
New Conjugatable Platinum(II) Chlorins: Synthesis, Reactivity and Singlet Oxygen Generation

 , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract

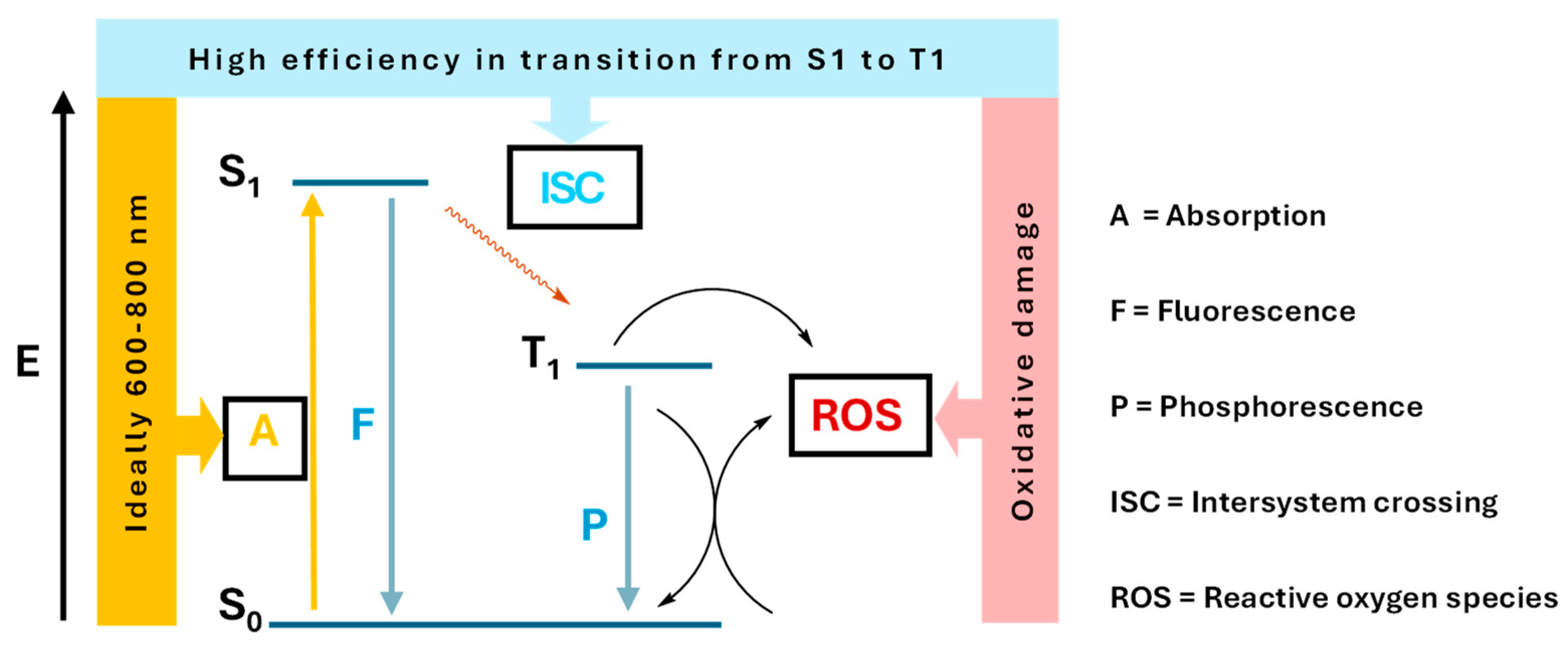
1. Introduction
2. Results and Discussion
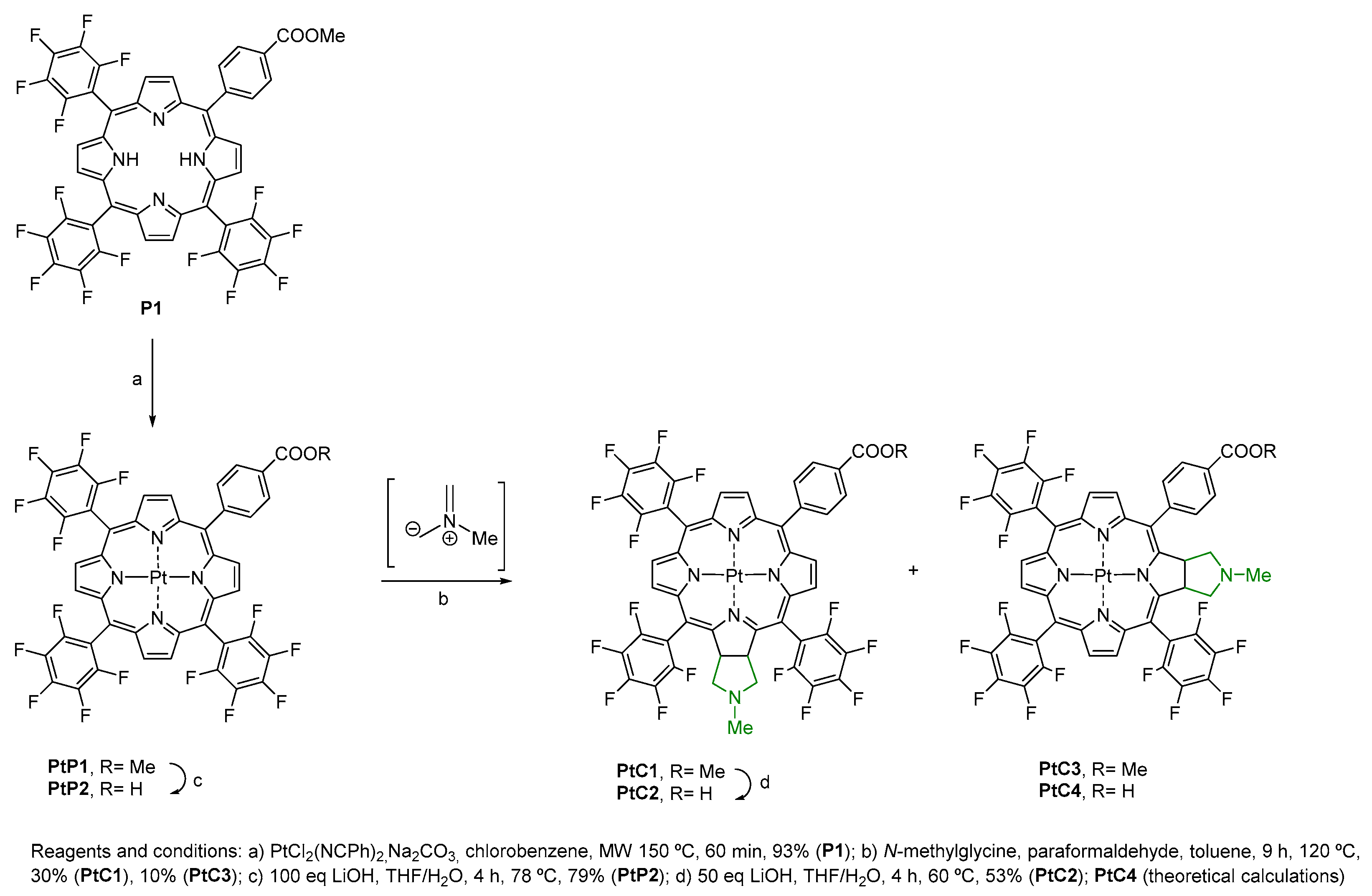
2.1. Synthesis
- (1)
- Replacing PtCl2 with a platinum salt less susceptible to oxidation (K2PtCl4). However, there was no better progression of the reaction, even increasing the reaction time to 2 h (entries 1–5, Table 1).
- (2)
- Verifying the release of HCl in the reaction mixture with PtCl2—we decided to use a base (Na2CO3) to reduce the temperature and increase the reaction time. However, a complete conversion of the porphyrin into the platinum(II) complex was not observed (entry 6, Table 1).
