
Investigation of the Effect of Molecules Containing Sulfonamide Moiety Adsorbed on the FAPbI3 Perovskite Surface: A First-Principles Study

 ,
,
Abstract
1. Introduction
2. Models and Computational Methods
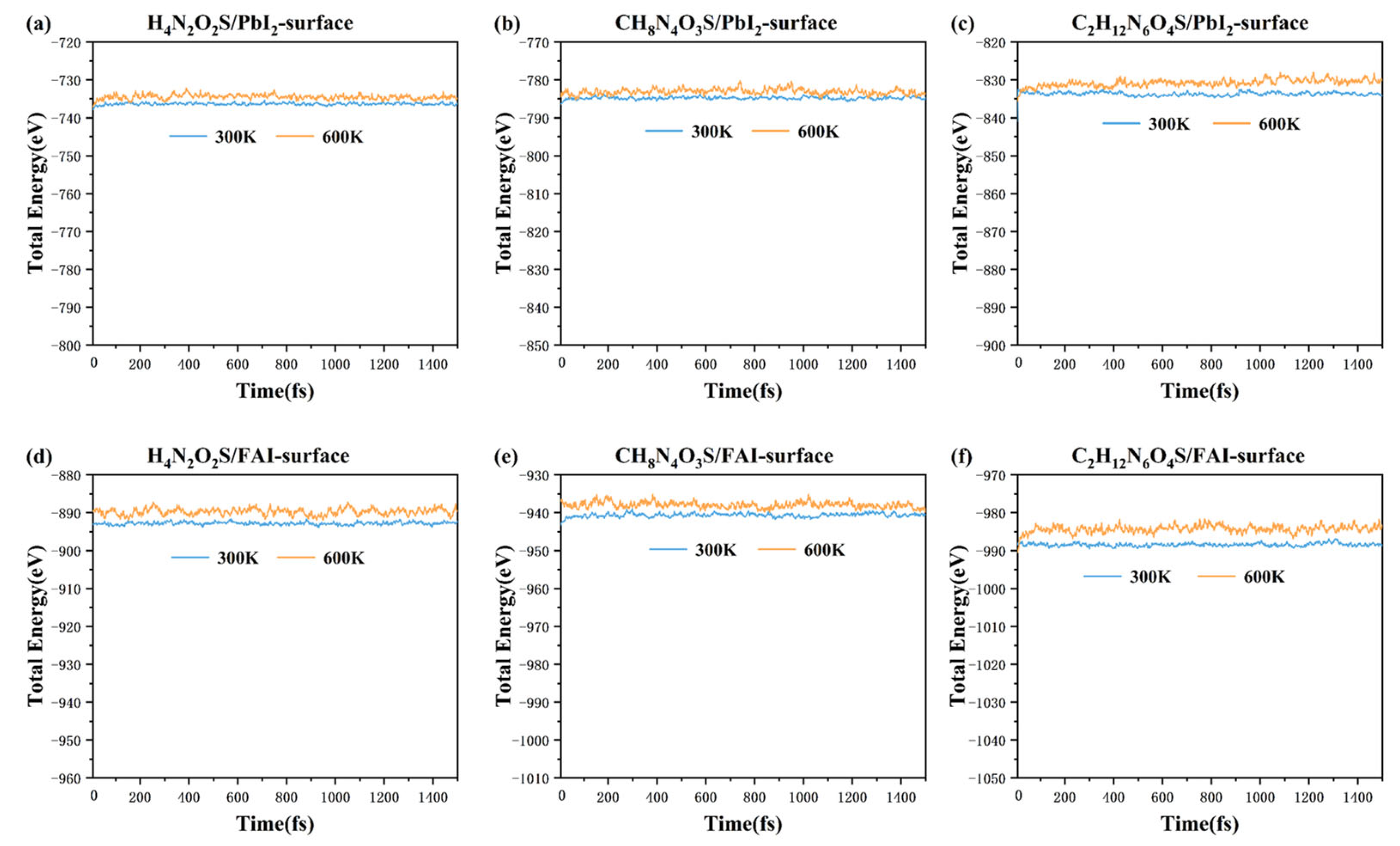
