


Caloric Restriction Mimetics as Priming Agents of Mesenchymal Stem Cells Secretome to Enhance Regenerative Responses to Parkinson’s Disease

 ,
, 
 and
and
Abstract
1. Introduction
2. Parkinson’s Disease
2.1. Pathophysiology
2.2. Cellular and Molecular Mechanisms
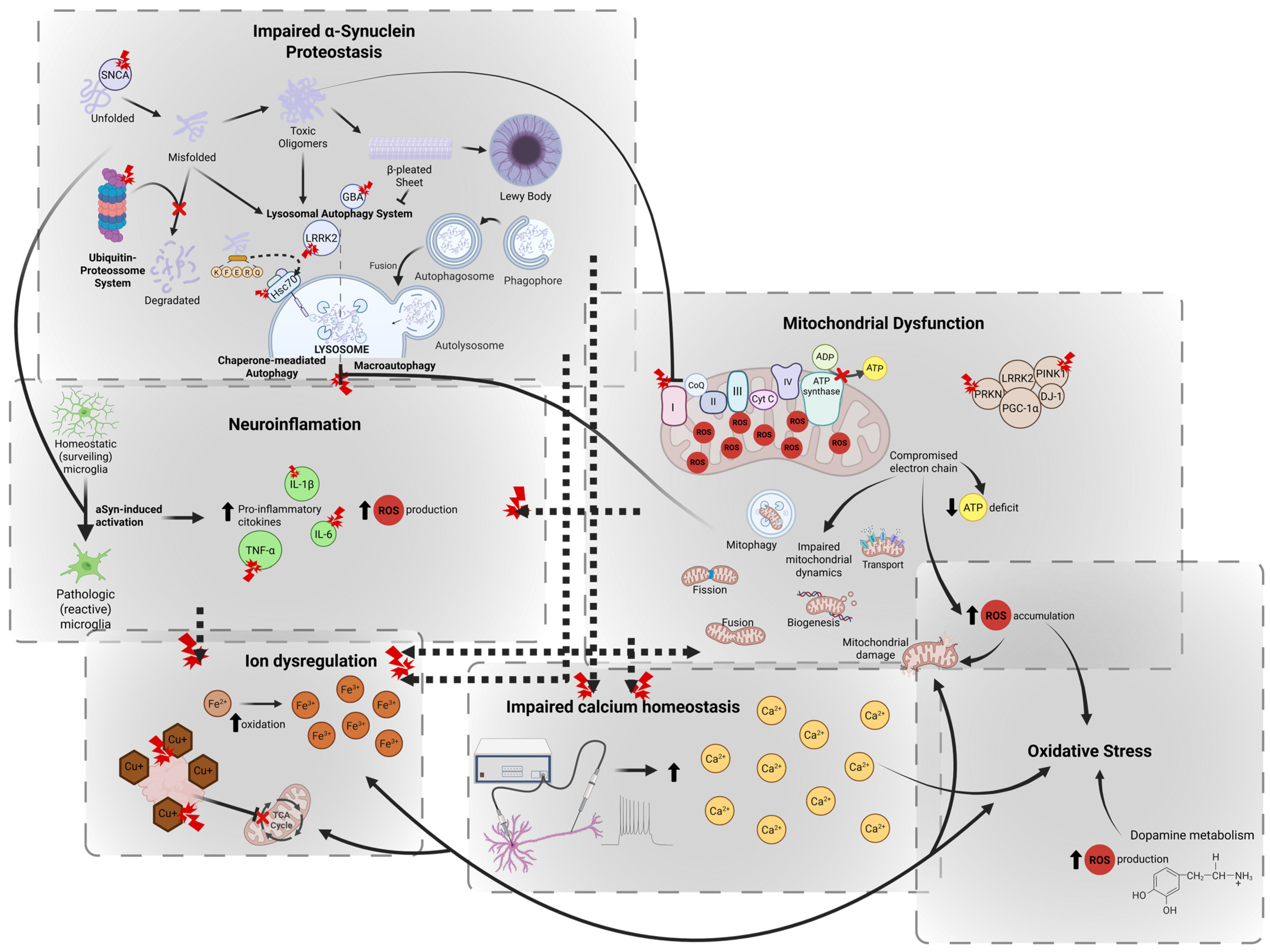
2.2.1. Impaired α-Synuclein Proteostasis
2.2.2. Mitochondrial Dysfunction
2.2.3. Oxidative Stress
2.2.4. Impaired Calcium Homeostasis
2.2.5. Ion Dysregulation
2.2.6. Neuroinflammation
2.3. Current Treatment Limitations
3. Mesenchymal Stem Cells
3.1. Mesenchymal Stem Cells Secretome
3.2. Mesenchymal Stem Cells’ Therapeutic Effects in Parkinson’s Disease
3.2.1. Neuroprotection
3.2.2. Clearance of α-Synuclein Aggregates
3.2.3. Immunomodulation
3.2.4. Mitochondrial Transfer and Bioenergetic Support
3.2.5. Blood−Brain Barrier Modulation
3.3. Current Limitations on MSCs Secretome
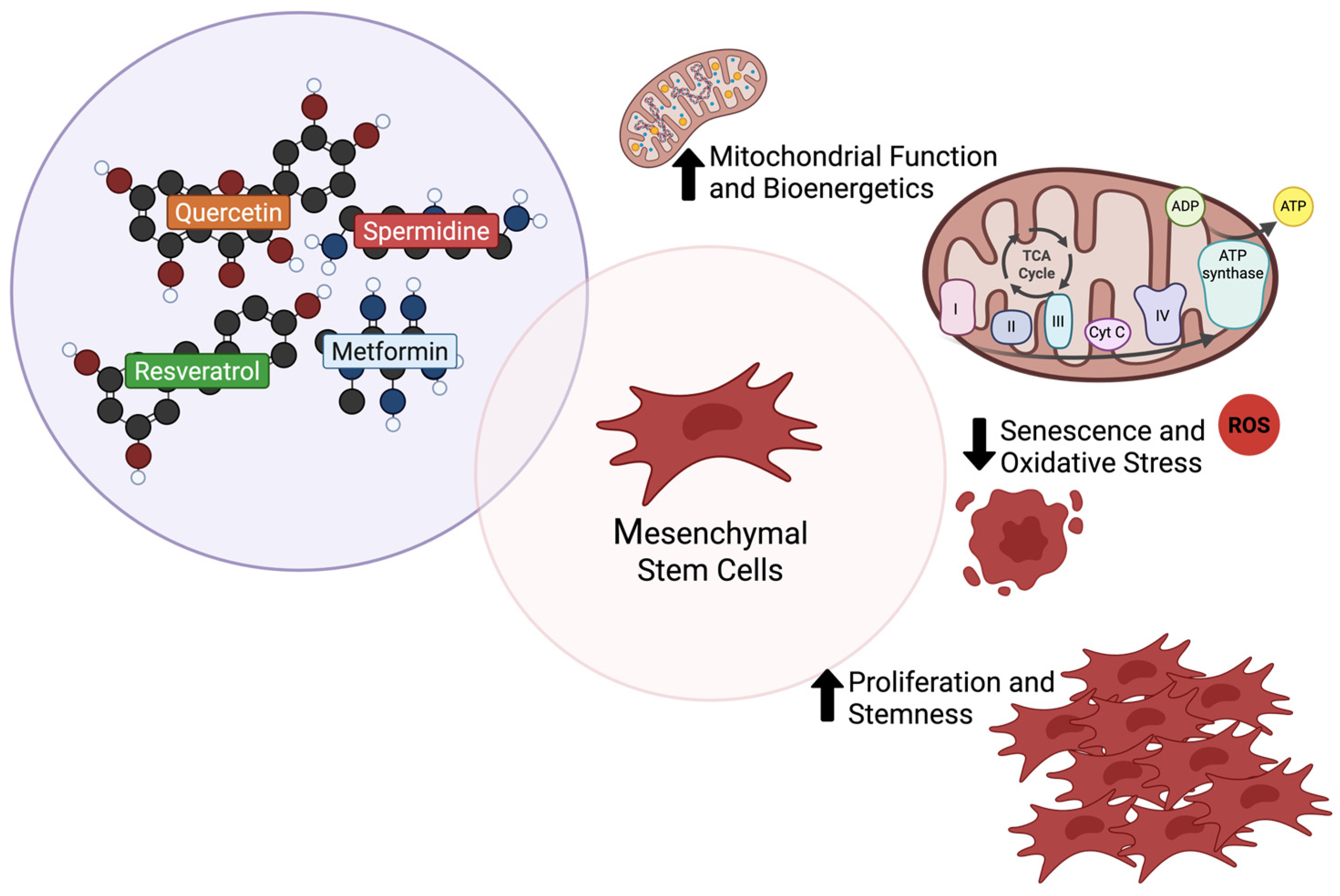
3.4. Enhancing Mesenchymal Stem Cells Secretome Efficiency
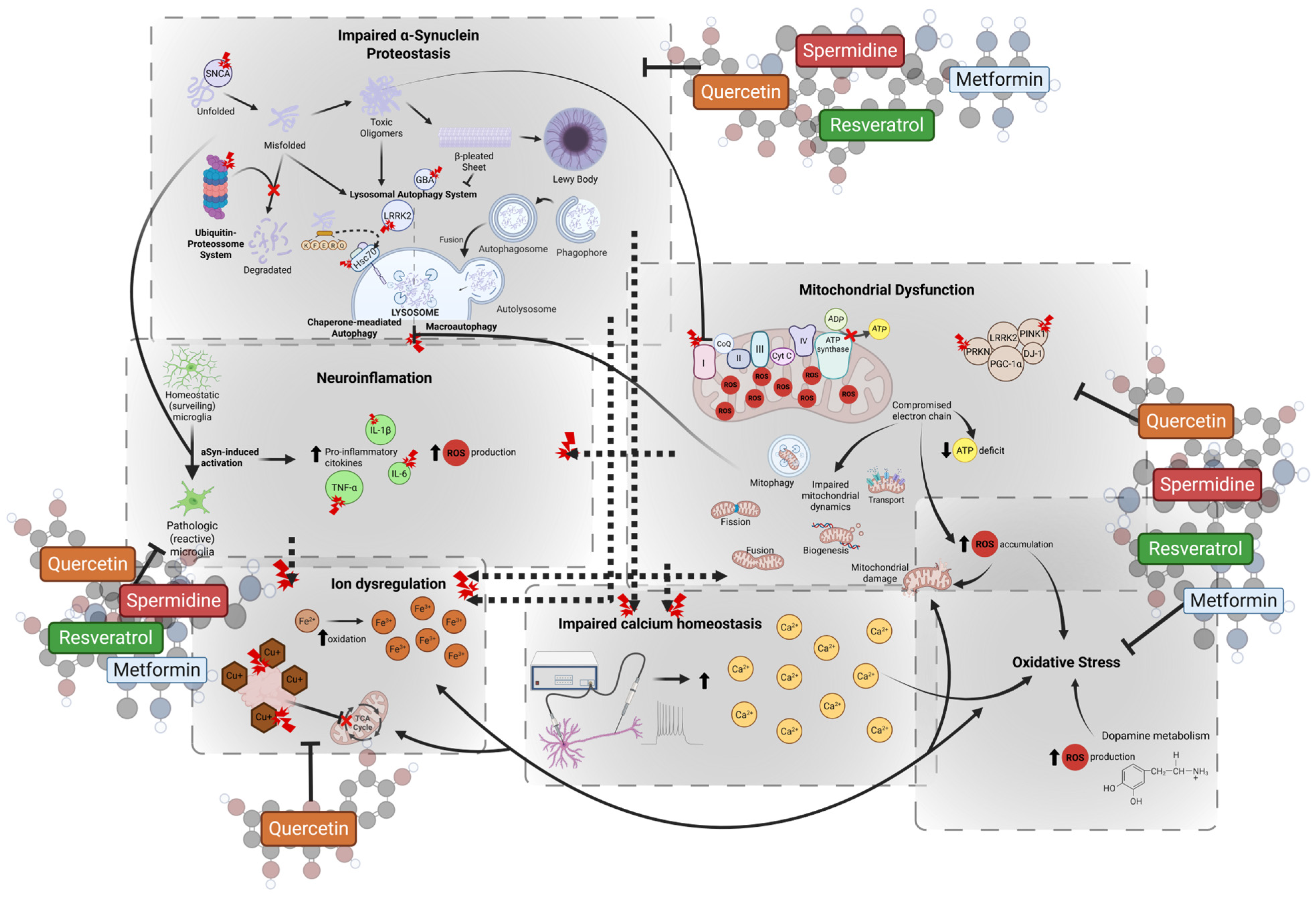
4. Caloric Restriction Mimetics
4.1. Quercetin
4.1.1. Quercetin and Parkinson’s Disease
4.1.2. Quercetin and Mesenchymal Stem Cells
4.2. Spermidine
4.2.1. Spermidine and Parkinson’s Disease
4.2.2. Spermidine and Mesenchymal Stem Cells
4.3. Resveratrol
4.3.1. Resveratrol and Parkinson’s Disease
4.3.2. Resveratrol and Mesenchymal Stem Cells
4.4. Metformin
4.4.1. Metformin and Parkinson’s Disease
4.4.2. Metformin and Mesenchymal Stem Cells
4.5. Strategic Priming of Mesenchymal Stem Cells with Caloric Restriction Mimetics
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| AMPK | adenosine-monophosphate-activated protein kinase |
| aSyn | α-synuclein |
| ATG | autophagy related |
