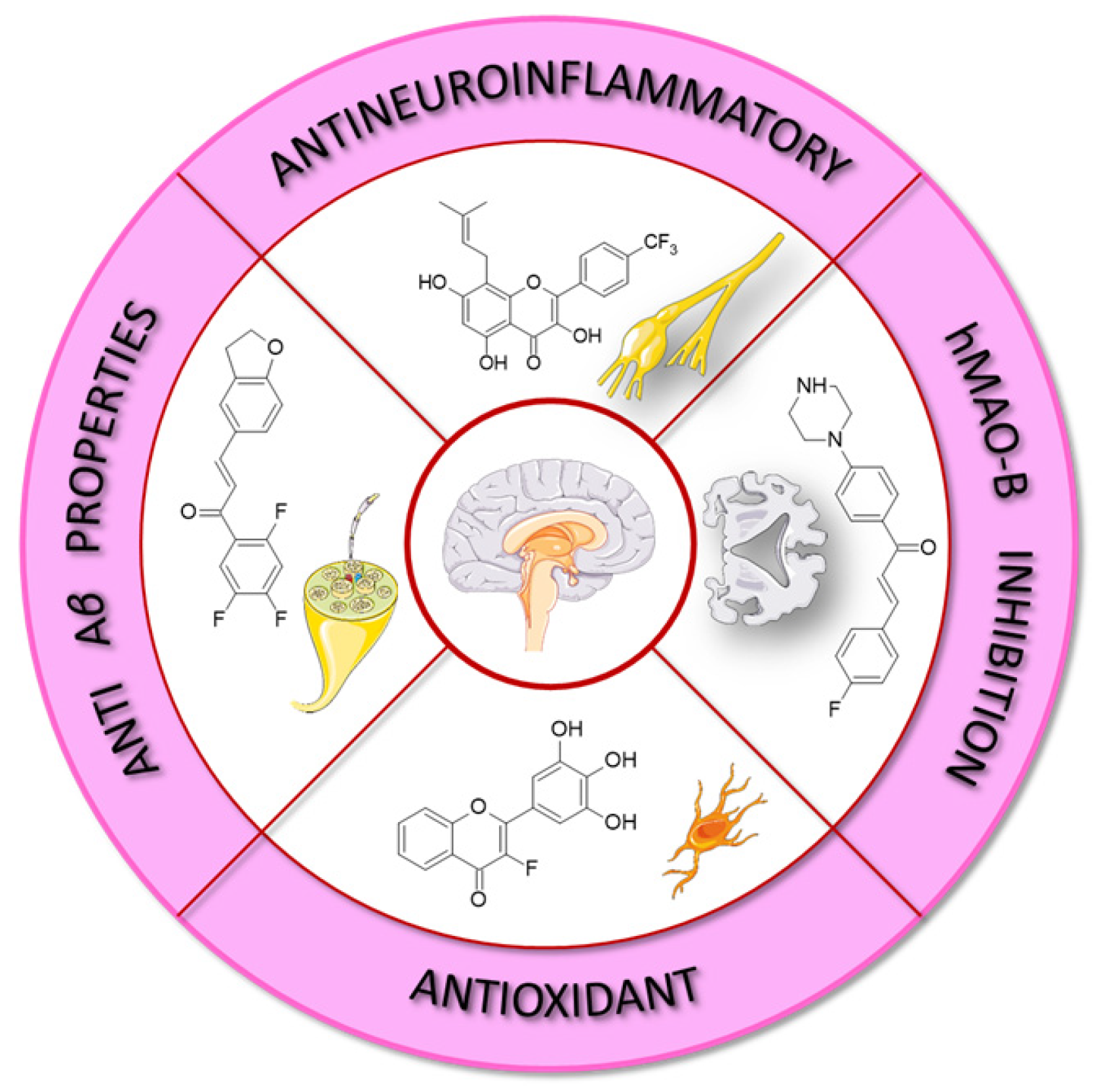
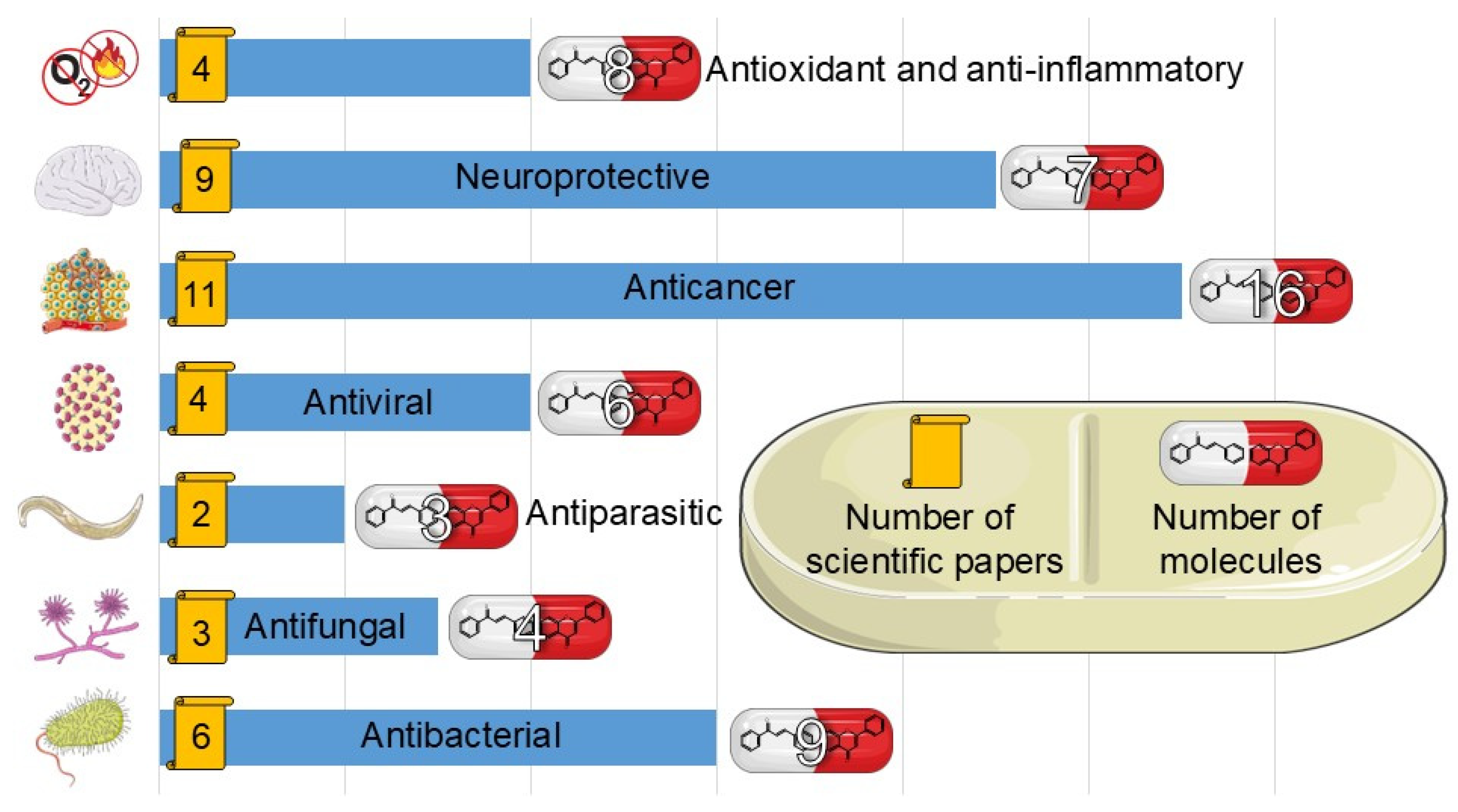
Fluorinated flavonoids constitute a significant area of research focused on discovering compounds that exhibit diverse properties. Inextricably bound to this topic are chalcones, which serve as key precursors in flavonoid synthesis.
4.1. Antibacterial, Antifungal and Antiparasitic Properties
As previously mentioned, flavonoids alone exhibit antibacterial activity, with several mechanisms proposed. These include the inhibition of bacterial cell membrane synthesis and biofilm formation, the disruption of the electron transport chain and ATP production, and the inhibition of specific enzymes. Chalcones, on the other hand, display strong antibacterial activity, especially against multidrug-resistant bacteria, by targeting enzymes such as DNA gyrase B, MurA transferase, and efflux pumps. Finally, both flavonoids and chalcones influence the host organism by modulating immune and inflammatory responses [
64,
104,
105].