3. Results and Discussion
3.1. Structural Properties of FAPbI3
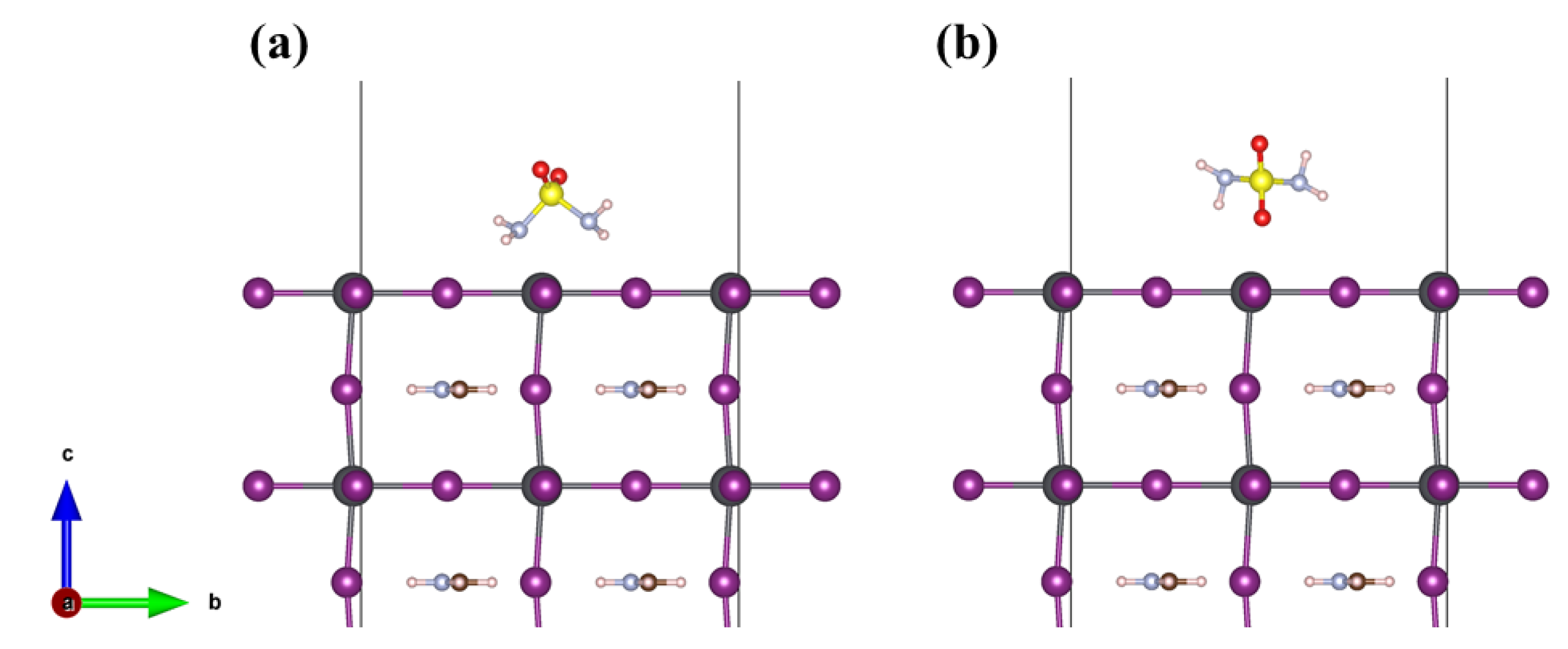
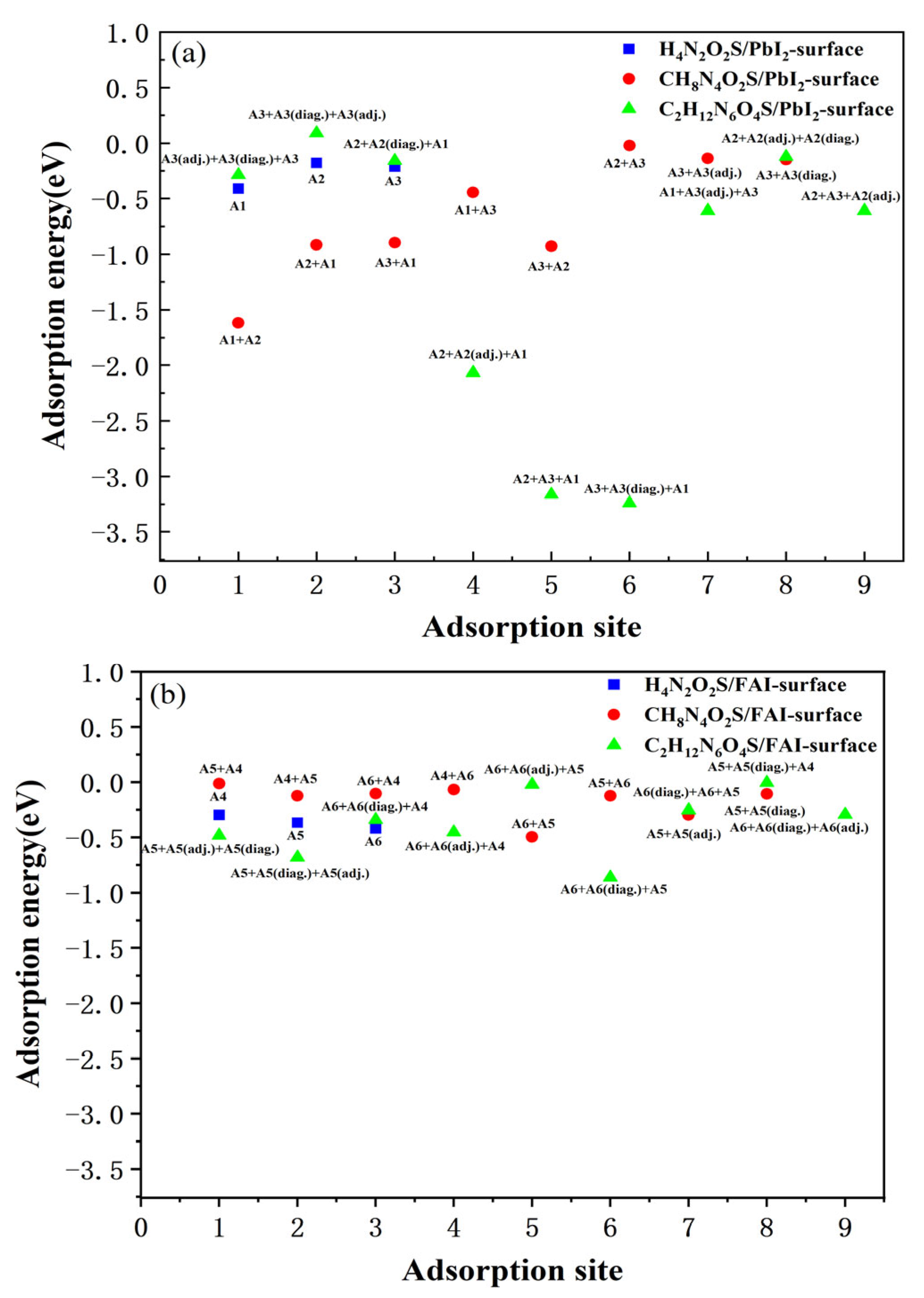
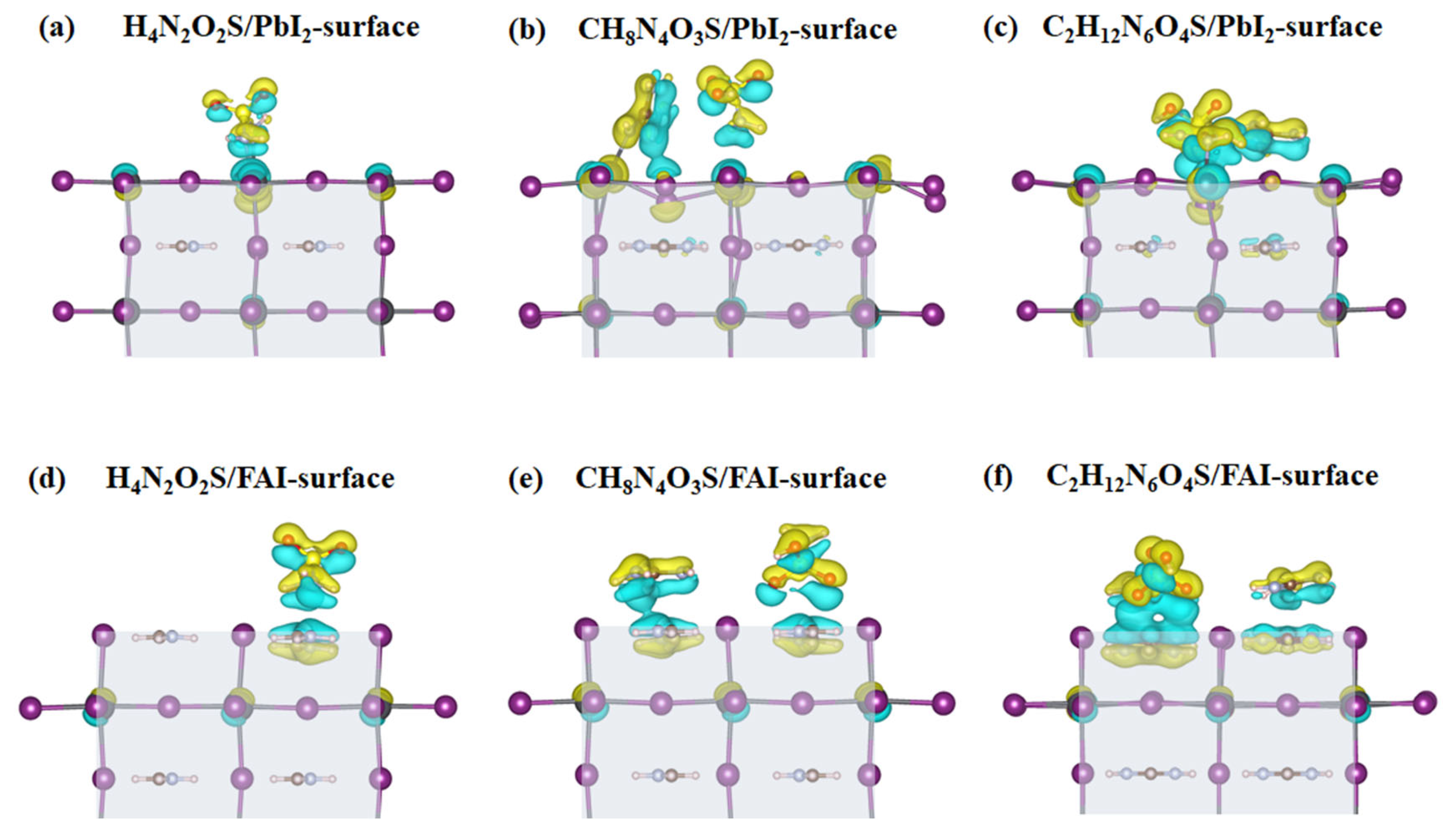
3.2. Adsorption Properties of Different Molecules on the FAPbI3(001) Surface
