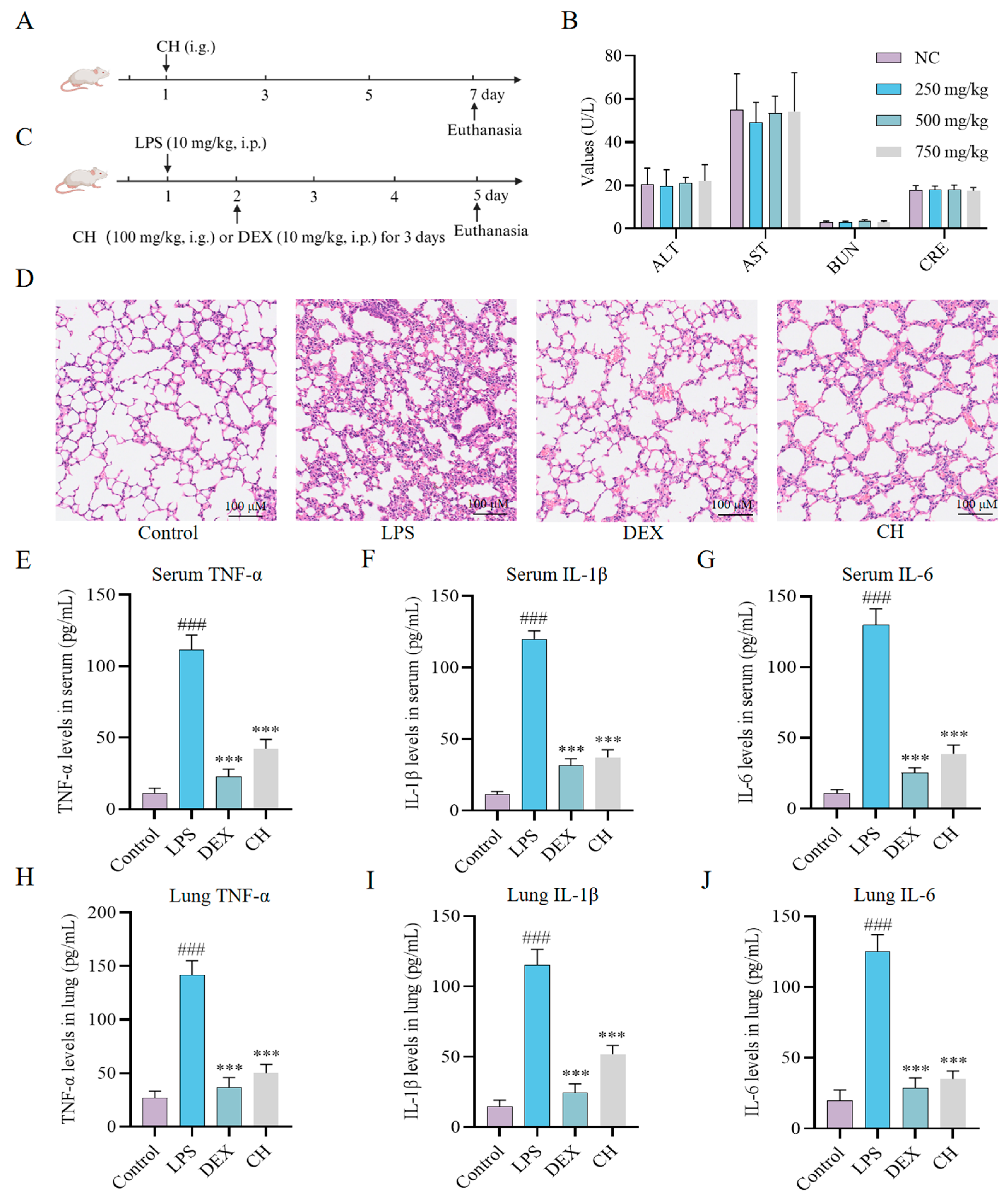
2.3. Therapeutic Impact of CH In Vivo
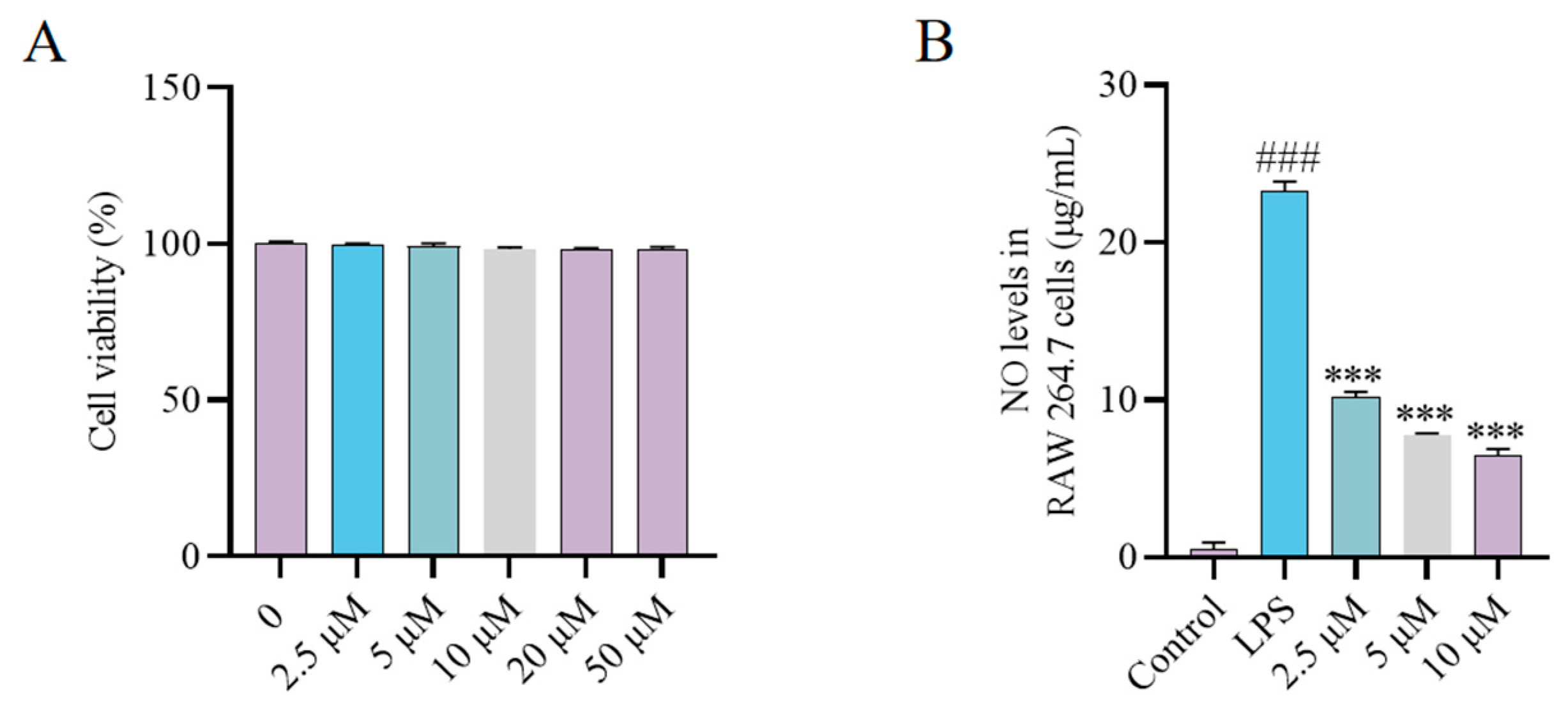
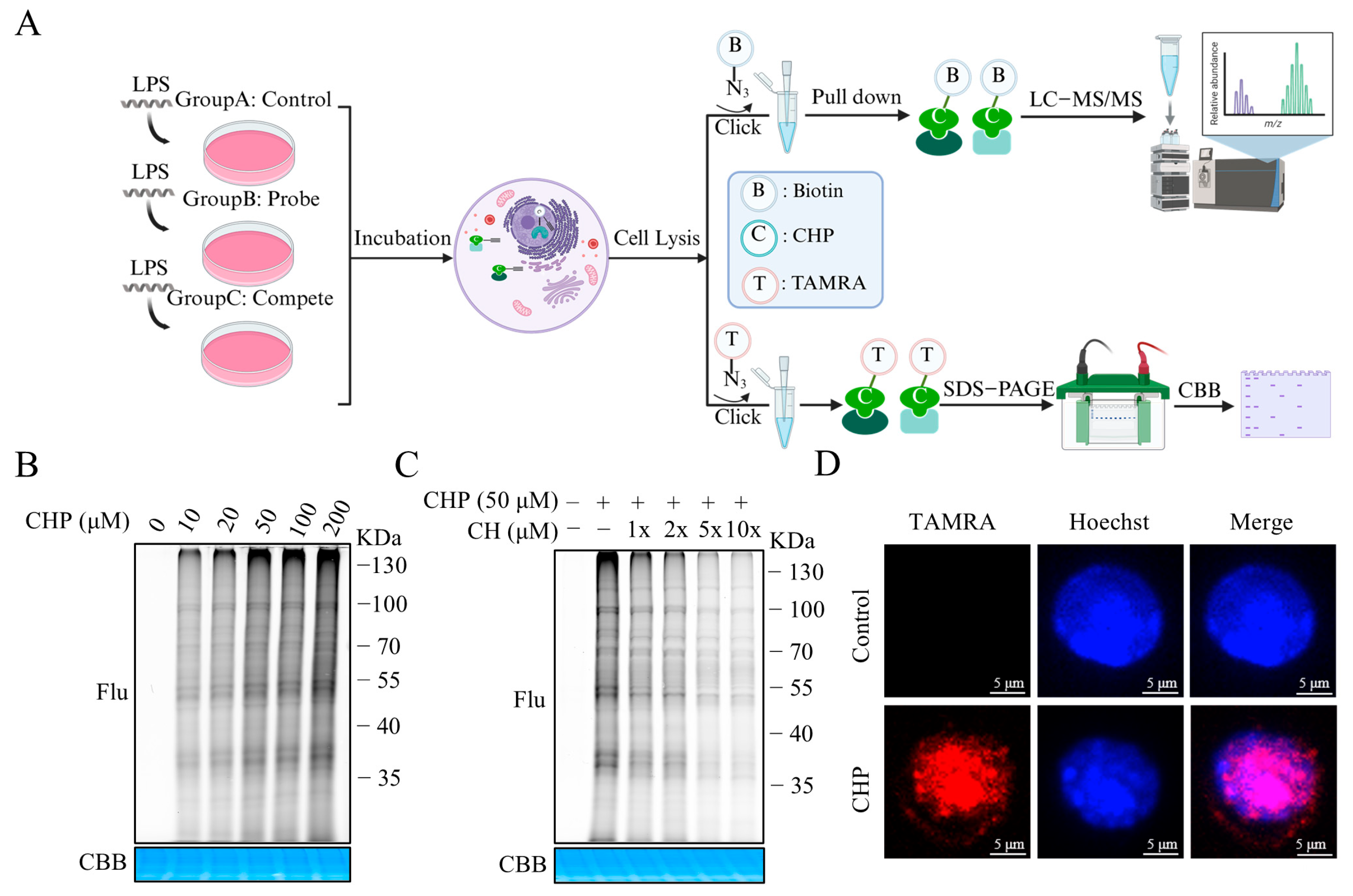
2.4. Synthesis and Bioactivity of the CH Probe
2.5. Identifying the Binding Proteins of CH by ABPP
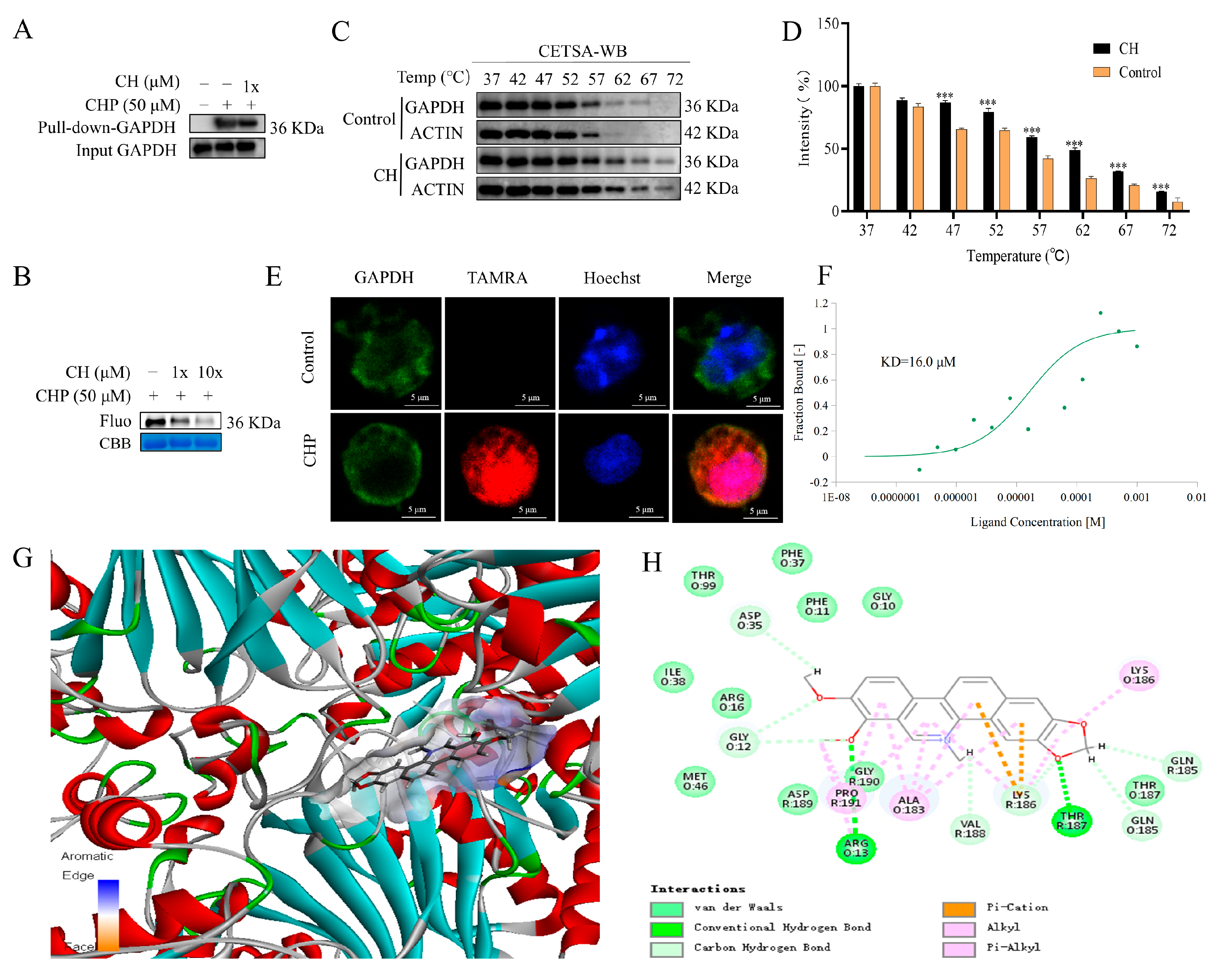
2.6. Validation of GAPDH as the Protein Target of CH
2.7. Inhibitory Effect of CH on the Glycolytic Pathway
3. Discussion
4. Materials and Methods
4.1. Agents and Chemicals
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Detection of NO Production
4.5. Measurement of Inflammatory Factors
4.6. Western Blot
4.7. Acute Toxicity in Mice
4.8. LPS-Induced ALI Mouse Model
4.9. Fluorescent Labeling and Competition
4.10. Target Identification and Pull Down
4.11. Cellular Thermal Shift Assay
4.12. Fluorescence Co-Localization
4.13. Microscale Thermophoresis
4.14. Assessment of GAPDH Enzymatic Activity
4.15. Measurement of Pyruvate Concentration
4.16. Seahorse Extracellular Flux Analyser Assays for OCR and ECAR
4.17. Molecular Docking
4.18. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABPP | activity-based protein profiling |
| ALI | acute lung injury |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| BUN | blood urea nitrogen |
| CETSA | cellular thermal shift assay |
| CH | chelerythrine chloride |
| COX-2 | cyclooxygenase-2 |
| CRE | creatinine |
| DEX | dexamethasone |
| ECAR | extracellular acidification rate |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| H&E | hematoxylin-eosin staining |
| IL | interleukin |
| iNOS | nitric oxide synthase |
| KD | dissociation constant |
| LD50 | half-lethal dose |
| LPS | lipopolysaccharide |