- (3)
- The use of a platinum salt soluble in organic solvents—bis(benzonitrile)platinum(II) dichloride—significantly improved the yield of complex PtP1. In this case, it was possible to replace benzonitrile with chlorobenzene as a solvent with a lower boiling point, facilitating the distillation step at the end of the reaction. Using these conditions in the presence of a base (NaOAc or Na2CO3), at a lower temperature (150 °C), it was possible to isolate platinum complex PtP1 (Scheme 1) in satisfactory yield (entries 7–8, Table 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Pt(II) Salt (Equiv.) | Solvent | Base (Equiv.) | P1 (mol/dm3) | Temp. (°C) | Time (min) | PtP1 (%) |
|---|---|---|---|---|---|---|---|
| 1 | K2PtCl4 (3) | benzonitrile | - | 0.005 | 250 | 20 | Mixture with P1 |
| 2 | K2PtCl4 (3) | benzonitrile | - | 0.004 | 250 | 20 | Mixture with P1 |
| 3 | K2PtCl4 (3) | benzonitrile | - | 0.004 | (1) 180; (2) 200 | 20 + 20 | Mixture with P1 |
| 4 | K2PtCl4 (3) | benzonitrile | - | 0.006 | 230 | 20 | Mixture with P1 |
| 5 | K2PtCl4 (3) | benzonitrile | - | 0.005 | 250 | 120 | Mixture with P1 |
| 6 | PtCl2 (3) | benzonitrile | Na2CO3 (19) | 0.011 | 150 | 60 + 120 | Mixture with P1 |
| 7 | PtCl2(PhCN)2 (2) | chlorobenzene | NaOAc (5) | 0.041 | 150 | 60 + 60 | 83 |
| 8 | PtCl2(PhCN)2 (2) | chlorobenzene | Na2CO3 (5) | 0.041 | 150 | 60 + 60 | 93 |
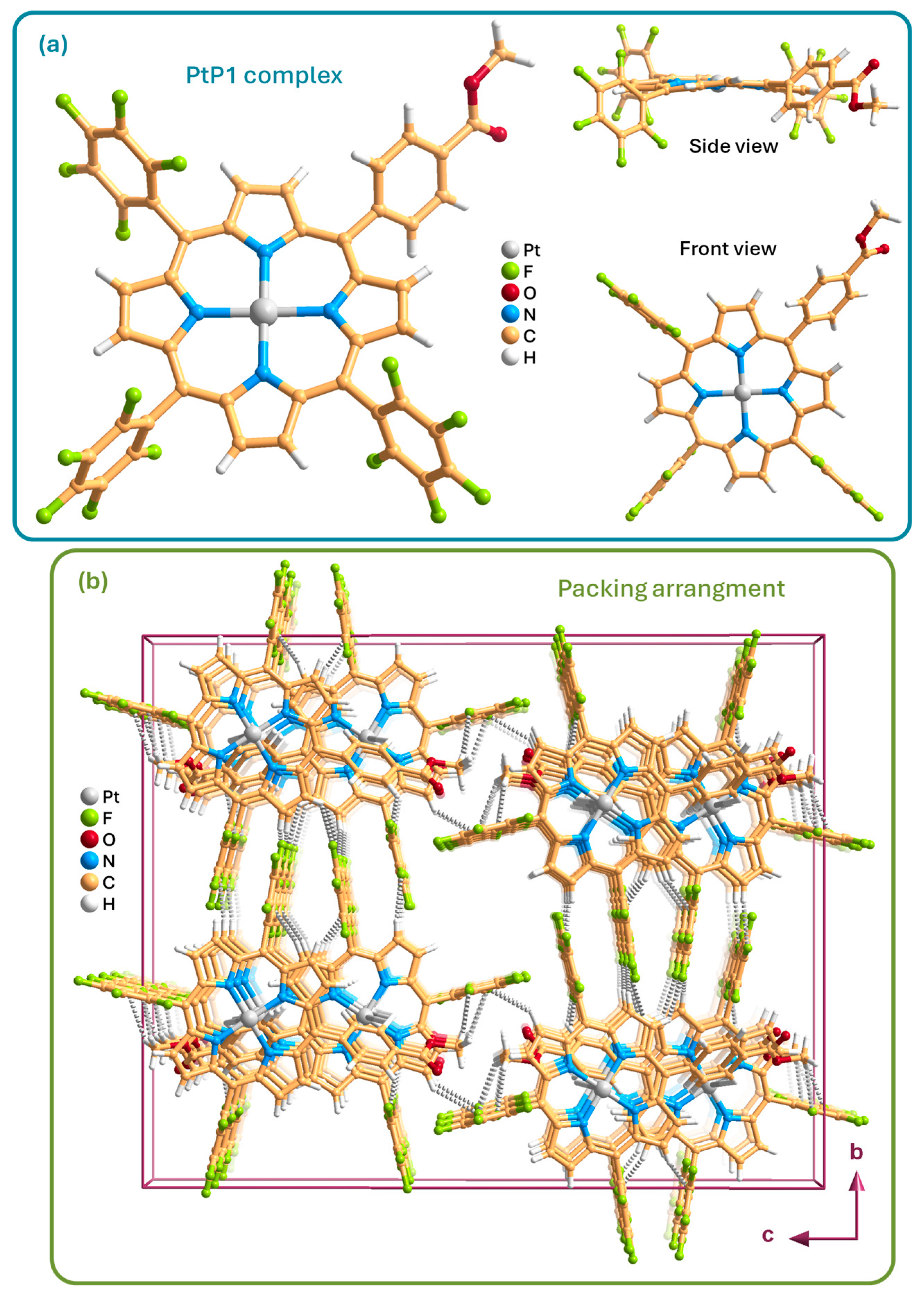
2.2. Characterization of Compounds
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of PtP1
3.3. 1,3-DC Reaction of PtP1 with Azomethine Ylide
3.4. Ester Hydrolysis
3.4.1. Synthesis of PtP2
3.4.2. Synthesis of PtC2
3.5. Conjugation with Indomethacin Derivative
3.5.1. Synthesis of PtP2-Ind
3.5.2. Synthesis of PtC2-Ind Conjugate
3.6. Singlet Oxygen Generation
3.7. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almeida, J.; Silva, A.M.G.; Rangel, M. Application of Photosensitizers in Photodynamic Diagnosis and Therapy of Cancer. In Interdisciplinary Cancer Research; Springer: Cham, Switzerland, 2024. [Google Scholar]
- Yanovsky, R.L.; Bartenstein, D.W.; Rogers, G.S.; Isakoff, S.J.; Chen, S.T. Photodynamic Therapy for Solid Tumors: A Review of the Literature. Photodermatol. Photoimmunol. Photomed. 2019, 35, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, L.; Wang, H.; Zhang, S.; Li, Y. Perspectives on Photodynamic Therapy Combined with Immunotherapy in Treatment of Colorectal Cancer: An Overview Based on Experimental Studies. Photodiagnosis Photodyn. Ther. 2025, 52, 104464. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tan, Q.; Sun, C.; Jia, Y.; Li, S.; Yang, X. Photodynamic Therapy for the Precise Treatment of Localized Prostate Cancer. Front. Oncol. 2025, 15, 1454392. [Google Scholar] [CrossRef]