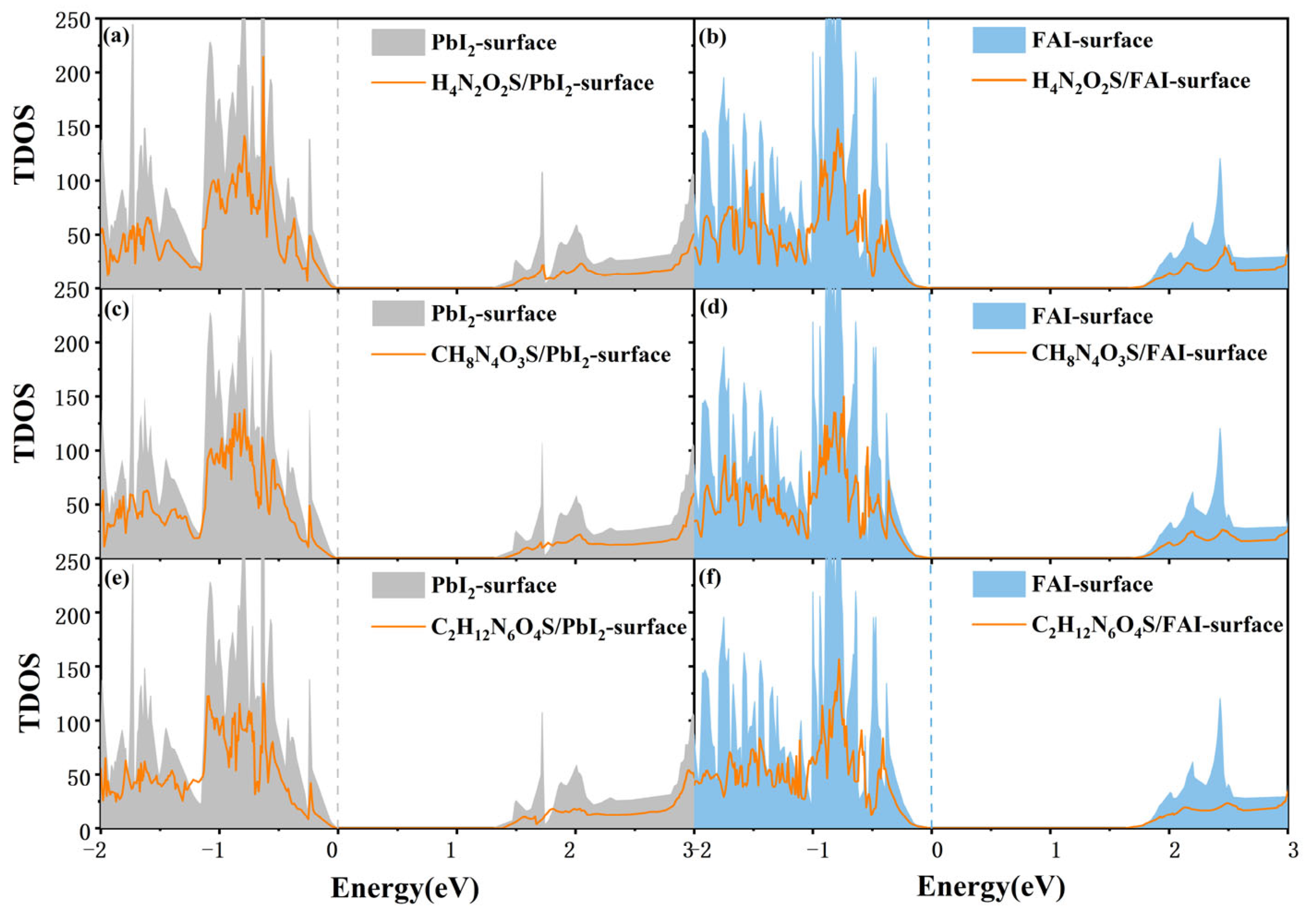
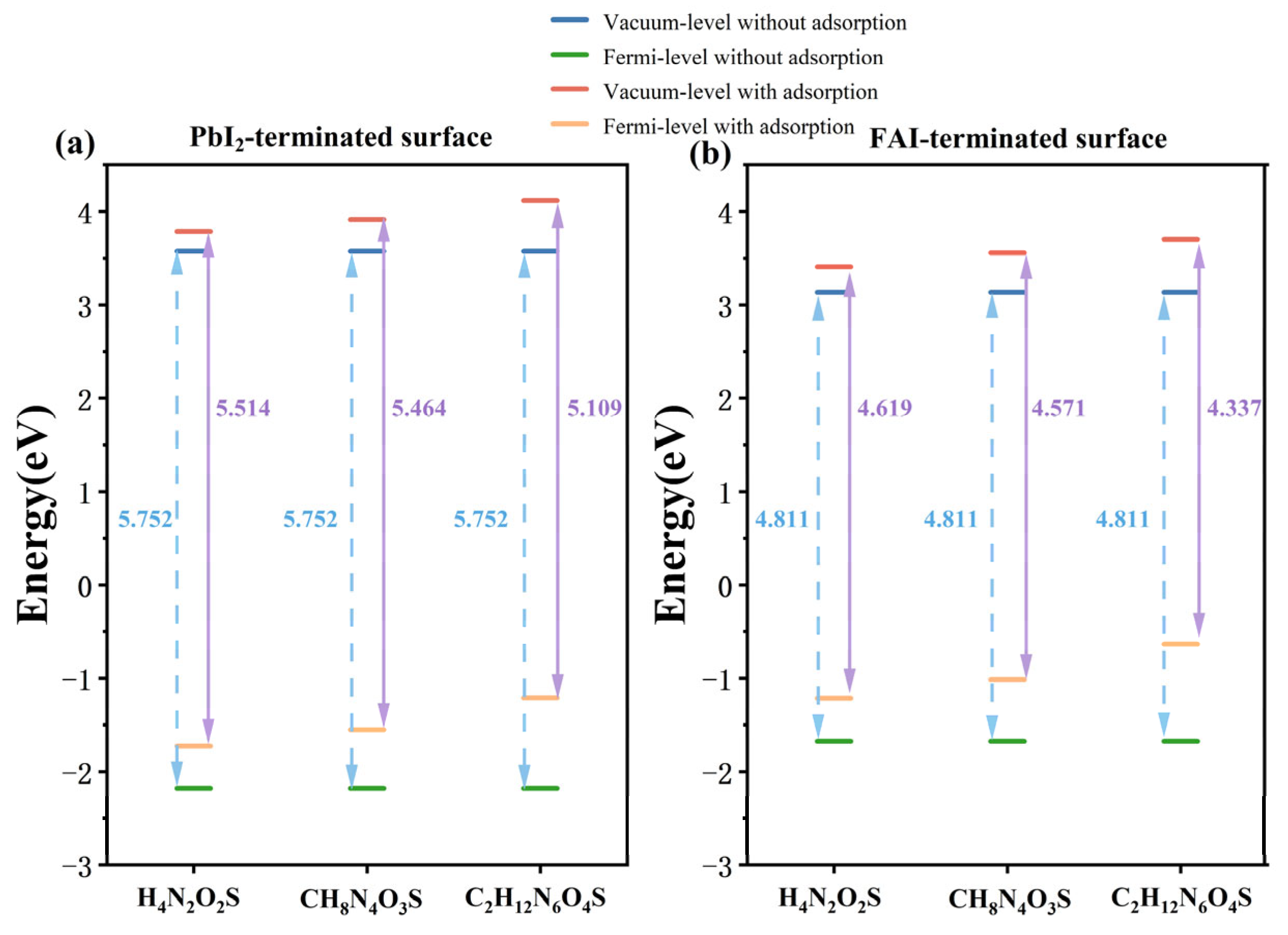
3.3. Electronic Properties of Different Molecules on the FAPbI3(001) Surface
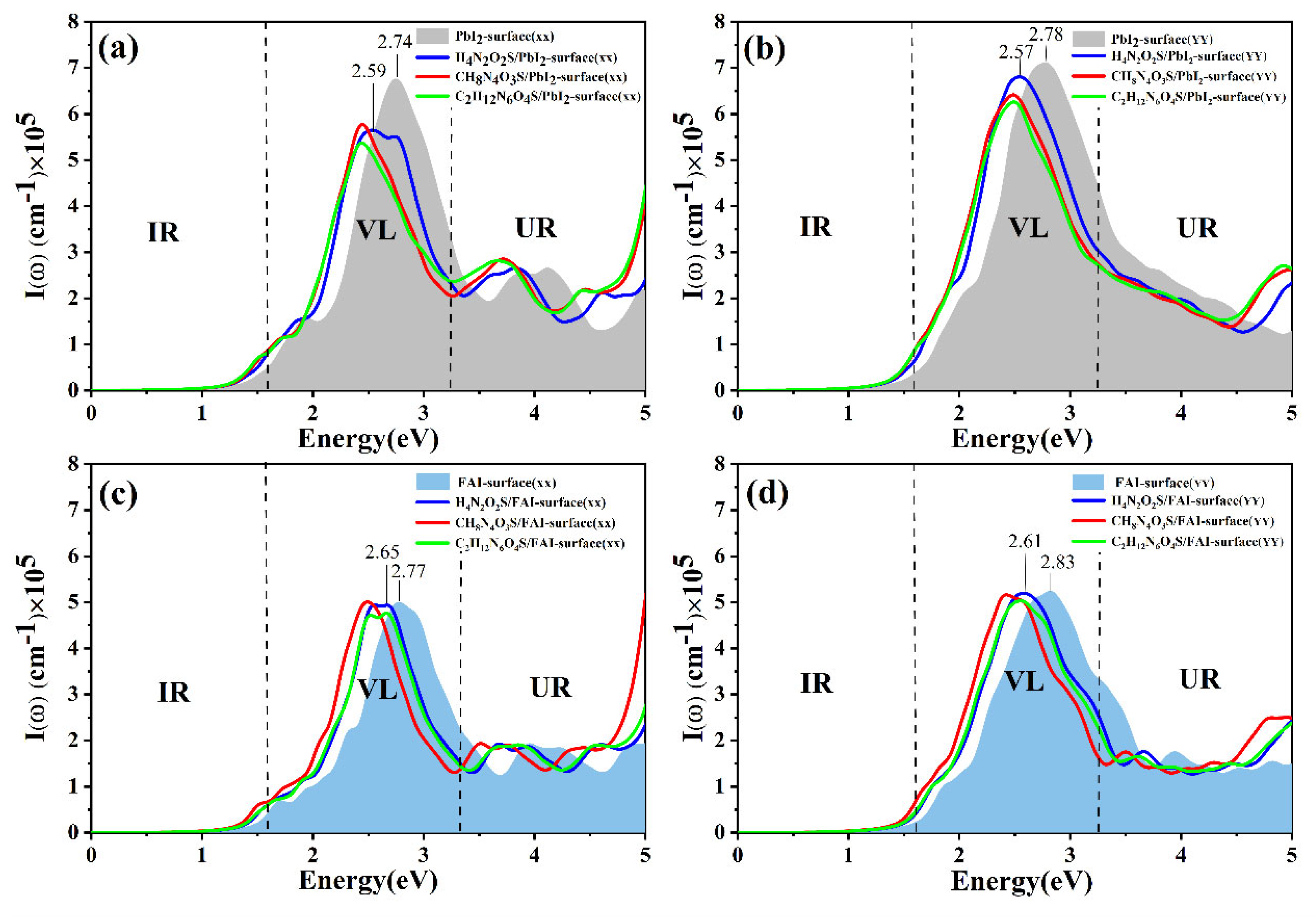
3.4. Optical Properties of Different Molecules Adsorbed on the FAPbI3(001) Surface
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, J.; Tong, S.; Zhou, T.; Lin, J.; Liu, Y. Research and development of modification strategies based on all inorganic perovskite solar cells. Nano Energy 2025, 138, 110815. [Google Scholar] [CrossRef]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- NREL. Research-Cell Lefficiency Records. Available online: https://www.nrel.gov/pv/cell-efficiency.html (accessed on 21 April 2025).