| Bax | B-cell lymphoma 2 associated X protein |
| BBB | blood−brain barrier |
| Bcl-2 | B-cell lymphoma 2 protein |
| BDNF | brain-derived neurotrophic factor |
| BECN1 | beclin 1 |
| CAT | catalase |
| CCL4 | chemokine ligand 4 |
| CFL1 | cofilin 1 |
| CLU | clusterin |
| CMA | chaperone-mediated autophagy |
| CR | caloric restriction |
| CREB | cAMP response-element binding protein |
| CRMs | caloric restriction mimetics |
| CST3 | cystatin C |
| DBS | deep brain stimulation |
| DFMO | difluoromethylornithine |
| DMT1 | divalent metal transporter 1 |
| DPSCs | dental pulp-derived MSCs |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| ERK | extracellular signal-regulated kinase |
| EVs | extracellular vesicles |
| FGF-9 | fibroblast growth factor 9 |
| FOXO | forkhead box O |
| GABA | gamma-aminobutyric acid |
| GABARAPL1 | GABA type A receptor-associated protein like 1 |
| GAD | glutamic acid decarboxylase |
| GBA | glucosylceramidase beta |
| GDNF | glial-derived neurotrophic factor |
| GLRX | glutaredoxin |
| GPX4 | glutathione peroxidase 4 |
| GSH | glutathione |
| GST | glutathione S-transferase |
| H3K9 | histone H3 lysine 9 |
| hBM | human bone marrow-derived |
| HGF | hepatocyte growth factor |
| HO-1 | heme oxygenate 1 |
| HSC70 | heat shock cognate 70 |
| HSPA8 | heat shock protein family A member 8 |
| hUC | human umbilical cord-derived |
| IDO | indoleamine 2,3-dioxygenase |
| IGF | insulin-like growth factor |
| IL | interleukin |
| IP-10 | interferon γ-induced protein 10 kDa |
| iPSCs | induced pluripotent stem cells |
| LAMP2A | lysosomal-associated membrane protein 2A |
| LAS | lysosomal autophagy system |
| LBs | Lewy bodies |
| l-DOPA | l-3,4-dihydroxyphenylalanine |