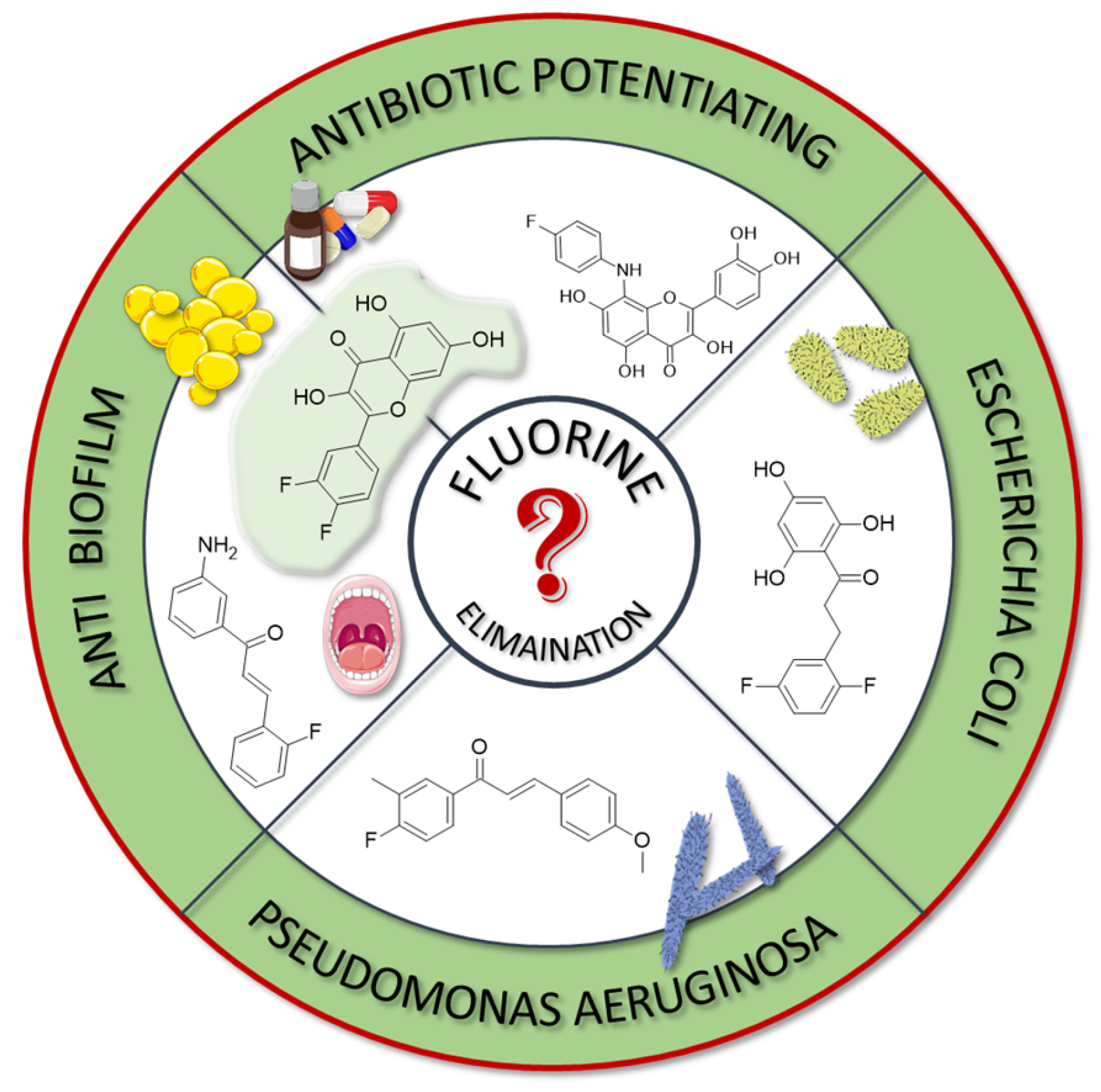
In many studies presented below, fluorinated derivatives of flavonoids and chalcones revealed potential antimicrobial properties (
Figure 8,
Table 1). The continuous evolution of treatment-resistant bacterial strains directs scientists to search for compounds that microorganisms have not yet adapted to and rendered harmless. In this regard, fluorinated flavonoids reveal some prospective antimicrobial potential. The influence of B-ring fluorination in this context was evaluated by Kho et al. [
106] in a series of tests on a fluorinated derivative of quercetin, 3′,4′-difluoroquercetin
1 (
Figure 9). The tests were conducted on various Gram-positive and Gram-negative bacterial strains. Compound
1 exhibited interesting antibacterial properties against Gram-positive bacteria, such as
Staphylococcus aureus and
Enterococcus sp. (MIC values ranging from 8 to 32 mg/L), compared to Gram-negative bacteria (MIC > 128 mg/L). Further research on
S. aureus biofilms revealed good antibiofilm activity, with IC
50 values ranging from 1.8 to 5.3 mg/L. The best activity was observed against methicillin-resistant
S. aureus (MRSA), while compound
1 was least effective against vancomycin-intermediate
S. aureus. The checkerboard synergy test evaluated the antibacterial activity of
1 in combination with the following antibiotics: aminopenicillin, ceftazidime, cefepime, meropenem, and vancomycin. The best antimicrobial properties (MIC
50 2 mg/L) were observed against
P. aeruginosa for the combination of ceftazidime and
1. The efflux activity of carbapenem-resistant
P. aeruginosa revealed that
1 could act as an inhibitor of transmembrane efflux pumps, but it was slightly weaker than the reference, carbonyl cyanide 3-chlorophenylhydrazone (CCCP). Next, in vivo tests were conducted on mice infected with methicillin-resistant
P. aeruginosa or carbapenem-susceptible
P. aeruginosa. The results showed significantly higher survival rates in animals treated with a combination of ceftazidime and
1 (10 and 40 mg/kg body mass) compared to those treated with the antibiotic alone or left untreated [
106]. Other bacterial efflux pump inhibitors were synthesised by Hurtová et al. [
107]. They studied quercetin and luteolin modified with fluorinated aniline (
4–
7). The aniline group was substituted at position C8, with a trifluoromethyl group or a fluorine atom located in the para or meta positions relative to the nitrogen atom. Assays conducted on MRSA with gentamicin (1 mg/L) showed that the best antimicrobial properties were exhibited by 8-(4-(trifluoromethyl)anilino)quercetin (
4), 8-(4-fluoroanilino)luteolin (
7), and 8-(4-(trifluoromethyl)anilino)luteolin (
6), with MIC values of approximately 8 μM. In comparison, the combination of gentamicin at breakpoint concentration with unmodified quercetin showed an MIC > 200 μM, while the combination with luteolin revealed an MIC equal to 84,7 μM. The same compounds also exhibited the most significant enhancement of the antibacterial properties of erythromycin, with MIC values ranging from 15 to 25 μM. Monofluorinated compounds were also tested for their ability to inhibit bacterial efflux pumps. For example, 8-(4-fluoroanilino)quercetin at 50 μM (
5) exhibited the same activity as CCCP at 100 μM, while compound
7 displayed even better properties. Notably, the monofluoroflavonoid derivatives were many times more effective at lower concentrations than CCCP. Further studies on modulating erythromycin resistance in MRSA revealed that 8-(4-(trifluoromethyl)anilino)quercetin (
4) and –luteolin (
6) can inhibit ribosomal methyltransferase. However, the precise mechanism of action of the studied compounds remains unclear [
107]. Trifluoromethyl substituents were reported to be more beneficial for increasing flavonoid antibacterial activity than chlorine. Shoaib et al. [
108] synthesised a flavone derivative with a trifluoromethyl substituent at the C4′ position and tested it against
Bacillus subtilis,
S. aureus, and
P. aeruginosa. The MIC values for this compound were 12.5, 25, and 25 µg/mL, respectively, whereas the unmodified flavonoid was studied at the concentrations of 25, 37.5, and 25 µg/mL. The addition of a methoxy group to a fluorinated flavone at the C7 position significantly weakened the antibacterial properties. In contrast, a derivative with a trifluoromethyl group at C3′ and a bromine atom at C6′ exhibited MIC values of 12.5, 6.25, and 6.25 µg/mL, respectively. These values were identical to those obtained for ciprofloxacin, except for
B. subtilis, where the antibiotic was twice as effective. Additionally, chlorinated flavonoid derivatives were also tested but displayed significantly weaker antibacterial activity. These results suggest that trifluoromethyl-substituted derivatives exhibit superior antibacterial properties compared to their chlorine-containing counterparts [
108].
Selected plants, such as
Glycyrrhiza inflata and
Piper sanctum, naturally produce chalcones with antibacterial properties, e.g., licochalcone A. For this reason, chalcone derivatives are frequently studied for their antibacterial properties. Based on the study by Vane et al. on the antibacterial properties of C4′-fluorinated chalcones, it can be concluded that the addition of fluorine and methoxy groups could be crucial for their antibacterial activity. The study was conducted on
S. aureus (MCC2408),
B. subtilis (MCC2010),
E. coli (MCC2412), and
P. aeruginosa (MCC2080). Notably, the compound containing fluorine along with two methoxy groups (at the C2′ and C4′ positions) (
3) exhibited superior antibacterial properties compared to streptomycin. Meanwhile, the compound with only one methoxy group at the C4′ (
2) position demonstrated the highest activity against the
P. aeruginosa strain. Compounds with a higher number of fluorine atoms or methoxy groups in the B-ring at other positions displayed significantly weaker or comparable antibacterial activity to streptomycin [
77].
A series of studies on 2,4,6-trimethoxy or hydroxyl and non-/mono- or difluoro-substituted chalcone derivatives was conducted by Burmaoglu et al. [
109]. Among all the tested compounds, only the compound with three hydroxyl groups in the A-ring (C2, C4, and C6) and two fluorine atoms in the B-ring (C2′ and C5′) (
8) exhibited antibacterial properties (MIC 7.8 µg/mL) against
S. aureus. Interestingly, these compounds, including
8, did not reveal promising activity against
S. pyogenes,
E. faecalis,
E. coli, or
P. aeruginosa (MIC values ranged between 30 and 125 µg/mL). In addition, ampicillin, as an antibiotic, presented moderate activity against
E. faecalis,
P. aeruginosa,
and E. coli, with MIC values of 62.5 µg/mL, 31.25 µg/mL, and 3.9 µg/mL, respectively [
109].
A very interesting study was conducted by Lima et al. [
110]. They evaluated 3-amino-2′-fluorochalcone (
9) suspended in a mucoadhesive hydrogel (at a concentration of 300 µg/mL) for its antimicrobial activity against
Aggregatibacter actinomycetemcomitans,
Fusobacterium periodonticum,
Prevotella intermedia,
Porphyromonas gingivalis, and
Tannerella forsythia—pathogens associated with periodontal disease and peri-implant infections. In vitro tests against these bacteria revealed MIC values ranging from 7.8 to 31.25 μg/mL for bare compound
9, and from 3.9 to 15.6 μg/mL for its hydrogel formulation, with SI between 8 and 32. For comparison, standard antibiotics, such as metronidazole and amphotericin B, exhibited significantly stronger antibacterial activity, with MIC values between 0.195 and 0.5 μg/mL. Further study, which was conducted on titanium conical implantable screws, demonstrated the excellent antibiofilm properties of the hydrogel formulation containing
9, which was comparable to 0.12% chlorhexidine. Toxicity assays performed on
Galleria mellonella larvae infected with
P. gingivalis indicated the low toxicity of
9, maintaining high larval survival rates [
110].
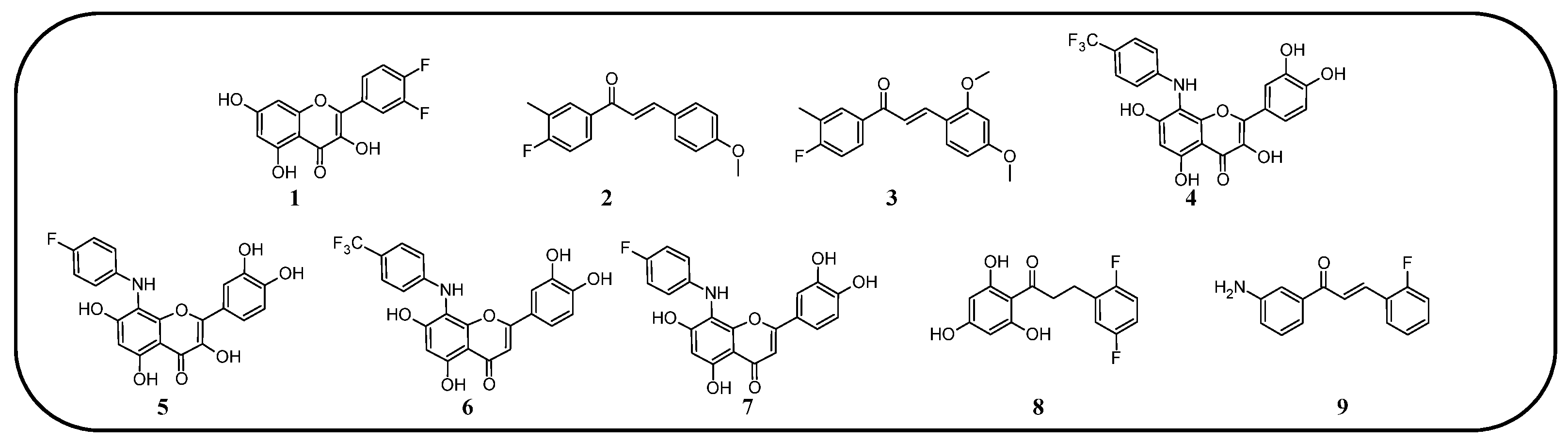
Figure 9.