- Le Corre, V.M.; Duijnstee, E.A.; El Tambouli, O.; Ball, J.M.; Snaith, H.J.; Lim, J.; Koster, L.J.A. Revealing Charge Carrier Mobility and Defect Densities in Metal Halide Perovskites via Space-Charge-Limited Current Measurements. ACS Energy Lett. 2021, 6, 1087. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zheng, L.; Hou, Y.; Nozariasbmarz, A.; Poudel, B.; Yoon, J.; Ye, T.; Yang, D.; Pogrebnyakov, A.V.; Gopalan, V.; et al. Overcoming Shockley-Queisser limit using halide perovskite platform. Joule 2022, 6, 756–771. [Google Scholar] [CrossRef]
- Kralj, S.; Morales-Masis, M. Growth and stabilization of cubic FAPbI3: On the interaction between phosphonic acids and co-evaporated perovskites. Matter 2024, 7, 3238–3240. [Google Scholar] [CrossRef]
- Qin, B.; Wang, D.; He, W.; Zhang, Y.; Wu, H.; Pennycook, S.J.; Zhao, L.-D. Realizing High Thermoelectric Performance in p-Type SnSe through Crystal Structure Modification. J. Am. Chem. Soc. 2019, 141, 1141–1149. [Google Scholar] [CrossRef]
- Qu, Y.; Li, X.; Zhang, H.; Huang, R.; Qi, W.; Su, R.; He, Z. Controllable synthesis of a sponge-like Z-scheme N,S-CQDs/Bi2MoO6@TiO2 film with enhanced photocatalytic and antimicrobial activity under visible/NIR light irradiation. J. Hazard. Mater. 2022, 429, 128310. [Google Scholar] [CrossRef]
- Zhang, P.; Xiong, J.; Chen, W.-H.; Du, P.; Song, L. Air-processed MAPbI3 perovskite solar cells achieve 20.87% efficiency and excellent bending resistance enabled via a polymer dual-passivation strategy. Dalton Trans. 2023, 52, 15974–15985. [Google Scholar] [CrossRef]
- Larciprete, R.; Agresti, A.; Pescetelli, S.; Pazniak, H.; Liedl, A.; Lacovig, P.; Lizzit, D.; Tosi, E.; Lizzit, S.; Di Carlo, A. Mixed Cation Halide Perovskite under Environmental and Physical Stress. Materials 2021, 14, 3954. [Google Scholar] [CrossRef]
- Vasilopoulou, M.; Fakharuddin, A.; Coutsolelos, A.G.; Falaras, P.; Argitis, P.; Yusoff, A.R.b.M.; Nazeeruddin, M.K. Molecular materials as interfacial layers and additives in perovskite solar cells. Chem. Soc. Rev. 2020, 49, 4496–4526. [Google Scholar] [CrossRef]
- Zhu, Z.; Ke, B.; Sun, K.; Jin, C.; Song, Z.; Jiang, R.; Li, J.; Kong, S.; Liu, C.; Bai, S.; et al. High-performance inverted perovskite solar cells and modules via aminothiazole passivation. Energy Environ. Sci. 2025, 18, 4120–4129. [Google Scholar] [CrossRef]
- Chen, H.; Sun, J.; Fan, K.; Zou, S.; Lin, Z.; Chen, J.; Zhang, Z.; Wang, K.; Jiang, Z.; Yan, K. Difunctional Polymerizable Additive Enables Efficient and Stable Wide-Bandgap Perovskites for Perovskite/Organic Tandems Solar Cells. Adv. Funct. Mater. 2025, 2502422. [Google Scholar] [CrossRef]
- Mo, H.; Wang, L.; Li, Y.; Zhu, T.; Sergeev, A.; Wang, J.; He, Y.; Ren, Z.; Rehman, A.U.; Ali, M.U.; et al. Multifunctional Universal Additive for Stable and Efficient Inverted Perovskite Solar Cells. Adv. Energy Mater. 2025, 2500088. [Google Scholar] [CrossRef]
- Gao, Y.; Gong, W.; Zhang, Z.; Guo, J.; Ma, J.; Li, X.; Zeng, Y.; Wu, M. Aminomethyl Phosphonic Acid as Highly Effective Multifunctional Additive for Modification of Electron Transport Layer and Perovskite in Photovoltaic Solar Cells. Angew. Chem. Int. Ed. 2025, 137, e202424479. [Google Scholar] [CrossRef]
- Liu, J.T.; Grignon, E.; Battaglia, A.M.; Imran, M.; Copeman, C.; Mills, H.A.; Howarth, A.J.; Sargent, E.H.; Seferos, D.S. An Investigation of Conjugated Sulfonamide Materials as Binders for Organic Lithium-Ion Batteries. Chem. Mater. 2023, 35, 9692–9701. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.; Su, H.; Zhao, Y.; Zeng, X.; Hu, M.; Xiao, W.; Mao, X. H-bonding effect of oxyanions enhanced photocatalytic degradation of sulfonamides by g-C3N4 in aqueous solution. J. Hazard. Mater. 2019, 366, 259–267. [Google Scholar] [CrossRef]
- Liu, H.; Xing, M.; Li, X. Novel superhard semiconductor carbon allotrope Pcc2 C24 under pressure: First-principles calculation. Chem. Phys. Lett. 2025, 860, 141823. [Google Scholar] [CrossRef]