- Kazemi, K.S.; Kazemi, P.; Mivehchi, H.; Nasiri, K.; Eshagh Hoseini, S.S.; Nejati, S.T.; Pour Bahrami, P.; Golestani, S.; Nabi Afjadi, M. Photodynamic Therapy: A Novel Approach for Head and Neck Cancer Treatment with Focusing on Oral Cavity. Biol. Proced. Online 2024, 26, 25. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, C.Y.; Gao, J.; Wang, Z. Recent Advances in Photodynamic Therapy for Cancer and Infectious Diseases. WIREs Nanomed. Nanobiotechnol. 2019, 11, e1560. [Google Scholar] [CrossRef]
- Jiang, J.; Lv, X.; Cheng, H.; Yang, D.; Xu, W.; Hu, Y.; Song, Y.; Zeng, G. Type I Photodynamic Antimicrobial Therapy: Principles, Progress, and Future Perspectives. Acta Biomater. 2024, 177, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, F.; Misba, L.; Khan, A.U. The Dual Role of Photodynamic Therapy to Treat Cancer and Microbial Infection. Drug Discov. Today 2024, 29, 104099. [Google Scholar] [CrossRef]
- Pham, T.C.; Nguyen, V.-N.; Choi, Y.; Lee, S.; Yoon, J. Recent Strategies to Develop Innovative Photosensitizers for Enhanced Photodynamic Therapy. Chem. Rev. 2021, 121, 13454–13619. [Google Scholar] [CrossRef]
- Obata, M.; Hirohara, S.; Tanaka, R.; Kinoshita, I.; Ohkubo, K.; Fukuzumi, S.; Tanihara, M.; Yano, S. In Vitro Heavy-Atom Effect of Palladium(II) and Platinum(II) Complexes of Pyrrolidine-Fused Chlorin in Photodynamic Therapy. J. Med. Chem. 2009, 52, 2747–2753. [Google Scholar] [CrossRef]
- Laranjo, M.; Aguiar, M.C.; Pereira, N.A.M.; Brites, G.; Nascimento, B.F.O.; Brito, A.F.; Casalta-Lopes, J.; Gonçalves, A.C.; Sarmento-Ribeiro, A.B.; Pineiro, M.; et al. Platinum(II) Ring-Fused Chlorins as Efficient Theranostic Agents: Dyes for Tumor-Imaging and Photodynamic Therapy of Cancer. Eur. J. Med. Chem. 2020, 200, 112468. [Google Scholar] [CrossRef]
- Pereira, N.A.M.; Laranjo, M.; Casalta-Lopes, J.; Serra, A.C.; Piñeiro, M.; Pina, J.; Seixas de Melo, J.S.; Senge, M.O.; Botelho, M.F.; Martelo, L.; et al. Platinum(II) Ring-Fused Chlorins as Near-Infrared Emitting Oxygen Sensors and Photodynamic Agents. ACS Med. Chem. Lett. 2017, 8, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Sperger, T.; Sanhueza, I.A.; Kalvet, I.; Schoenebeck, F. Computational Studies of Synthetically Relevant Homogeneous Organometallic Catalysis Involving Ni, Pd, Ir, and Rh: An Overview of Commonly Employed DFT Methods and Mechanistic Insights. Chem. Rev. 2015, 115, 9532–9586. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Verma, P.; Cramer, C.J.; Gagliardi, L.; Truhlar, D.G. Combining Wave Function Methods with Density Functional Theory for Excited States. Chem. Rev. 2018, 118, 7249–7292. [Google Scholar] [CrossRef]
- Gusev, D.G. Assessing the Accuracy of M06-L Organometallic Thermochemistry. Organometallics 2013, 32, 4239–4243. [Google Scholar] [CrossRef]
- Butera, V.; D’Anna, L.; Rubino, S.; Bonsignore, R.; Spinello, A.; Terenzi, A.; Barone, G. How the Metal Ion Affects the 1 H NMR Chemical Shift Values of Schiff Base Metal Complexes: Rationalization by DFT Calculations. J. Phys. Chem. A 2023, 127, 9283–9290. [Google Scholar] [CrossRef]
- Ballester, F.J.; Hernández-García, A.; Santana, M.D.; Bautista, D.; Ashoo, P.; Ortega-Forte, E.; Barone, G.; Ruiz, J. Photoactivatable Ruthenium Complexes Containing Minimal Straining Benzothiazolyl-1,2,3-Triazole Chelators for Cancer Treatment. Inorg. Chem. 2024, 63, 6202–6216. [Google Scholar] [CrossRef]
- Bonsignore, R.; Trippodo, E.; Di Gesù, R.; Carreca, A.P.; Rubino, S.; Spinello, A.; Terenzi, A.; Barone, G. Novel Half Salphen Cobalt(III) Complexes: Synthesis, DNA Binding and Anticancer Studies. Dalton Trans. 2024, 53, 6311–6322. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, H.; Yan, M.; Xue, J.; Chen, J. A Novel Zinc Phthalocyanine-Indometacin Photosensitizer with “Three-in-One” Cyclooxygenase-2-Driven Dual Targeting and Aggregation Inhibition for High-Efficient Anticancer Therapy. Dye Pigment. 2022, 198, 109997. [Google Scholar] [CrossRef]
- Siriwibool, S.; Wangngae, S.; Chansaenpak, K.; Wet-osot, S.; Lai, R.-Y.; Noisa, P.; Sukwattanasinitt, M.; Kamkaew, A. Indomethacin-Based near-Infrared Photosensitizer for Targeted Photodynamic Cancer Therapy. Bioorg Chem. 2022, 122, 105758. [Google Scholar] [CrossRef]
- Kassab, A.E.; Gedawy, E.M. Repurposing of Indomethacin and Naproxen as Anticancer Agents: Progress from 2017 to Present. RSC Adv. 2024, 14, 40031–40057. [Google Scholar] [CrossRef]