Flavonoids, chalcones, and their fluorinated derivatives (1–9) with antibacterial properties.
Figure 9.
Flavonoids, chalcones, and their fluorinated derivatives (1–9) with antibacterial properties.
Table 1.
Short summary of the antibacterial properties of fluorinated derivatives of flavonoids and chalcones.
Table 1.
Short summary of the antibacterial properties of fluorinated derivatives of flavonoids and chalcones.
| To Sum Up |
|---|
| Pros |
|---|
Fluorinated flavonoids and chalcones exhibit good antibacterial activity, with MIC values comparable to traditional antibiotics [ 109]. Fluorinated flavonoids enhance the antibacterial potential of antibiotics by interacting with efflux pumps and demonstrate a synergistic effect, even against antibiotic-resistant bacteria [ 106, 107]. 3′,4′-Difluoroquercetin exhibits good antibiofilm properties against S. aureus [ 106].
|
| Cons |
Certain mechanisms allow bacteria to eliminate fluorine from the molecule, thereby weakening the effect of the xenobiotic [ 111].
|
Interestingly, certain natural mechanisms were found in microorganisms that are related to the removal of a fluorine atom from the structure of a fluorinated flavonoid. The study by Seeger et al. [
111] was conducted on recombinant
E. coli cells expressing the biphenyl-2,3-dioxygenase (BphA) genes of
Burkholderia sp. (strain LB400). It was found that 2′-fluoro-7-hydroxy-8-methylisoflavanone was enzymatically converted to 7,2′,3′-trihydroxy-8-methylisoflavone (
Figure 10) [
111].
Flavonoids and chalcones exhibit antifungal activity by disrupting cell membranes and mitochondria, interfering with cell division, and inhibiting protein and RNA synthesis [
72,
112]. Similarly, these compounds also demonstrate significant antiparasitic properties. Their mechanisms of action are varied and include the induction of oxidative stress in parasitic cells, inhibition of crucial parasitic enzymes (e.g., proteases, reductases), disruption of cellular membranes and mitochondrial integrity, and modulation of the host immune response. Moreover, flavonoids have been reported to induce parasite paralysis, disturb calcium homeostasis, alter glucose and glycogen metabolism, and increase nitric oxide synthase activity [
74,
96,
112].
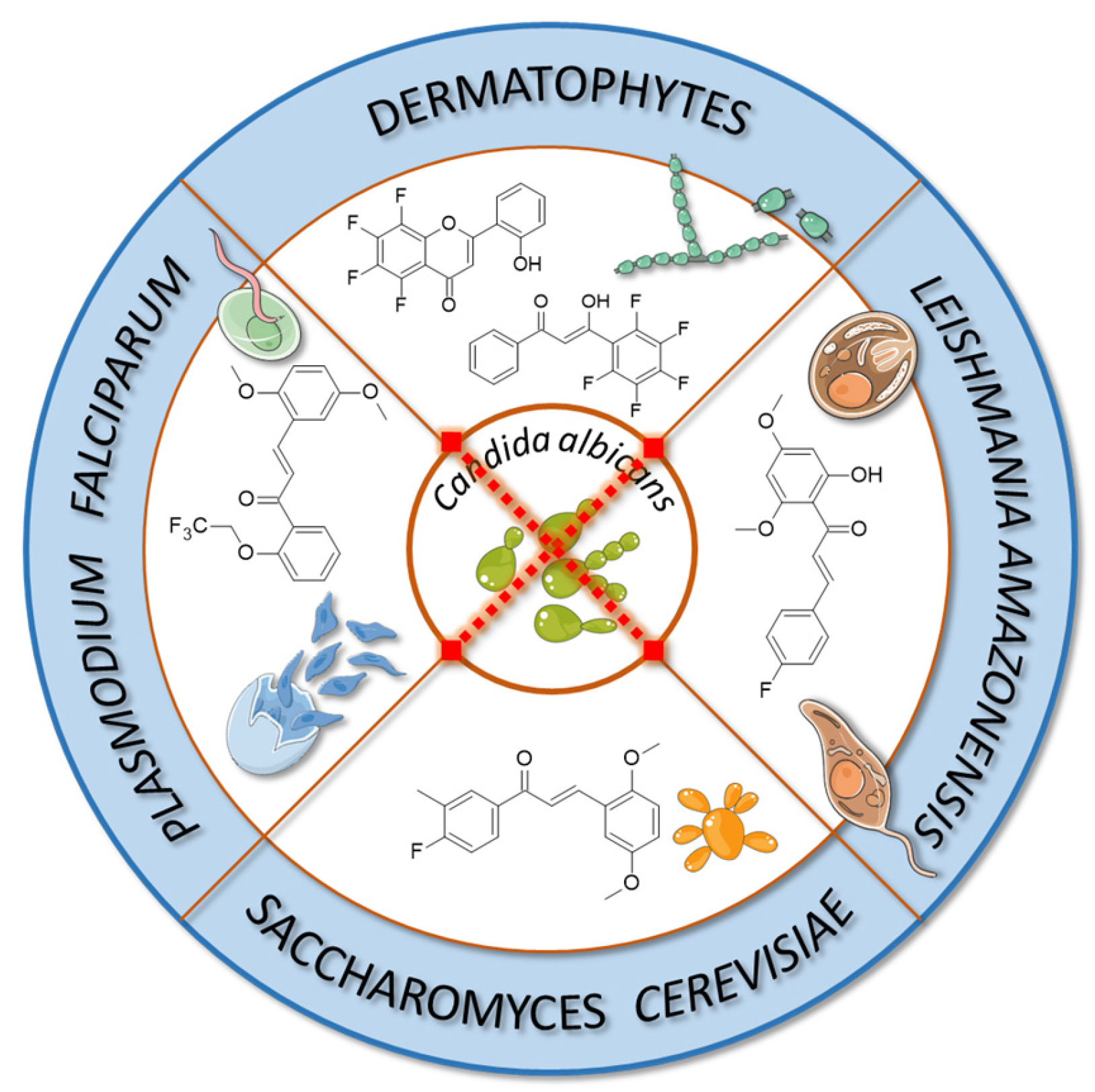
Fluorinated derivatives of flavonoids and chalcones revealed potential antifungal and antiparasitic properties (
Figure 11,
Table 2). There are not many studies on the antifungal properties of fluorinated flavonoids and chalcones. One such study, presented by Shcherbakov et al. [
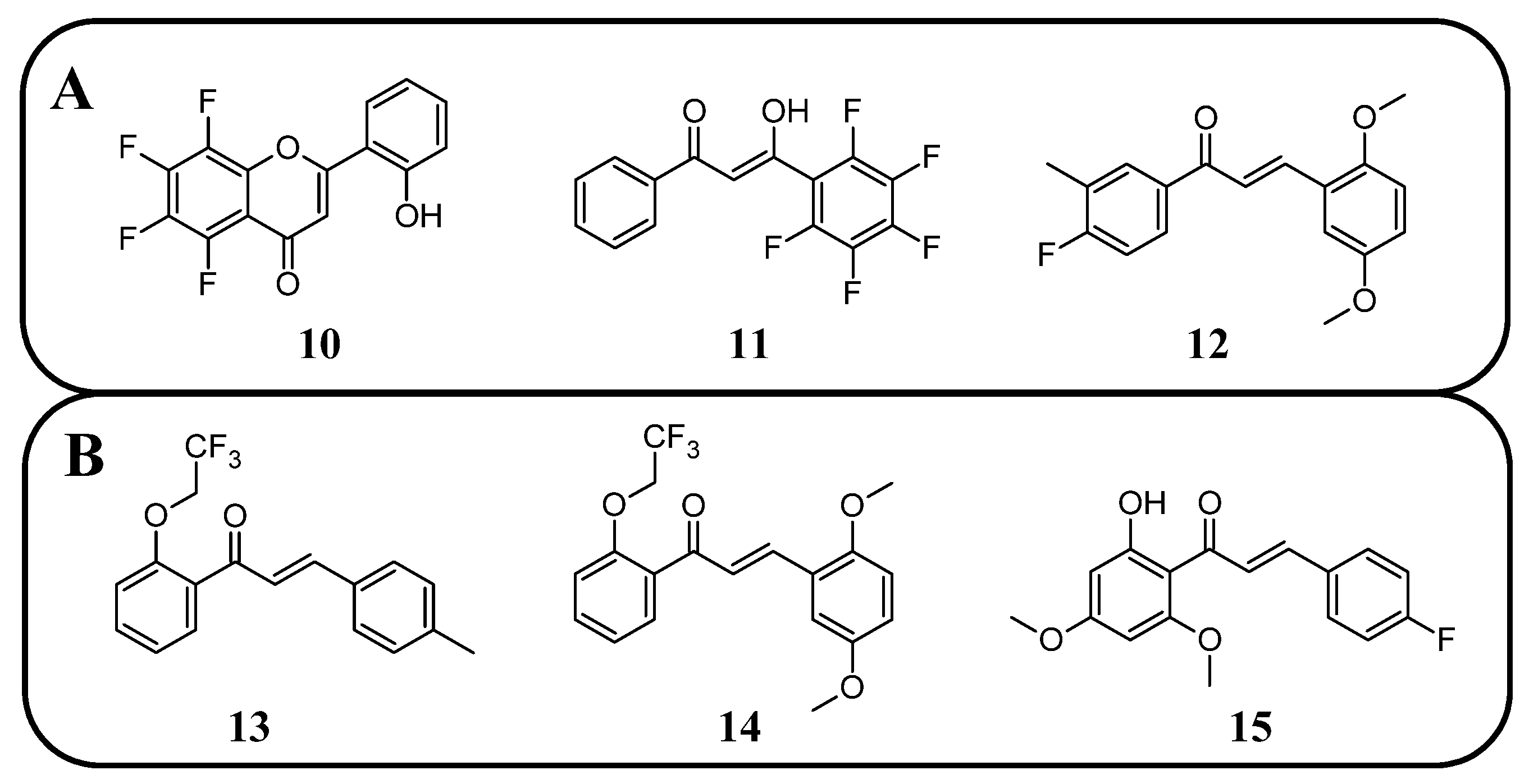
89], clearly demonstrated that flavones with a fluorinated B-ring exhibited little to no antifungal activity. In contrast, fluorination of the A-ring
10 (
Figure 12) resulted in compounds with strong antifungal properties. However, these compounds were not universally effective, as they specifically targeted species such as
Trichophyton tonsurans,
T. rubrum, and
Epidermophyton floccosum. These are the fungi primarily responsible for skin fungal infections—dermatophytes. Fluorinated chalcones also exhibited excellent antifungal properties, particularly the chalcone containing five fluorine atoms in the B-ring (
11), with an MIC < 1.56 µM for nearly all tested species. However, no clear correlation was observed between the number of fluorine atoms and their fungistatic effect. Additionally, the compounds did not reveal activity against
Candida albicans. This study underscores the promising antifungal potential of fluorinated chalcones and the selective activity of flavonoids with fluorinated A-rings, suggesting targeted applications rather than broad-spectrum use [
89].