| MST | microscale thermophoresis |
| NO | nitric oxide |
| OCR | oxygen consumption rate |
| TNF | tumor necrosis factor |
| WB | western blot |
References
- Mollaei, M.; Abbasi, A.; Hassan, Z.M.; Pakravan, N. The intrinsic and extrinsic elements regulating inflammation. Life Sci. 2020, 260, 118258. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Schurman, S.H.; Bektas, A.; Kaileh, M.; Roy, R.; Wilson, D.M.; Sen, R.; Ferrucci, L. Aging and inflammation. Cold Spring Harb. Perspect. Med. 2024, 14, a041197. [Google Scholar] [CrossRef] [PubMed]
- da Silva, P.R.; do Espírito Santo, R.F.; Melo, C.O.; Pachú Cavalcante, F.E.; Costa, T.B.; Barbosa, Y.V.; E Silva, Y.M.S.M.; de Sousa, N.F.; Villarreal, C.F.; de Moura, R.O.; et al. The compound (E)-2-cyano-N,3-diphenylacrylamide (JMPR-01): A potential drug for treatment of inflammatory diseases. Pharmaceutics 2022, 14, 188. [Google Scholar] [CrossRef]
- Zarrin, A.A.; Bao, K.; Lupardus, P.; Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 2021, 20, 39–63. [Google Scholar] [CrossRef]
- Long, M.E.; Mallampalli, R.K.; Horowitz, J.C. Pathogenesis of pneumonia and acute lung injury. Clin. Sci. 2022, 136, 747–769. [Google Scholar] [CrossRef]
- Yin, Z.; Song, R.; Yu, T.; Fu, Y.; Ding, Y.; Nie, H. Natural compounds regulate macrophage polarization and alleviate inflammation against ALI/ARDS. Biomolecules 2025, 15, 192. [Google Scholar] [CrossRef]
- Zhou, X.; Liao, Y. Gut-lung crosstalk in sepsis-induced acute lung injury. Front. Microbiol. 2011, 12, 779620. [Google Scholar] [CrossRef]