- Kumar, S.; Zain Mehdi, S.M.; Taunk, M.; Kumar, S.; Aherwar, A.; Singh, S.; Singh, T. Synergistic effects of polymer integration on the properties, stability, and applications of MXenes. J. Mater. Chem. A 2025, 13, 11050. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, B.; Xiao, J.; Zhou, L.; Chen, M. Application of First Principles Computations Based on Density Functional Theory (DFT) in Cathode Materials of Sodium-Ion Batteries. Batteries 2023, 9, 86. [Google Scholar] [CrossRef]
- Prasad, C.V.; Park, J.H.; Hoai Ta, Q.T.; Kim, K.J.; Jeon, H.J.; Mir, M.A.; Raza, M.; Tri, N.N.; Kim, H.; Labed, M.; et al. p-CuGaO2/β-Ga2O3 interfaces: A high-throughput approach for interface prediction and generation for power device applications. Mater. Today Adv. 2025, 26, 100577. [Google Scholar] [CrossRef]
- Hussain, S.; Rehman, J.U.; Hussain, A.; Tahir, M.B.; Iqbal, F. Study of double perovskite materials RbX2Y3O10 (XMg, Ca, YTi, Zr) for photocatalytic applications: A DFT insights. Int. J. Hydrogen Energy 2024, 62, 739–748. [Google Scholar] [CrossRef]
- Ghorpade, U.V.; Suryawanshi, M.P.; Shin, S.W.; Wang, X.; Jo, E.; Bae, H.; Park, K.; Ha, J.-S.; Kolekar, S.S.; Kim, J.H. Eutectic solvent-mediated selective synthesis of Cu–Sb–S-based nanocrystals: Combined experimental and theoretical studies toward highly efficient water splitting. J. Mater. Chem. A 2018, 6, 19798–19809. [Google Scholar] [CrossRef]
- Choudhary, A.; Malakkal, L.; Siripurapu, R.K.; Szpunar, B.; Szpunar, J. First principles calculations of hydrogen storage on Cu and Pd-decorated graphene. Int. J. Hydrogen Energy 2016, 41, 17652–17656. [Google Scholar] [CrossRef]
- Herbold, M.; Behler, J. Machine learning transferable atomic forces for large systems from underconverged molecular fragments. Phys. Chem. Chem. Phys. 2023, 25, 12979–12989. [Google Scholar] [CrossRef]
- Ullah, A.; Rahman, N.; Husain, M.; Almalki, W.M.; Hussien, M.; Tirth, V.; Abualnaja, K.M.; Alosaimi, G.; Alfaifi, A.H.; Almutib, E.; et al. DFT study of structural, elastic and optoelectronic properties of binary X2Se (X = Cd, Zn) chalcogenides. Inorg. Chem. Commun. 2025, 174, 113985. [Google Scholar] [CrossRef]
- Yang, C.; Chen, C.; Bian, T.; Xu, C.; Che, X.; Li, D.; Liang, K.; Dong, X.; Yin, J.; Li, G.; et al. Anisotropic δ-to-α Phase Transition in Formamidinium Lead Iodide Thin Films. ACS Nano 2025, 19, 9225–9231. [Google Scholar] [CrossRef]
- Shuang, F.; Ji, R.; Xiong, L.; Gao, W. Effect of periodic image interactions on kink pair activation of screw dislocation. Comput. Mater. Sci. 2023, 228, 112369. [Google Scholar] [CrossRef]
- Williams, S.T.; Rajagopal, A.; Jo, S.B.; Chueh, C.C.; Tang, T.F.L.; Kraeger, A.; Jen, A.K.Y. Realizing a new class of hybrid organic–inorganic multifunctional perovskite. J. Mater. Chem. A 2017, 5, 10640–10650. [Google Scholar] [CrossRef]
- Chang, Z.; Cao, X.; Chang, J. Effects of palladium doping on structural, mechanical, and opto-electronic behavior of Cs2Pt1-xPdxBr6 double perovskites. J. Solid State Chem. 2024, 337, 124828. [Google Scholar] [CrossRef]
- Bhattarai, S.; Hossain, M.K.; Pandey, R.; Madan, J.; Samajdar, D.P.; Rahman, M.F.; Ansari, M.Z.; Amami, M. Perovskite Solar Cells with Dual Light Absorber Layers for Performance Efficiency Exceeding 30%. Energy Fuels 2023, 37, 10631–10641. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, H.; Almalki, M.; Xu, J.; Krishna, A.; Eickemeyer, F.T.; Gao, J.; Wu, Y.M.; Zakeeruddin, S.M.; Chu, J.; et al. Stabilization of FAPbI3 with Multifunctional Alkali-Functionalized Polymer. Adv. Mater. 2023, 35, 2211619. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.T.; Weber, O.J.; Frost, J.M.; Walsh, A. Cubic Perovskite Structure of Black Formamidinium Lead Iodide, α-[HC(NH2)2]PbI3, at 298 K. J. Phys. Chem. Lett. 2015, 6, 3209–3212. [Google Scholar] [CrossRef]