Antifungal tests conducted using chalcone derivatives fluorinated at the C4′ position showed that none of them exhibited better activity against
C. albicans (MCC1439) than fluconazole. However, the chalcone derivative with methoxy groups at the C3′ and C6′ positions, a methyl group at C3, and a fluorine atom at C4 (
12) demonstrated robust activity against
S. cerevisiae (MCC1033), being twice as effective as fluconazole [
77]. Unfortunately, the studies do not contain information about the selectivity of these compounds in the presence of healthy cells.
The previously mentioned study by Lima et al. indicates that the synthesised compound
9 (
Figure 10), suspended in a hydrogel (300 µg/mL), exhibits good antibiofilm properties against
C. albicans (MIC 7.8 µM), comparable to 0.12% chlorhexidine. This property was investigated on titanium conical implantable screws [
110].
Fluorinated chalcones are also being researched for their antiparasitic properties. Devi et al. [
85] studied 2,2,2-trifluoroethoxychalcone and 2-fluoroethoxychalcone derivatives against
P. falciparum (3D7). The key finding was that monofluoroethoxy derivatives consistently exhibited up to five-fold greater parasite-killing ability compared to their corresponding trifluoroethoxy derivatives. However, two trifluoroethoxy derivatives stood out for their superior activity: (
E)-3-(4-methylphenyl)-1-(2-(2,2,2-trifluoroethoxy)phenyl)prop-2-en-1-one
13 (
Figure 12) (IC
50 9.4 µM) and (
E)-3-(2,5-dimethoxyphenyl)-1-(2-(2,2,2-trifluoroethoxy)phenyl)prop-2-en-1-one (
14) (IC
50 2.2 6 µM). In contrast, other trifluoro derivatives failed to achieve IC
50 values below 10 µM. The selectivity assay conducted on Vero cells revealed that these two compounds had the highest selectivity indexes (8.6 and 8.2, respectively). No strict correlation was observed between the presence of the fluoroethoxy group in the chalcone structure and its antiparasitic properties [
85].
Boeck et al. [
86] evaluated the properties of fluorinated chalcones for combating
Leishmania amazonensis, a flagellate parasite transmitted by mosquitoes that causes leishmaniasis (Dum-Dum fever). The compounds were tested in vitro in cultures containing insect-stage promastigotes and intramacrophage amastigotes of
L. amazonensis (Josefa strain). Among the tested compounds, 4′-fluoro-2-hydroxy-4,6-dimethoxychalcone (
15) was the only fluorinated chalcone examined. Its IC
50 values were ~0.8 µM against promastigote forms and 4.3 µM against amastigote forms. Additionally, the compound showed minimal toxicity to macrophages (IC
50 > 100 µM), making it one of the most promising antiparasitic agents among those studied [
86].
4.2. Antiviral Properties
Natural chalcones and their derivatives exhibit potent antiviral properties by selectively inhibiting viral enzymes such as lactate dehydrogenase, fumarate reductase, protein kinases, and integrase/protease. In contrast, the antiviral activities of flavonoids are not as extensively documented. These compounds show limited ability to interact with viral neuraminidase, proteases, and DNA/RNA polymerases. Nevertheless, both flavonoids and chalcones appear to be promising agents in combating HIV-1 and SARS-CoV-2 infections. In addition to their direct antiviral effects, these compounds may also enhance the host immune response [
71,
105].
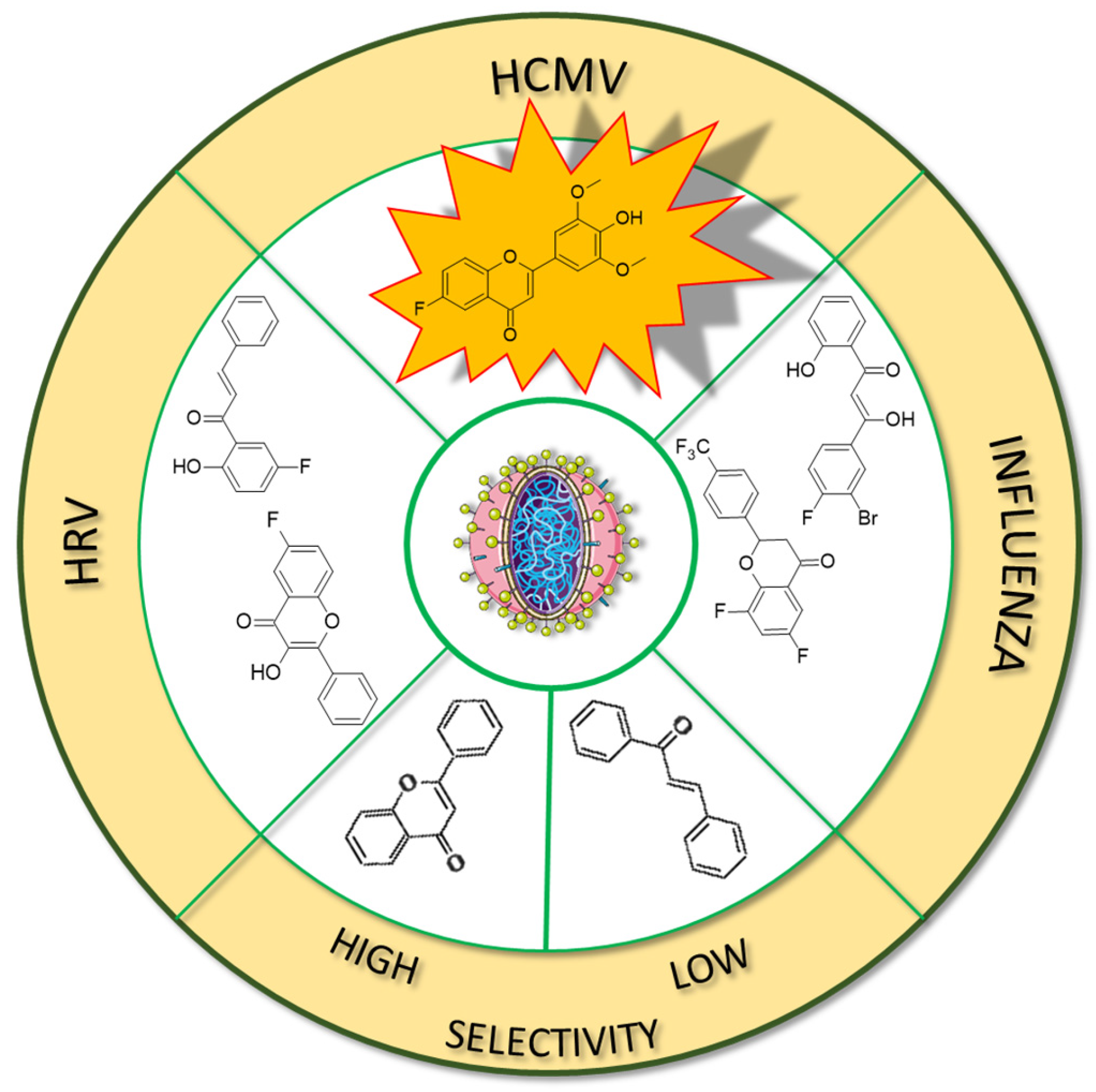
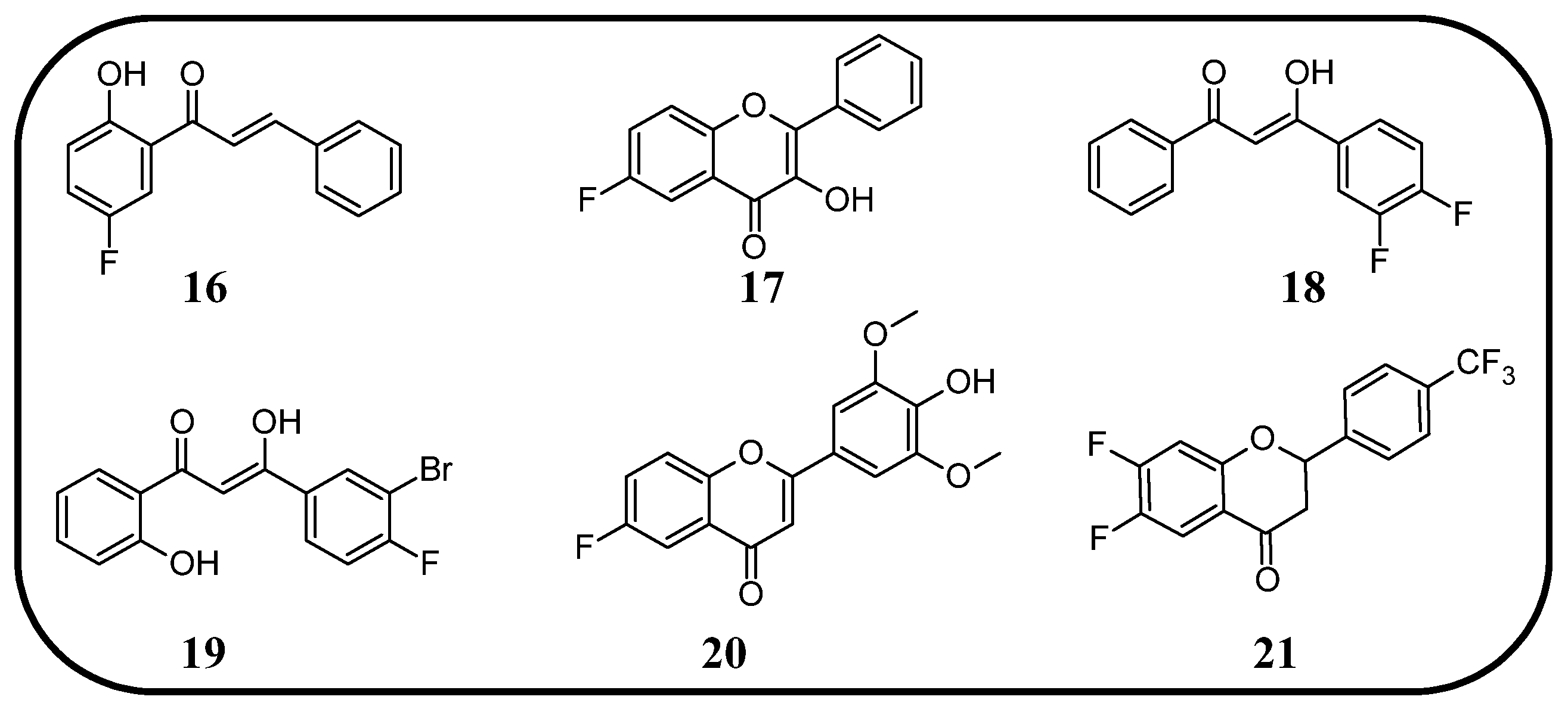
In many studies presented below, fluorinated derivatives of flavonoids and chalcones revealed potential antiviral properties (
Figure 13). The introduction of fluorine into chalcone scaffolds is an interesting strategy for improving antiviral activity. Many studies have explored the antiviral potential of fluorinated chalcones against respiratory viruses. The series of compounds tested by Conti et al. [
76] included fluoro-substituted flavonoids, 2-styrylchromones, and chalcones. The antiviral properties were assessed on HeLa cell lines infected with human rhinovirus (HRV) serotypes 1B and 14. It turns out that fluorinated compounds (including flavonoids with a fluorine atom at position C6) exhibit weak activity against HRV 1B. The most promising compound was the chalcone 1-(5-fluoro-2-hydroxyphenyl)-3-phenylpropen-1-one
16 (
Figure 14,
Table 3), with a concentration required to reduce the HRV 1B virus plaque count by 50% at 5.84 µM. The 6-fluoro-3-hydroxyflavone (
17) demonstrated the best activity against HRV 14 serotype A (IC
50 5.56 µM). This compound also showed high cytotoxicity, with CC
50 values of 25 µM. In addition, the tested flavonoids and chalcones caused a reduction in the size of viral plaques (from 50% to 70%) for both HRV serotypes, suggesting a slowdown in viral replication kinetics. In the case of fluorostyrylchromones, no significant size changes were observed, or they were negligible, whereas the compound BW683C (a dichloroflavan with strong antiviral properties) had an IC
50 of approx. 0.025 µM against HRV 1B and presented no activity against HRV 14 [
76,
113]. Other fluorinated chalcones, particularly 1,3-diaryl-1,3-diketones, were tested by Shcherbakov et al. [
89]. The analysis focused on their interaction against influenza type A/Puerto Rico/8/34 (H1N1) in infected Madin–Darby canine kidney (MDCK) cell lines. The B-ring was fluorinated in the tested compounds. Low selectivity values were observed for fluorinated compounds, especially for fluorinated flavones (selectivity index below 5). A similar trend was observed for chalcones containing four fluorine atoms in the B-ring and chalcones with methylated hydroxyl groups. Interestingly, the IC