- Dushianthan, A.; Grocott, M.P.W.; Postle, A.D.; Cusack, R. Acute respiratory distress syndrome and acute lung injury. Postgrad. Med. J. 2011, 87, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol.-Mech. Dis. 2011, 6, 147–163. [Google Scholar] [CrossRef]
- Pereira-Leite, C.; Nunes, C.; Jamal, S.K.; Cuccovia, I.M.; Reis, S. Nonsteroidal anti-inflammatory therapy: A journey toward safety. Med. Res. Rev. 2017, 37, 802–859. [Google Scholar] [CrossRef]
- Chang, Y.C.; Kim, C.H. Molecular research of glycolysis. Int. J. Mol. Sci. 2022, 23, 5052. [Google Scholar] [CrossRef] [PubMed]
- Minhas, P.S.; Latif-Hernandez, A.; McReynolds, M.R.; Durairaj, A.S.; Wang, Q.; Rubin, A.; Joshi, A.U.; He, J.Q.; Gauba, E.; Liu, L.; et al. Restoring metabolism of myeloid cells reverses cognitive de cline in ageing. Nature 2021, 590, 122–128. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Liao, S.T.; Han, C.; Xu, D.Q.; Fu, X.W.; Wang, J.S.; Kong, L.Y. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 2019, 10, 5091. [Google Scholar] [CrossRef]
- Wen, L.; Liao, L.P.; Song, N.; Liu, Y.J.; Ding, Y.L.; Zhang, Y.Y.; Zhou, X.R.; Sun, Z.Y.; Xiao, S.H.; Wang, H.B.; et al. Natural product 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose is a reversible inhibitor of glyceraldehyde 3-phosphate dehydrogenase. Acta Pharmacol. Sin. 2022, 43, 470–482. [Google Scholar]
- Yoo, H.J.; Choi, D.W.; Roh, Y.J.; Lee, Y.M.; Lim, J.H.; Eo, S.; Lee, H.J.; Kim, N.Y.; Kim, S.; Cho, S.; et al. MsrB1-regulated GAPDH oxidation plays programmatic roles in shaping metabolic and inflammatory signatures during macrophage activation. Cell Rep. 2022, 41, 111598. [Google Scholar] [CrossRef] [PubMed]