| LGALS | galectin |
| LIF | leukemia inhibitory factor |
| LRRK2 | leucine-rich repeat kinase 2 |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| MDA | malonaldehyde |
| miRNA | micro-RNA |
| MMP-2 | metalloproteinase-2 |
| MPP+ | 1-methyl-4-phenylpyridinium ion |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MSCs | mesenchymal stem cells |
| mTOR | mechanistic target of rapamycin |
| NCOA4 | nuclear receptor coactivator 4 |
| NF-M | neurofilament M |
| NF-κB | nuclear factor κ B |
| Ngn | neurogenin |
| NO | nitric oxide |
| Nrf2 | nuclear factor erythroid-related factor 2 |
| NSE | neuron-specific enolase |
| NT-3 | neurotrophin-3 |
| OCR | oxygen consumption rate |
| ODC1 | ornithine decarboxylase 1 |
| OPG | osteoprotegerin |
| PD | Parkinson’s disease |
| PGC-1α | peroxisome proliferator-activated receptor-γ coactivator 1-α |
| PI3K | phosphoinositide 3-kinase |
| PINK1 | phosphatase and tensin homolog induced novel kinase 1 |
| PKD1 | protein kinase D1 |
| PMCA | plasma membrane Ca2+ ATPase |
| PP2A | protein phosphatase 2A |
| PRKN | Parkin |
| RANTES | regulated upon activation, normal T cell expressed and secreted |
| ROS | reactive oxygen species |
| SA-β-gal | senescence-associated β-galactosidase |
| SERCA | sarcoplasmic/endoplasmic reticulum Ca2+-ATPase |
| SHEDs | stem cells derived from human exfoliated deciduous teeth |
| SIRT | sirtuin |
| SLC7A11 | solute carrier family 7 member 11 |
| SN | substantia nigra |
| SNCA | synuclein alpha |
| SNpc | substantia nigra pars compacta |
| SOD | superoxide dismutase |
| SQSTM | sequestosome |
| STAT | signal transducer and activator of transcription |
| TCA | tricarboxylic acid |