50, indicating the reduction in viral production, was lowest among chalcones with the fewest fluorine atoms. The compound containing fluorine atoms at C3′ and C4′ positions had an IC
50 value of 7 µM and SI of 16 (
18), while the compound with fluorine at C4′ and bromine at C3′ presented an IC
50 of 6 µM with an SI of 33 (
19). The IC
50 values of other chalcones ranged between 10 and 16 µM, still significantly outclassing fluorinated flavonoids, which had IC
50 values between 200 and 300 µM. Regarding cytotoxicity towards MDCK cells, a fully fluorinated B-ring posed the most significant risk, with a CC
50 of 57 µM, and the chalcone with the C3, C4, and C5 positions fluorinated had a CC
50 of 43 µM. The CC
50 values for fluorinated chalcones ranged between 43 and 300 µM, while those for flavonoids were around 1000 µM. For comparison, ribavirin, an antiviral drug, showed a CC
50 of approx. 2000 µM, an IC
50 of 36 µM, and a selectivity index > 59 [
89].
Fluorinated flavonoids were tested against influenza viruses and human cytomegalovirus (HCMV). Tricin (4′,5,7-trihydroxy-3′,5′-dimethoxyflavone) and its fluorinated derivative 6-fluoro-4′-hydroxy-3′,5′-dimetoxyflavone (
20) were compared to ganciclovir by evaluating antiviral activity on human embryonic lung (HEL) fibroblast cells infected with HCMV. Previous studies have shown that tricin has anti-HCMV properties based on binding with cyclin-dependent kinase 9 (CDK9), so its mechanism of action is different from that of ganciclovir. The antiviral activity was not as strong as that of ganciclovir, but as a result of fluorination, tricin showed stronger binding to CDK9 and enhanced anti-HCMV properties. The EC
50 of
20 was 0.126 nM, significantly better than that of ganciclovir (EC
50 27.5 nM) and tricin (54.3 nM). Neither
20 nor tricin showed cytotoxicity against HEL cells. In the study, 7-fluoro-3′,5′-dimethoxyflavone was also applied. It was found that the anti-HCMV effect of
20 (fluorine group at C6 position) was up to 1000 times stronger than that of tricin and the compound with fluorine at the C7 position [
114]. Troshkova et al. [
75] studied the properties of fluorinated flavanones against influenza virus A/Puerto Rico/8/34 (H1N1) in the MDCK cell line. They noted that the SI of the fluorinated compounds was very low, around 10 or less, and it was accompanied by high cytotoxicity towards MDCK cells (CC
50 between 9 and 107 µM). Simultaneously, fluorinated compounds showed good antiviral properties, with IC
50 values between 3 and 33 µM. Only flavanones with fluorine at positions C7 and C6 or C7 and C4′ exhibited weak antiviral properties. The compound, 6,8-difluoro-4′-trifluoromethylflavanone (
21), revealed high CC
50 (>915 µM), a selectivity index of 150, and a low IC
50 (6 µM). For comparison, the values for oseltamivir carboxylate, the primary drug used for the treatment of influenza type A and B infections, were CC
50 > 100 µM, IC
50 = 0.18 µM, and SI = 556. Therefore, flavonoid derivative
21 was selected for further studies against influenza virus A/mallard/Pennsylvania/10218/84 (H5N2) and B/Florida/04/06, also in the MDCK cell line. The compound showed good but slightly weaker antiviral properties, with SI values of 53 and 42, respectively [
75].
4.3. Anticancer Properties
Flavonoids and chalcones have shown promise in preventing and slowing the progression of various cancers, including breast, colon, liver, and lung cancers. Their anticancer effects include modulation of angiogenesis, inflammation, and oxidative stress, as well as the induction of apoptosis and cell cycle arrest. These actions are mediated through key signalling pathways, such as NF-
κB, and by targeting specific enzymes such as xanthine oxidase. Some of these compounds also influence non-coding RNAs involved in tumour regulation. In certain cases, flavones can act as pro-oxidants and induce ROS-mediated apoptosis in cancer cells [
31,
41,
115].
The anticancer properties of fluorinated derivatives of flavonoids and chalcones are widely studied and tested (
Figure 15,
Table 4). An in vitro analysis of inhibitory activity conducted on A549 (lung) and HepG2 (liver) carcinoma cell lines showed that fluorination and trifluoromethylation at the C3′ and C4′ positions of the B-ring in chrysin and its derivatives resulted in higher IC
50 values than unmodified chrysin, indicating weaker cytotoxicity. No toxic effect was observed against A549 cells, while IC
50 values ranged from 33.5 to 59.4 μM against HepG2. An assay conducted on the Cellosaurus cell line (SGC-7901) revealed that trifluoromethylation at position C4′ reduced the anticancer properties of chrysin (IC
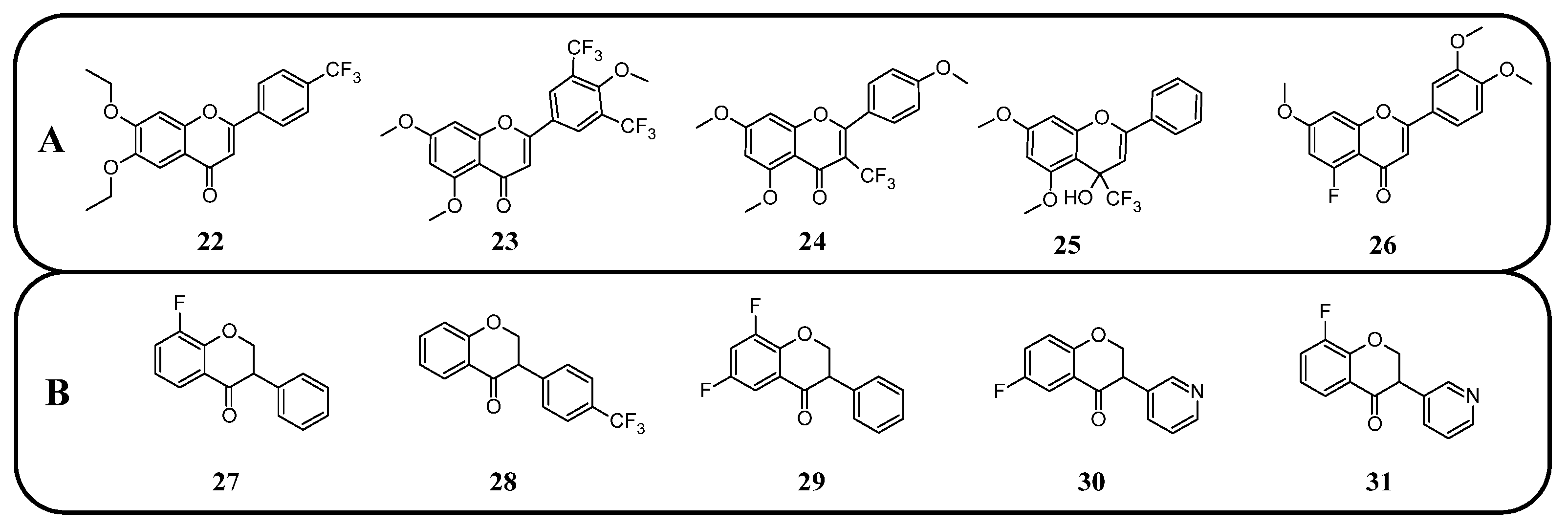
50 increased from 5.8 to 6.6 μM). However, only after the trifluoromethylation and ethylation of both hydroxyl groups, the product 22 was obtained with superior properties (IC
50 = 2.7 μM) (
Figure 16). Methylated chrysin without a fluorine group revealed an IC
50 of 3.7 μM, suggesting that the improved activity could be linked to increased liposolubility. However, methylated chrysin with a trifluoromethyl group at position C8 exhibited antibacterial properties similar to unmodified chrysin. In vivo assays on MCF-7 (human breast cancer), HeLa (cervical cancer), and KSP (kinesin spindle protein) cell lines using chrysin derivatives demonstrated that fluorination impaired anticancer properties of compounds [
78].