- Maji, A.K.; Pratim-Banerji, P.B. Chelidonium majus L. (greater celandine)—A review on its phytochemical and therapeutic perspectives. Int. J. Herb. Med. 2015, 3, 10–27. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, L.C.; Zhao, R.J.; Pu, L.M.; Chen, K.Y.; Nasim, A.A.; Leung, E.L.; Fan, X.X. Chelerythrine ameliorates rheumatoid arthritis by modulating the AMPK/mTOR/ULK-1 signaling pathway. Phytomedicine 2022, 104, 154140. [Google Scholar] [CrossRef]
- Niu, X.F.; Zhou, P.; Li, W.F.; Xu, H.B. Effects of chelerythrine, a specific inhibitor of cyclooxygenase-2, on acute inflammation in mice. Fitoterapia 2011, 82, 620–625. [Google Scholar] [CrossRef]
- Liang, D.; Liu, L.; Zheng, Q.; Zhao, M.; Zhang, G.; Tang, S.; Tang, J.; Chen, N. Chelerythrine chloride inhibits the progression of colorectal cancer by targeting cancer-associated fibroblasts through intervention with WNT10B/β-catenin and TGFβ2/Smad2/3 axis. Phytother. Res. 2023, 37, 4674–4689. [Google Scholar] [CrossRef]
- Fan, L.; Fan, Y.; Liu, L.; Tao, W.; Shan, X.; Dong, Y.; Li, L.; Zhang, S.; Wang, H. Chelerythrine attenuates the inflammation of lipopolysaccharide-induced acute lung inflammation through NF-κB signaling pathway mediated by Nrf2. Front.Pharmacol. 2018, 9, 1047. [Google Scholar] [CrossRef] [PubMed]
- Mutreja, V.; Kumar, A.; Sareen, S.; Pathania, K.; Sandhu, H.; Kataria, R.; Park, J. Aggregation-induced quenching of carbon dots for detection of nitric oxide. ChemistrySelect 2022, 7, e202200448. [Google Scholar] [CrossRef]
- Yang, G.; Gao, M.; Sun, Y.; Wang, C.; Fang, X.; Gao, H.; Diao, W.; Yu, H. Design, synthesis and anti-inflammatory activity of 3-amino acid derivatives of ocotillol-type sapogenins. Eur. J. Med. Chem. 2020, 202, 112507. [Google Scholar] [CrossRef] [PubMed]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife 2014, 3, e03342. [Google Scholar] [CrossRef]