- Almeida, J.; Zhang, G.; Wang, M.; Queirós, C.; Cerqueira, A.F.R.; Tomé, A.C.; Barone, G.; Vicente, M.G.H.; Hey-Hawkins, E.; Silva, A.M.G.; et al. Synthesis, Characterization, and Cellular Investigations of Porphyrin– and Chlorin–Indomethacin Conjugates for Photodynamic Therapy of Cancer. Org. Biomol. Chem. 2021, 19, 6501–6512. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Hou, Y.-J.; Chan, M.C.W.; Guo, J.; Liu, Y.; Wang, Y. [Meso-Tetrakis(Pentafluorophenyl)Porphyrinato]Platinum(Ii) as an Efficient, Oxidation-Resistant Red Phosphor: Spectroscopic Properties and Applications in Organic Light-Emitting Diodes. J. Mater. Chem. 2003, 13, 1362. [Google Scholar] [CrossRef]
- Palma, M.; Cárdenas-Jirón, G.I.; Menéndez Rodríguez, M.I. Effect of Chlorin Structure on Theoretical Electronic Absorption Spectra and on the Energy Released by Porphyrin-Based Photosensitizers. J. Phys. Chem. A 2008, 112, 13574–13583. [Google Scholar] [CrossRef] [PubMed]
- Pushpan, S.; Venkatraman, S.; Anand, V.; Sankar, J.; Parmeswaran, D.; Ganesan, S.; Chandrashekar, T. Porphyrins in Photodynamic Therapy—A Search for Ideal Photosensitizers. Curr. Med. Chem. Anti-Cancer Agents 2002, 2, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.K.; Lin, M.; Sen, A.; Gretz, E. Bis(Benzonitrile)Dichloro Complexes of Palladium and Platinum; Inorganic Syntheses; Wiley: Hoboken, NJ, USA, 1990; pp. 60–63. ISBN 9780470132593. [Google Scholar]
- Almeida, J.; Silva, A.M.N.; Rebelo, S.L.H.; Cunha-Silva, L.; Rangel, M.; De Castro, B.; Leite, A.; Silva, A.M.G. Synthesis and Coordination Studies of 5-(4′-Carboxyphenyl)-10,15,20-Tris(Pentafluorophenyl)Porphyrin and Its Pyrrolidine-Fused Chlorin Derivative. New J. Chem. 2018, 42, 8169–8179. [Google Scholar] [CrossRef]
- Kottke, T.; Stalke, D. Crystal Handling at Low Temperatures. J. Appl. Crystallogr. 1993, 26, 615–619. [Google Scholar] [CrossRef]
- Bruker AXS. SAINT+, Data Integration Engine v. 7.23a ©, 1997–2005; Bruker AXS: Madison, WI, USA, 2006. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- van der Sluis, P.; Spek, A.L. BYPASS: An Effective Method for the Refinement of Crystal Structures Containing Disordered Solvent Regions. Acta Crystallogr. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Spek, A.L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.G.; Dige, N.C.; Vanjare, B.D.; Kim, C.-H.; Seo, S.-Y.; Lee, K.H. Design and Synthesis of New Porphyrin Analogues as Potent Photosensitizers for Photodynamic Therapy: Spectroscopic Approach. J. Fluoresc. 2020, 30, 397–406. [Google Scholar] [CrossRef]