| TFAM | mitochondrial transcription factor A |
| TGF-β2 | transforming growth factor beta 2 |
| TIMP-1 | tissue inhibitor of metalloproteinase |
| TLR-3 | toll-like receptor 3 |
| TNF-α | tumor necrosis factor α |
| TNTs | tunneling nanotubes |
| UCHL1 | ubiquitin C-terminal hydrolase L1 |
| UPS | ubiquitin-proteasome system |
| VEGF | vascular endothelial growth factor |
| VSP35 | vacuolar protein sorting 35 |
References
- Su, D.; Cui, Y.; He, C.; Yin, P.; Bai, R.; Zhu, J.; Lam, J.S.T.; Zhang, J.; Yan, R.; Zheng, X.; et al. Projections for Prevalence of Parkinson’s Disease and Its Driving Factors in 195 Countries and Territories to 2050: Modelling Study of Global Burden of Disease Study 2021. BMJ 2025, 388, e080952. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.u.A. Parkinson’s Disease: Mechanisms, Translational Models and Management Strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Pires, A.O.; Teixeira, F.G.; Mendes-Pinheiro, B.; Serra, S.C.; Sousa, N.; Salgado, A.J. Old and New Challenges in Parkinson’s Disease Therapeutics. Prog. Neurobiol. 2017, 156, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.R.; Marote, A.; Mendes-Pinheiro, B.; Teixeira, F.G.; Salgado, A.J. Cell Secretome Based Approaches in Parkinson’s Disease Regenerative Medicine. Expert Opin. Biol. Ther. 2018, 18, 1235–1245. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Carvalho, M.M.; Panchalingam, K.M.; Rodrigues, A.J.; Mendes-Pinheiro, B.; Anjo, S.; Manadas, B.; Behie, L.A.; Sousa, N.; Salgado, A.J. Impact of the Secretome of Human Mesenchymal Stem Cells on Brain Structure and Animal Behavior in a Rat Model of Parkinson’s Disease. Stem Cells Transl. Med. 2017, 6, 634–646. [Google Scholar] [CrossRef]