- Fan, X.G.; Pei, S.Y.; Zhou, D.; Zhou, P.C.; Huang, Y.; Hu, X.W.; Li, T.; Wang, Y.; Huang, Z.B.; Li, N. Melittin ameliorates inflammation in mouse acute liver failure via inhibition of PKM2-mediated Warburg effect. Acta Pharmacol. Sin. 2021, 42, 1256–1266. [Google Scholar] [CrossRef]
- Gao, C.L.; Hou, G.G.; Liu, J.; Ru, T.; Xu, Y.Z.; Zhao, S.Y.; Ye, H.; Zhang, L.Y.; Chen, K.X.; Guo, Y.W.; et al. Synthesis and target identification of benzoxepane derivatives as potential anti-neuroinflammatory agents for ischemic stroke. Angew. Chem. Int. Edit. 2020, 59, 2429–2439. [Google Scholar] [CrossRef]
- Broderick, L.; DeNardo, D.; Franklin, B.S.; Hoffman, H.M.; Latz, E. The inflammasomes and autoinflammatory syndromes. Annu. Rev. Pathol.-Mech. 2015, 10, 395–424. [Google Scholar] [CrossRef]
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.; Ferguson, N.; Caldwell, E.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Nanchal, R.S.; Truwit, J.D. Recent advances in understanding and treating acute respiratory distress syndrome. F1000Research 2018, 7, 1322. [Google Scholar] [CrossRef]
- Da-Silva-Santos, J.E.; Santos-Silva, M.C.; Cunha-Fde, Q.; Assreuy, J. The role of ATP-sensitive potassium channels in neutrophil migration and plasma exudation. J. Pharmacol. Exp. Ther. 2002, 300, 946–951. [Google Scholar] [CrossRef]
- Janz, D.R.; Ware, L.B. Biomarkers of ALI/ARDS: Pathogenesis, discovery, and relevance to clinical trials. Semin. Respir. Crit Care Med. 2013, 34, 537–548. [Google Scholar] [PubMed]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute respiratory distress syndrome: Advances in diagnosis and treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef]