Another study presented the activity of trifluoromethylated compounds against the U2OS (osteosarcoma) cell line. Modified flavones with trifluoromethyl substituents attached to the B-ring presented a stronger inhibitory effect on U2OS cells compared to other modifications of the C-ring. Notably, 3′,5′-ditrifluoromethyl-4′,5,7-trimethoxyflavone (
23) showed the strongest inhibitory activity towards cells in the G2/M phase of the cell cycle, while the G1 and S phases were reduced to approximately 50%. Trifluoromethylation at C3 in methylated apigenin (3-trifluoromethyl-4′,5,7-trimethoxyflavone) (
24) resulted in weaker inhibition of the G2/M phase (7%). Flavonoid derivatives with a trifluoromethyl group at the C4 position (
25) exhibited strong cytotoxic effects, killing all U2OS cells at a concentration of just 500 nM. This suggests that the simultaneous presence of both an OH and a CF
3 group at the C4 position leads to exceptionally high cytotoxicity. The presented compounds were not tested on healthy cells [
101].
The flavonoid derivative, 5-fluoro-3′,4′,7-trimethoxyflavone (
26), synthesised by Tsunekawa et al. [
91], was tested in a highly specific context to evaluate the potential to reverse drug resistance mediated by breast cancer resistance protein (BCRP)/ATP-binding cassette subfamily G member 2. Its ability to reverse resistance to 7-ethyl-10-hydroxycamptothecin (SN-38) was investigated using human chronic myelogenous leukaemia cell lines expressing BCRP (K562/BCRP) and non-expressing control cells (K562). The concentration required to achieve a two-fold reduction in drug sensitivity (RI
50) for
26 was determined to be 25 µM. Unfortunately, the fluorinated compound showed a weaker reversal effect compared to the compound with a hydroxyl group instead of the fluorine atom, which had a much lower RI
50 value of 7.4 µM. This experiment demonstrates that replacing a hydroxyl group with a fluorine atom does not always result in improved properties [
91].
Fluorinated isoflavanones and their derivatives obtained by Amato et al. [
94] were tested for anticancer activity on oestrogen-dependent MCF-7 (human breast cancer) cell lines. The IC
50 values of isoflavonone modified with one or two fluorine substituents ranged between 15 and 35 μM, with 8-fluoroisoflavanone
27 (
Figure 16) showing the best activity (IC
50 15 μM) and logP values between 3 and 3.7. No clear correlation was observed between fluorine positioning in the molecule and anticancer properties. Compounds that did not exhibit anticancer activity included 4′-trifluoromethylisoflavanone (
28) and 6,8-difluoroisoflavanone (
29). Unlike the standard anticancer drug letrozole, the modified isoflavanones did not show significant mutagenic, tumorigenic, irritating, or reproductive toxicity in in silico tests. Only compounds with fluorination of the B-ring demonstrated a moderate influence on cell reproduction. The most potent isoflavonoid derivative, with an IC
50 of 0.8 μM and a logP of 1.83, was 6-fluoro-3-(pyridin-3-yl)chroman-4-one (
30). Interestingly, 8-fluoro-3-(pyridin-3-yl)chroman-4-one (
31) exhibited the same logP value but significantly lower activity (IC
50 67 μM). The commercially available drug letrozole demonstrated significantly superior activity, with an IC
50 of 0.0028 μM, despite having slightly worse lipophilicity (logP of 2.15). Fluorinated isoflavanones and their derivatives appear to be promising candidates for further investigation into reducing hormone-dependent breast cancer cell proliferation [
94].
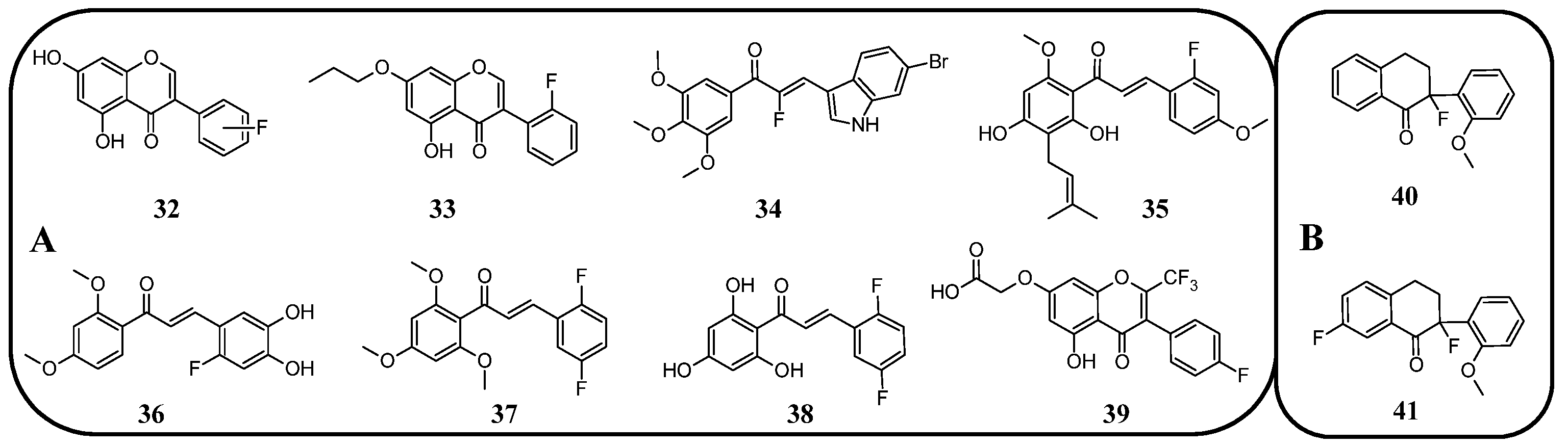
Further analysis of the natural isoflavonoid genistein and its fluorinated derivatives was conducted by Yang et al. [
95]. The study focused on the relationship between fluorine localisation in the B-ring and the properties of the molecule. Additionally, the hydroxyl group in position C7 was alkylated with different simple alkyl chains. The anticancer properties of the obtained genistein analogues
32 (
Figure 17) were tested against three cancer cell lines: MCF-7 (breast cancer), MDA-MB-231 (triple-negative breast cancer), and MDA-MB-435 (melanoma). For comparison, genistein showed IC
50 values ranging between 20 and 24 μM, while tamoxifen (a clinical anti-breast cancer drug) exhibited IC
50 values of 9.13, 10.94, and 5.87 μM, respectively. The analysis revealed that the presence of a hydroxyl group increased the toxicity of the derivatives in each series of compounds. Notably, derivatives with a single fluorine atom demonstrated strong anticancer properties. For 4′-fluoro-5,7-dihydroxyisoflavone, the IC
50 values were 6.80, 9.82, and 13.24 μM, and for 5′-fluoro-5,7-dihydroxyisoflavone, the IC
50 values were 6.79, 13.27, and 12.22 μM, respectively. The alkylated flavonoid 2′-fluoro-5-hydroxy-7-propoxyisoflavone (
33) exhibited similarly potent antineoplastic properties, especially against melanoma (IC
50 6.18 μM). The study also revealed that the elongation of the C7 alkoxy group is accompanied by a decrease in anticancer properties. However, the toxicity increased again when the chain exceeded seven carbon atoms. Moreover, no clear correlation was observed between other fluorine substitution patterns (number of fluorinated carbons: C2′, C2′ and C4′, C3′ and C4′, or C3′ and C4′) and anticancer properties [
95].
A broad study of the antiproliferative properties of
α-fluorinated chalcones was conducted by Sun et al. [
116]. The compounds were tested on six cell lines: A549, HeLa, MCF-7, U937, MGC-803, and HepG2. In particular, one of the derivatives
34, exhibited good anticancer properties, although it had very weak effects on HepG2 cells. A comparison with combretastatin A4 (4 µM) showed that the fluorinated derivative presented weaker antiproliferative action (IC
50 values ranging from 0.025 to 0.254 µM, compared to the IC
50 of combretastatin, which ranges from 0.011 to 0.0003 µM). However, the derivative still presented a higher selectivity ratio (11.5 vs. 2.5 for the drug). Detailed analysis revealed that the fluorinated compound induced cell cycle arrest in MGC-803 cells at the G2/M phase by regulating the expression of proteins (p-Cdc2, Cyclin B1, and p21). The induction of apoptosis in MGC-803 cells may be related to the activation of caspases-3/-7/-9. Studies on HUVECs (human umbilical vein endothelial cells) clearly demonstrated that the tested compound, even at lower doses of 2.5 μM, was capable of inhibiting tumour blood vessel formation, which results in the suppression of angiogenesis [
116]. To evaluate its antiangiogenic activity, an attempt was also made to modify the fluorinated natural chalcone xanthohumol, a compound found in hops (
Humulus lupulus). In vitro assays conducted on HUVECs with different concentrations of the compounds showed their weak activity at 5 µM. However, at 20 µM, a sudden increase in cell mortality was observed for xanthohumol with the methoxy group at the para position and fluorine at C2′ in the B-ring: (
E)-1-(2,4-dihydroxy-6-methoxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(2-fluoro-4-methoxyphenyl)prop-2-en-1-one (
35). This compound most effectively inhibited cell migration and adhesion. Migration assays revealed that the presence of a hydroxyl group at the C4 position significantly reduced cell motility, but this effect did not appear to depend on the fluorine position in the B-ring. Additionally, compounds containing fluorine and two MOM (methoxymethyl) groups in the A-ring exhibited strong antiproliferative and cell growth-inhibitory properties. Morphogenesis studies in vitro at 10 µM showed that, after 6 h, the xanthohumol derivative with a para-methoxy group and fluorine at C2′ again demonstrated the strongest activity. All fluorinated derivatives inhibited morphogenesis at least twice as effectively as unmodified xanthohumol [
117]. An analysis of several mono- and difluorinated chalcones with two methoxy groups in the A-ring and two hydroxyl groups in the B-ring identified 2,4-dimethoxy-6′-fluoro-3′,4′-dihydroxychalcone (
36) as the most potent antiproliferative compound. This chalcone was tested using a human cancer cell (HCC) panel consisting of 39 cell lines. The results showed strong antiproliferative activity against certain cell lines, including MCF7, HBC-4 and HBC-5 (breast), NCI-H522 (lung), OVCAR-3 (ovarian), and MKN128 (gastric), with GI
50 values between 0.5 and 3 µM. However, some cancer types showed resistance, particularly renal, melanoma, and prostate HCC, as well as most ovarian and CNS-derived HCC lines. The most resistant was MDA-MB-231 (breast cancer), with a GI
50 of 38 µM. A key finding was that adding a second fluorine atom at C5′ drastically reduced or eliminated antiproliferative activity [
79].