- Eperon, G.E.; Stranks, S.D.; Menelaou, C.; Johnston, M.B.; Herz, L.M.; Snaith, H.J. Formamidinium lead trihalide: A broadly tunable perovskite for efficient planar heterojunction solar cells. Energy Environ. Sci. 2014, 7, 982–988. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Gao, Y.; Yu, Y.; Yang, J.; Wang, P.; Duo, Y.; Yang, J.; Huo, Z.; Ran, J.; Wang, J.; Wei, Z.; et al. Correction to “Polarization-Sensitive Solar-Blind Ultraviolet Photodetectors Based on Semipolar (112̅2) AlGaN Film”. ACS Appl. Mater. Interfaces 2025, 17, 17651–17652. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PbI2-Terminated Surface | FAI-Terminated Surface | |||
|---|---|---|---|---|
| Molecule | Adsorption Sites | Adsorption Energy (eV) | Adsorption Sites | Adsorption Energy (eV) |
| H4N2O2S | A1 | −0.41 | A4 | −0.296 |
| A2 | −0.178 | A5 | −0.367 | |
| A3 | −0.211 | A6 | −0.419 | |
| CH8N4O3S | A1 + A2 | −1.618 | A5 + A4 | −0.015 |
| A2 + A1 | −0.917 | A4 + A5 | −0.125 | |
| A3 + A1 | −0.896 | A6 + A4 | −0.105 | |
| A1 + A3 | −0.444 | A4 + A6 | −0.069 | |
| A3 + A2 | −0.928 | A6 + A5 | −0.495 | |
| A2 + A3 | −0.021 | A5 + A6 | −0.124 | |
| A3 + A3(adj.) | −0.139 | A5 + A5(adj.) | −0.298 | |
| A3 + A3(diag.) | −0.15 | A5 + A5(diag.) | −0.107 | |
| C2H12N6O4S | A3(adj.) + A3(diag.) + A3 | 0.285 | A5 + A5(adj.) + A5(diag.) | −0.482 |
| A3 + A3(diag.) + A3(adj.) | 0.092 | A5 + A5(diag.) + A5(adj.) | −0.682 | |
| A2 + A2(diag.) + A1 | −0.158 | A6 + A6(diag.) + A4 | −0.341 | |
| A2 + A2(adj.) + A1 | −2.068 | A6 + A6(adj.) + A4 | −0.454 | |
| A2 + A3 + A1 | −3.163 | A6 + A6(adj.) + A5 | −0.023 | |
| A3 + A3(diag.) + A1 | −3.242 | A6 + A6(diag.) + A5 | −0.862 | |
| A1 + A3(adj.) + A3 | −0.608 | A6(diag.) + A6 + A5 | −0.253 | |
| A2 + A2(adj.) + A2(diag.) | −0.122 | A5 + A5(diag.) + A4 | −0.007 | |
| A2 + A3 + A2(adj.) | −0.609 | A6 + A6(diag.) + A6(adj.) | −0.293 | |
| PbI2-Terminated Surface | FAI-Terminated Surface | |||
|---|---|---|---|---|
| Molecule | Atom | Bader Charge (e) | Atom | Bader Charge (e) |
| H4N2O2S | Pb(surface) | +0.93 | Pb(surface) | +0.84 |
| O | −0.25 | O | −0.25 | |
| N | −0.36 | N | −0.31 | |
| N(surface) | −0.10 | N(surface) | −0.11 | |
| S | −0.12 | S | −0.13 | |
| I(surface) | −0.02 | I(surface) | −0.07 | |
| CH8N4O3S | Pb(surface) | +0.97 | Pb(surface) | +0.86 |
| O | −0.27 | O | −0.27 | |
| N | −0.25 | N | −0.22 | |
| N(surface) | −0.11 | N(surface) | −0.12 | |
| S | −0.49 | S | −0.4 | |
| I(surface) | −0.04 | I(surface) | −0.08 | |
| C2H12N6O4S | Pb(surface) | +0.98 | Pb(surface) | +0.91 |
| O | −0.33 | O | −0.31 | |
| N | −0.36 | N | −0.35 | |
| N(surface) | −0.12 | N(surface) | −0.14 | |
| S | −0.64 | S | −0.59 | |
| I(surface) | −0.05 | I(surface) | −0.09 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Zhuang, Y.; Dou, Y.; Wang, J.; Zhang, H.; Lu, W.; Zhang, Q.; Zhang, X.; Wu, Y.; Jiang, X. Investigation of the Effect of Molecules Containing Sulfonamide Moiety Adsorbed on the FAPbI3 Perovskite Surface: A First-Principles Study. Molecules 2025, 30, 2463. https://doi.org/10.3390/molecules30112463
Yang S, Zhuang Y, Dou Y, Wang J, Zhang H, Lu W, Zhang Q, Zhang X, Wu Y, Jiang X. Investigation of the Effect of Molecules Containing Sulfonamide Moiety Adsorbed on the FAPbI3 Perovskite Surface: A First-Principles Study. Molecules. 2025; 30(11):2463. https://doi.org/10.3390/molecules30112463
Chicago/Turabian StyleYang, Shiyan, Yu Zhuang, Youbo Dou, Jianjun Wang, Hongwen Zhang, Wenjing Lu, Qiuli Zhang, Xihua Zhang, Yuan Wu, and Xianfeng Jiang. 2025. "Investigation of the Effect of Molecules Containing Sulfonamide Moiety Adsorbed on the FAPbI3 Perovskite Surface: A First-Principles Study" Molecules 30, no. 11: 2463. https://doi.org/10.3390/molecules30112463
APA StyleYang, S., Zhuang, Y., Dou, Y., Wang, J., Zhang, H., Lu, W., Zhang, Q., Zhang, X., Wu, Y., & Jiang, X. (2025). Investigation of the Effect of Molecules Containing Sulfonamide Moiety Adsorbed on the FAPbI3 Perovskite Surface: A First-Principles Study. Molecules, 30(11), 2463. https://doi.org/10.3390/molecules30112463