- He, Y.Q.; Zhou, C.C.; Yu, L.Y.; Wang, L.; Deng, J.L.; Tao, Y.L.; Zhang, F.; Chen, W.S. Natural product derived phytochemicals in managing acute lung injury by multiple mechanisms. Pharmacol. Res. 2021, 163, 105224. [Google Scholar] [CrossRef]
- Niu, X.; Mu, Q.; Li, W.; Huang, H.; Yao, H.; Li, H. Protective effects of chelerythrine against lipopolysaccharide-induced endotoxic shock in mice. Inflammation 2014, 37, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wen, L.; Shi, Q.; Gao, F.; Huang, B.; Wang, C. Chelerythrine ameliorates pulmonary fibrosis via activating the Nrf2/ARE signaling pathway. Cell Biochem. Biophys. 2021, 79, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Green, D.; Ricci, J.E. Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 2009, 16, 1573–1581. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Xu, S.; Wang, Y.; Li, J.; Wang, S.; Fu, L.; Jiang, M.; Bai, G. Phillyrin and its metabolites exert antipyretic effects by targeting the NAD+ binding domain of GAPDH, MDH2 and IDH2. Phytomedicine 2024, 134, 155955. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O'Sullivan, D.; Huang, S.C.; Vander-Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Nakano, T.; Goto, S.; Takaoka, Y.; Tseng, H.P.; Fujimura, T.; Kawamoto, S.; Ono, K.; Chen, C.L. A novel moonlight function of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for immunomodulation. Biofactors 2018, 44, 597–608. [Google Scholar] [CrossRef]