Burmaoglu et al. [
80] conducted further studies on approximately 70 fluorinated chalcones across HEK293, A549, A498, HeLa, A375, and HepG2 cell lines, yielding key findings. They concluded that the removal of the ketone or double bond in the chalcone linker can enhance antiproliferative activity, depending on A-ring substitutions (methoxy/hydroxyl groups). Moreover, they noted that substitution at the B-ring increases antitumour potency, especially with two fluoro groups at positions 2 and 5 against A549 and A498 cell lines. It is worth noting that HEK293 cells showed strong resistance to fluorinated chalcones. The most potent and selective compound was (
E)-3-(2,5-difluorophenyl)-1-(2,4,6-trimethoxyphenyl)prop-2-en-1-one (
37), with IC
50 values ranging from 0.03 µM (A498) to 0.12 µM (A549) and an SI between 10 (A549) and 20 (A498). The highest specificity, combined with strong antitumour activity (SI 25 against HeLa and HepG2), was observed for (
E)-3-(2,5-difluorophenyl)-1-(2,4,6-trihydroxyphenyl)prop-2-en-1-one (
38) [
80].
Fluorinated isoflavone derivatives of daidzein synthesised by Ayoup et al. [
118] did not reveal improved biological activity. The IC
50 value of daidzein against MCF-7 breast cancer cells was 11.87 μM, with an SI of 6.5. Substitution of the hydroxyl group at the 4′ position with a fluorine atom and the introduction of a trifluoromethyl group at the C2 position resulted in a derivative with reduced anticancer activity (IC
50 15.43 μM; SI 5.3). Additionally, it was shown that daidzein and its derivatives are only weakly active against B16F10 melanoma cells. The only compound with slightly improved anticancer properties with IC
50 11.23 μM and SI 6.4) was derivative
39 [
118].
Other research investigating the antineoplastic activity of monofluoroisoflavonoids utilised 1-carbaisoflavanone derivatives (
Figure 17). The 1-carba-3-fluoro-6′-methoxy-isoflavanone (
40) exhibited the best anticancer properties, with the highest selectivity against MCF-7 breast cancer cells (IC
50 60 μM). Additionally, 1-carba-3,6-difluoro-6′-methoxy-isoflavanone (
41) exhibited anticancer activity against chronic myeloid leukaemia cell lines, such as K562, Lucena I, and FEPS, with IC
50 values ranging from 27 to 63 μM. These two studies highlight the superiority of fluorinated isoflavonoids over carbaisoflavonoids in the treatment of breast cancer [
96].
4.4. Neuroprotective Properties
Flavonoids were reported to cross the blood–brain barrier (BBB). A low molecular weight, high lipophilicity, and small topological polar surface area (TPSA) are factors that promote BBB permeation of flavonoids. Therefore, naringenin and quercetin are examples of flavonoids with good and moderate permeability, respectively, while rutin and hesperidin are those with low potential, mainly because of the presence of saccharides, which increase molecular weight and TPSA and lower lipophilicity [
119,
120,
121]. Fluorination is interesting because of the augmentation of two aspects: activity [
122,
123,
124,
125] and BBB permeation [
126]. The introduction of fluorine, which is a small, electronegative atom, affects lipophilicity and electronic properties, which facilitates the interaction with the BBB [
126].
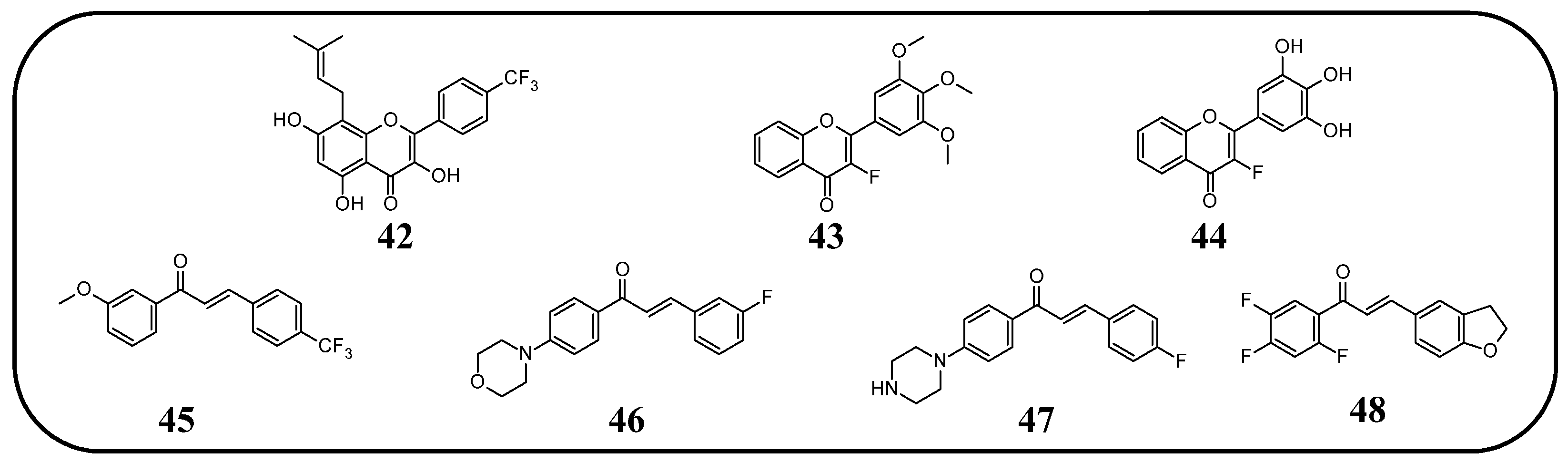
In many studies presented below, fluorinated derivatives of flavonoids and chalcones revealed potential neuroprotective properties (
Figure 18). Jia et al. [
122,
123,
124,
125] performed a study on fluorinated icaritin and explored the neuroprotective properties of these compounds. Trifluoroicaritin (
42) (
Figure 19,
Table 5) was evaluated in vivo using a rat model of spared nerve injury (SNI)-induced neuropathic pain. The effective dose of compound
42 was 5.0 mg/kg, and its therapeutic efficacy became evident from day 1, persisting until day 21. The movement assay (CatWalk-automated gait analysis) demonstrated that
42 increased paw pressure and improved motor coordination, indicating a reduction in pain hypersensitivity. The rotarod test confirmed an improvement in balance and motor abilities. A positive correlation was also observed between contact pressure intensity and rotational speed, suggesting that
42 influences both sensory and motor functions. Compound
42 effectively reduced mechanical allodynia (a condition where pain is caused by stimuli that normally do not cause pain) but had no effect on thermal hyperalgesia (increased sensitivity to painful heat) [
122]. The results suggest that
42 exerts an analgesic effect by modulating
α7nAChR-dependent pathways (
α7 nicotinic acetylcholine receptors), which inhibit the BDNF/TrkB/KCC2 signalling cascade, crucial for neuroinflammatory processes and pain hypersensitivity in SNI rats. Additionally, the flavonoid increased the levels of anti-inflammatory markers (IL-10, CD206) and reduced pro-inflammatory cytokines (IL-1
β, CD40). The optimal dose of
42 was 5 mg/kg body mass, effectively alleviating pain and anxiety symptoms [
123]. Other in vivo tests conducted on rat models with complete Freund’s adjuvant (CFA)-induced chronic inflammatory pain showed the impact of
42 on microglia. Administration of the xenobiotic at doses of 3 mg/kg and 10 mg/kg equally reduced motor dysfunction and pain perception. Flavonoid derivative
42 demonstrated the ability to activate microglia, as confirmed by its capacity to increase CB2 receptor levels and decrease P2Y12 receptor levels (suppressing neuroinflammatory responses). The flavonoid further reduced the inflammatory response by influencing the expression of CD11b and iNOS, blocking the release of cytokines such as IL-1
β, IL-6, and TNF-
α. Finally, compound
42 promoted the transformation of microglia towards the anti-inflammatory (M2) phenotype by activating the IL-10/
β-endorphin pathway, alleviating chronic inflammatory pain in CFA rats [
125].
Alshammari et al. [
90] studied the antioxidant properties of monofluoroflavones using the 1,1-diphenyl-2-picryl-hydrazyl (DPPH) antioxidant assay, which revealed better radical-scavenging activity compared to non-fluorinated flavones. The EC
50 of 3-fluoro-3′,4′,5′-trimethoxyflavone (
43) was 37 μg/mL and 0.24 μg/mL for 3-fluoro-3′,4′,5′-trihydroxyflavone (
44). In comparison, the EC
50 values for non-fluorinated counterparts were 71 μg/mL and 0.33 μg/mL, respectively. In this test, the EC
50 value of vitamin C was determined to be 0.16 μg/mL. The neuroprotective properties of the fluoroflavones were assessed using a Sprague-Dawley rat cortical neurons in an oxidative glutamate toxicity assay. The test measures glutamate toxicity resulting from the induction of oxidative stress caused by depletion of intracellular glutathione levels. First, cells are exposed to glutamate; then, ROS are added to enhance oxidative stress, followed by assessment of cell viability. The data indicate that in vitro, C3-fluorinated hydroxyflavonoids exhibit two-fold greater neuroprotective activity than their fluorinated methoxy derivatives [
90].
Fluorinated chalcones, due to their structural similarity to drugs such as fluoxetine and melperone, have become a subject of interest for neurologists. Mathew et al. [
81,
82] examined a series of different 5′-methoxychalcones with fluorine in the B-ring in terms of their ability to inhibit human monoamine oxidase B and A (hMAO-B and hMAO-A). hMAO-B is an enzyme potentially playing a role in the development of neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease [
127]. hMAO inhibitors prolong the activity of dopamine, making them preferred treatments in the early stages of Parkinson’s disease (e.g., selegiline). The fluorinated chalcones showed very weak binding to hMAO-A but significantly better affinity for hMAO-B. The most active compounds were those with fluorinated groups at position C4, particularly the compound with a trifluoromethyl group (
45). Along with the activity, there was a high selectivity index between the two enzymes (hMAO-B/hMAO-A equals 0.05 for the chalcone and 0.04 for selegiline). Reversibility tests showed, however, that chalcones bind weakly to the enzyme. Of the initial 87% hMAO-B inhibition, only 11% remained after washing, whereas for selegiline, the value remained stable at around 47% [
82]. In subsequent studies, fluorinated chalcones were tested with morpholine or imidazole at the C4′ position. They were examined in the context of their ability to inhibit hMAO-A, hMAO-B, and acetylcholinesterase (AChE). Among the morpholine derivatives, the compound with fluorine at the C3 position—(2
E)-3-(3-fluorophenyl)-1-[4-(morpholin-4-yl)-phenyl]prop-2-en-1-one (
46)—demonstrated the best hMAO-B inhibitory activity, comparable to commercial drugs (IC
50 0.087 µM, which is better than pargyline). The selectivity index of this compound was very high (hMAO-A/hMAO-B—512). This compound was found to be a reversible inhibitor in further studies. The compound with fluorine at the C2 position exhibited slightly weaker properties. The presence of fluorine or a trifluoromethyl group at the C4 position resulted in reduced MAO-B inhibitory activity. However, fluorinated morpholine-containing derivatives still demonstrated significantly better properties than imidazole-containing derivatives. The chalcones showed no affinity for AChE. Additionally, the parallel artificial membrane permeation assay (PAMPA) was conducted to determine the blood–brain barrier (BBB) permeation potential of the morpholine-containing chalcones. All derivatives exhibited permeability similar to that of testosterone (approximately 19 × 10⁻⁶ cm/s) [
83].