- Menezes, J.C.; Faustino, M.A.F.; De Oliveira, K.T.; Uliana, M.P.; Ferreira, V.F.; Hackbarth, S.; Röder, B.; Teixeira Tasso, T.; Furuyama, T.; Kobayashi, N.; et al. Synthesis of New Chlorin e6 Trimethyl and Protoporphyrin IX Dimethyl Ester Derivatives and Their Photophysical and Electrochemical Characterizations. Chem. Eur. J. 2014, 20, 13644–13655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic Compact Effective Potentials and Efficient, Shared-Exponent Basis Sets for the Third-, Fourth-, and Fifth-Row Atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set. for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W. Gaussian, Revision C.016; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]


 | |||||
|---|---|---|---|---|---|
| λmax/nm (logε) | |||||
| Soret Band | Q Bands | ||||
| P2 | 413 (5.44) | 507 (4.23) | 536 (3.46) | 581 (3.73) | 652 (3.35) |
| PtP2 | 394 (5.31) | 507 (4.23) | 539 (4.18) | ||
| C2 | 408 (5.14) | 503 (4.08) | 530 (3.79) | 594 (3.68) | 648 (4.64) |
| PtC1 | 394 (5.25) | 476 (3.89) | 549 (4.03) | 591 (4.78) | |
| PtC2 | 394 (5.16) | 476 (3.81) | 549 (3.97) | 591 (4.72) | |
| PtC3 | 395 (5.27) | 481 (4.03) | 560 (4.19) | 598 (4.80) | |
| PtP2-Ind | 393 (5.44) | 507 (4.29) | 539 (4.29) | ||
| PtC2-Ind | 394 | 476 | 549 | 591 | |
| PtP2 triplet | +154.5 kJ/mol compared to the energy of the singlet. |
| PtC2 triplet | +137.4 kJ/mol compared to the energy of the singlet. |
| PtC4 triplet | +138.1 kJ/mol compared to the energy of the singlet. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, J.; Barone, G.; Cunha-Silva, L.; Cerqueira, A.F.R.; Tomé, A.C.; Rangel, M.; Silva, A.M.G. New Conjugatable Platinum(II) Chlorins: Synthesis, Reactivity and Singlet Oxygen Generation. Molecules 2025, 30, 2496. https://doi.org/10.3390/molecules30122496
Almeida J, Barone G, Cunha-Silva L, Cerqueira AFR, Tomé AC, Rangel M, Silva AMG. New Conjugatable Platinum(II) Chlorins: Synthesis, Reactivity and Singlet Oxygen Generation. Molecules. 2025; 30(12):2496. https://doi.org/10.3390/molecules30122496
Chicago/Turabian StyleAlmeida, José, Giampaolo Barone, Luís Cunha-Silva, Ana F. R. Cerqueira, Augusto C. Tomé, Maria Rangel, and Ana M. G. Silva. 2025. "New Conjugatable Platinum(II) Chlorins: Synthesis, Reactivity and Singlet Oxygen Generation" Molecules 30, no. 12: 2496. https://doi.org/10.3390/molecules30122496
APA StyleAlmeida, J., Barone, G., Cunha-Silva, L., Cerqueira, A. F. R., Tomé, A. C., Rangel, M., & Silva, A. M. G. (2025). New Conjugatable Platinum(II) Chlorins: Synthesis, Reactivity and Singlet Oxygen Generation. Molecules, 30(12), 2496. https://doi.org/10.3390/molecules30122496