- Garcin, E.D. GAPDH as a model non-canonical AU-rich RNA binding protein. Semin. Cell Dev. Biol. 2019, 86, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Luo, P.; Xia, F.; Tang, H.; Chen, J.; Zhang, J.; Liu, D.; Zhu, Y.; Liu, Y.; Gu, L.; et al. Capsaicin ameliorates inflammation in a TRPV1-independent mechanism by inhibiting PKM2-LDHA-mediated warburg effect in sepsis. Cell Chem. Biol. 2022, 29, 1248–1259.e6. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Tan, L.; Tao, H.; Li, Y.; Liu, H. CETSA and thermal proteome profiling strategies for target identification and drug discovery of natural products. Phytomedicine 2023, 116, 154862. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Liu, D.; Wang, M.; Zhang, S.; Kuang, W.; Yuan, L.; Wang, J.; Liu, G. Celastrol induces premature ovarian insufficiency by inducing apoptosis in granulosa cells. Biomed. Pharmacother. 2023, 169, 115815. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Kuang, W.; Xu, L.; He, Y.; Zhou, L.; Zhang, Y.; Chen, R.; Li, H.; Fan, T.; et al. Discovery and evolution of berberine analogues as anti-Helicobacter pylori agents with multi-target mechanisms. Bioorg. Chem. 2024, 151, 107628. [Google Scholar] [CrossRef]
- Bai, J.; Qin, Q.; Li, S.; Cui, X.; Zhong, Y.; Yang, L.; An, L.; Deng, D.; Zhao, J.; Zhang, R.; et al. Salvia miltiorrhiza inhibited lung cancer through aerobic glycolysis suppression. J. Ethnopharmacol. 2024, 331, 118281. [Google Scholar] [CrossRef]
- Yan, Y.; Zang, X.; Jamieson, C.S.; Lin, H.C.; Houk, K.N.; Zhou, J.; Tang, Y. Biosynthesis of the fungal glyceraldehyde-3-phosphate dehydrogenase inhibitor heptelidic acid and mechanism of self-resistance. Chem. Sci. 2020, 11, 9554–9562. [Google Scholar] [CrossRef]