An attempt was also made to introduce a piperazine moiety at the C3 position. Compounds containing fluorine (
47) or trifluoromethyl group at the C4′ position exhibited the lowest IC
50 values against hMAO-B among all tested compounds (0.65 and 0.71 µM). These were the only compounds that showed activity against butyrylcholinesterase- BChE (approximately 36 µM) and AChE (approximately 27 µM). Unfortunately, their selectivity index—around 49, was lower than that of the morpholine derivatives. Piperazine derivatives also proved to be reversible inhibitors. Additionally, fluorinated compounds demonstrated the best properties among the halogenated derivatives [
84].
Sánchez et al. [
128] examined the neuroprotective properties of chalcones with a halogenated A-ring and a benzodihydrofuran moiety instead of the classical C-ring. Tests were conducted on the PC-12 cell line, derived from a rat adrenal medulla pheochromocytoma. The cells were incubated for 24 h at various concentrations, with a separate assessment of cells treated with amyloid
β peptide (A
β). Once again, fluorinated compounds outperformed other halogen derivatives. Notably, the compound with fluorine atoms at positions C2, C4, and C6- (
E)-3-(2,3-dihydrobenzofuran-5-yl)-1-(2,4,5-trifluorophenyl)prop-2-en-1-one (
48) exhibited low cytotoxicity and provided up to 150% cell viability in A
β-treated cells, whereas without this compound, cell survival was approximately 70% [
128].
Figure 19.
Fluorinated flavonoids and chalcones (42–48) with neuroprotective activity.
Figure 19.
Fluorinated flavonoids and chalcones (42–48) with neuroprotective activity.
Table 5.
Short summary of the neuroprotective properties of fluorinated derivatives of flavonoids and chalcones.
Table 5.
Short summary of the neuroprotective properties of fluorinated derivatives of flavonoids and chalcones.
| To Sum Up |
|---|
| Pros |
|---|
Fluorinated flavonoids exhibit neuroprotective properties through a complex mechanism. They act as antioxidants, suppress neuroinflammation, inhibit the activity of MAO-B enzymes, and protect cells from the effects of amyloid β peptide [ 128]. Fluorinated chalcones modified with a morpholine group are capable of good blood–brain barrier permeation [ 83, 84].
|
| Cons |
Fluorinated chalcones present only weak affinities for cholinesterases [ 84].
|
4.5. Other Properties
The remaining studies describe the potential anti-inflammatory and antioxidant properties of fluorinated derivatives of flavonoids and chalcones, as well as their applications in diagnostics. Flavonoids are known to exhibit anti-inflammatory activities via different mechanisms, especially by the inhibition of the synthesis and actions of specific pro-inflammatory mediators such as eicosanoids, cytokines, adhesion molecules, and C-reactive protein, as well as transcription factors and regulatory enzymes. It is important that flavonoids can inhibit the onset and development of inflammatory diseases [
17,
18,
19,
20]. These molecules can downregulate mast cell activation, which is responsible for secreting inflammatory mediators such as histamine and pro-inflammatory cytokines [
18]. Flavonoids as polyphenols present high antioxidant activity and can scavenge free radicals, donate hydrogen atoms, and chelate metal cations. The main antioxidant mechanism of action relies on transferring hydrogen atoms to free radicals [
12]. These natural products, as potent antioxidants, reveal the potential to attenuate tissue damage or fibrosis [
17].
Below, the anti-inflammatory and antioxidant properties of fluorinated derivatives of flavonoids and chalcones, as well as their applications in diagnostics, are briefly discussed and summarised (
Table 6). Stadlbauer et al. [
93] highlighted the anti-inflammatory properties of polyphenols found in green tea. The activity of 3-
O-gallates, specifically epicatechin-3-
O-gallate (ECG), the second most prevalent polyphenol in green tea, and its fluorinated derivative (−)-5,7-difluoro-epicatechin-3-
O-gallate
49 (
Figure 20), was investigated. Both compounds contain the flavanol catechin. Assays demonstrated that
49 reduced the viability of human lymphocyte cells. The proliferation of activated human lymphocytes was inhibited to almost 0% at a concentration of 30 µM, whereas ECG achieved a similar effect only at 100 µM. However, the reduction in cell proliferation was due to the induction of apoptosis. The antiproliferative properties of
49 indicate that it could be used to treat chronic inflammation [
93].
Fluorinated chalcones were also examined for their anti-inflammatory potential, particularly their ability to inhibit nitric oxide production. NO plays a crucial role as a vasodilator, contributing to oedema formation, leukocyte activation, and cytokine production. Additionally, its reaction with superoxide anions leads to the formation of peroxynitrite, a compound known to cause tissue damage. In the study by Rojas et al. [
129], the A-ring of the chalcones was modified with one or two fluorine atoms or a trifluoromethyl moiety, while the B-ring contained either two or three methoxy groups. The results indicated that trimethoxychalcone derivatives with a fluorine atom at the C4 position were significantly more effective at inhibiting NO production compared to those containing a trifluoromethyl group. Interestingly, the trifluoromethyl moiety provided good inhibition when positioned at C2, regardless of the number or positioning of methoxy groups in the B-ring. The IC
50 values of the tested compounds ranged from 0.28 to 0.91 µM, with monofluorinated derivatives consistently exhibiting stronger NO inhibition than their trifluoromethyl analogues. However, chalcones containing two fluorine atoms, including one at C2, showed slightly weaker activity compared to their monofluorinated counterparts. The most potent compound in limiting NO production was 4-fluoro-3′,4′,5′-trimethoxychalcone (
50), which demonstrated an IC
50 of 0.03 µM and achieved 94.1% inhibition of nitrite formation. Moreover, findings suggest that the inhibition of NO production by these fluorinated chalcones in macrophages may occur at the enzyme expression level, further highlighting their potential as anti-inflammatory agents [
129].
Flavonoids and chalcones are well known for their antioxidant properties. Bist et al. [
88] analysed nearly 40 fluorinated chalcone derivatives for their inhibitory activity on ROS production stimulated by lipopolysaccharide in RAW 264.7 macrophages. The analysis showed that a hydroxyl group at the
para position in the A-ring, along with a fluorinated moiety at the
meta position in the B-ring, are essential for inhibiting ROS production in macrophages. The IC
50 values for ROS inhibition were lowest for the trifluoromethyl moiety (1.44 µM) (53), higher for the fluorine group (9.15 µM) (54), and the weakest activity was observed for the trifluoromethoxy group (>10 µM) (55). Chalcones containing the trifluoromethoxy group exhibited strong activity when positioned at the
ortho position. Among these compounds, the highest activity was observed for chalcones with a hydroxyl group at C4 (IC
50 1.34 µM) (56), followed by C3 (IC
50 1.61 µM). However, a hydroxyl group at the
ortho position did not inhibit ROS production. Additionally, the presence of a hydroxyl group at the
ortho position of the A-ring, combined with a fluorinated B-ring, was often associated with macrophage toxicity. It was also observed that B-ring-modified chalcones with fluorine-containing groups at the C3 position exhibited no significant ROS inhibition when lacking a hydroxyl group [
88].
In another study, several chalcones with two methoxy groups in the A-ring and two hydroxyl groups in the B-ring, modified with one or two fluorine atoms in the B-ring, were tested. Their activity was evaluated against Fe
3+- ADP-induced NADPH-dependent lipid peroxidation in rat liver microsomes. The results indicated that fluorine substitution at C2′ (
57) and C5′ (
58), with a methoxy group at C4 and C2, provided the strongest antioxidant properties. Additionally, an analysis conducted on rat basophilic leukemia-1 (RBL-1) cells assessed the inhibitory action on 5-lipoxygenase. Compound
56 exhibited the highest ability to inhibit the enzyme, with an IC
50 value of 0.012 µM. This value was the same for the analogue with fluorine at position C6′ or at both C6′ and C5′ (with methoxy moieties at C2 and C5) [
79].
Figure 20.
Other fluorinated flavonoids and chalcones (49–58).
Figure 20.
Other fluorinated flavonoids and chalcones (49–58).
A fluorine group has also been applied as a labelling agent in diagnostics. The
18F isotope is widely used in positron emission tomography (PET) imaging (e.g., in the structure of fluorodeoxyglucose-FDG). An attempt was made to introduce this isotope into the chalcone structure, resulting in (
E)-3-(4-(methylamino)phenyl)-1-(4-fluorophenyl)-2-propen-1-one (
51) and (
E)-3-(4-(dimethylamino)phenyl)-1-(4-fluorophenyl)-2-propen-1-one (
52). First, in vivo biodistribution studies in healthy mice showed that the brain uptake of the radiopharmaceuticals after 2 min was similar for
51 (5.47% ID/g brain, injected dose per gram of brain) and florbetapir, a commonly used radiopharmaceutical (4.90% ID/g brain). For
52, the uptake was 4.43% ID/g brain. However, after 30 min, the retention of both fluorinated chalcones was nearly three times lower compared to the commercial radiopharmaceutical. The in vitro studies focused on the ability of the compounds to bind
β-amyloid (A
β) plaques. The dissociation constant (K
d) values ranged from 4.5 to 6.5 nM, indicating a high affinity for A
β1–42 aggregates. For comparison, [
3H]PiB has a K
d 4.7 nM. Subsequently, in vitro autoradiography (ARG) was performed using human brain sections with Alzheimer’s disease pathology. The compounds with intensely labelled A
β plaques and did not accumulate in regions without A
β plaques. The conclusions drawn from these experiments suggest that
52 and
51 may be useful in the PET imaging of A
β plaques in the diseased brain. However, one of the major concerns regarding the use of these compounds is their rapid clearance from the body [
87].
Table 6.
Short summary of the anti-inflammatory and antioxidant properties of fluorinated derivatives of flavonoids and chalcones, as well as their applications in diagnostics.
Table 6.
Short summary of the anti-inflammatory and antioxidant properties of fluorinated derivatives of flavonoids and chalcones, as well as their applications in diagnostics.
| To Sum Up |
|---|
| Pros |
|---|
Fluorinated flavonoids exhibit anti-inflammatory properties by reducing lymphocyte viability and inhibiting nitric oxide production [ 93, 129]. Due to the ability of fluorinated chalcone derivatives to cross the BBB and their strong binding to β-amyloid (A β) plaques, they are promising candidates for neurodegenerative disease imaging [ 87]. Fluorination of hydroxyl-containing flavonoids enhances their antioxidant properties [ 88].
|
| Cons |
A significant drawback in using chalcones for imaging is their rapid elimination from the body [ 87].
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}