- Fan, T.Y.; Wang, Y.X.; Tang, S.; Hu, X.X.; Zen, Q.X.; Pang, J.; Yang, Y.S.; You, X.F.; Song, D.Q. Synthesis and antibacterial evaluation of 13-substituted cycloberberine derivatives as a novel class of anti-MRSA agents. Eur. J. Med. Chem. 2018, 157, 877–886. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Fan, T.; Zhuge, R.; Li, H.; Li, G.; Zhou, L.; Xu, L.; Hao, X.; Gu, W.; Wang, J. Chelerythrine Chloride Alleviated Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting Glycolytic Pathway Through Targeting Glyceraldehyde-3-Phosphate Dehydrogenase. Molecules 2025, 30, 2572. https://doi.org/10.3390/molecules30122572
He Y, Fan T, Zhuge R, Li H, Li G, Zhou L, Xu L, Hao X, Gu W, Wang J. Chelerythrine Chloride Alleviated Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting Glycolytic Pathway Through Targeting Glyceraldehyde-3-Phosphate Dehydrogenase. Molecules. 2025; 30(12):2572. https://doi.org/10.3390/molecules30122572
Chicago/Turabian StyleHe, Yuting, Tianyun Fan, Ruishen Zhuge, Huiying Li, Guanjun Li, Lirun Zhou, Liting Xu, Xiaojiang Hao, Wei Gu, and Jigang Wang. 2025. "Chelerythrine Chloride Alleviated Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting Glycolytic Pathway Through Targeting Glyceraldehyde-3-Phosphate Dehydrogenase" Molecules 30, no. 12: 2572. https://doi.org/10.3390/molecules30122572
APA StyleHe, Y., Fan, T., Zhuge, R., Li, H., Li, G., Zhou, L., Xu, L., Hao, X., Gu, W., & Wang, J. (2025). Chelerythrine Chloride Alleviated Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting Glycolytic Pathway Through Targeting Glyceraldehyde-3-Phosphate Dehydrogenase. Molecules, 30(12), 2572. https://doi.org/10.3390/molecules30122572