- Mendes-Pinheiro, B.; Anjo, S.I.; Manadas, B.; Da Silva, J.D.; Marote, A.; Behie, L.A.; Teixeira, F.G.; Salgado, A.J. Bone Marrow Mesenchymal Stem Cells’ Secretome Exerts Neuroprotective Effects in a Parkinson’s Disease Rat Model. Front. Bioeng. Biotechnol. 2019, 7, 294. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Vilaça-Faria, H.; Domingues, A.V.; Campos, J.; Salgado, A.J. Preclinical Comparison of Stem Cells Secretome and Levodopa Application in a 6-Hydroxydopamine Rat Model of Parkinson’s Disease. Cells 2020, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.R.; Pereira-Sousa, J.; Teixeira, F.G.; Sousa, R.A.; Teixeira-Castro, A.; Salgado, A.J. Mesenchymal Stem Cell Secretome Protects against Alpha-Synuclein-Induced Neurodegeneration in a Caenorhabditis Elegans Model of Parkinson’s Disease. Cytotherapy 2021, 23, 894–901. [Google Scholar] [CrossRef]
- Vilaça-Faria, H.; Marote, A.; Lages, I.; Ribeiro, C.; Mendes-Pinheiro, B.; Domingues, A.V.; Campos, J.; Lanceros-Mendez, S.; Salgado, A.J.; Teixeira, F.G. Fractionating Stem Cells Secretome for Parkinson’s Disease Modeling: Is It the Whole Better than the Sum of Its Parts? Biochimie 2021, 189, 87–98. [Google Scholar] [CrossRef]
- Mendes-Pinheiro, B.; Campos, J.; Marote, A.; Soares-Cunha, C.; Nickels, S.L.; Monzel, A.S.; Cibrão, J.R.; Loureiro-Campos, E.; Serra, S.C.; Barata-Antunes, S.; et al. Treating Parkinson’s Disease with Human Bone Marrow Mesenchymal Stem Cell Secretome: A Translational Investigation Using Human Brain Organoids and Different Routes of In Vivo Administration. Cells 2023, 12, 2565. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.R.; de Fuzeta, M.A.; dos Santos Cunha, R.M.; Pereira-Sousa, J.; Silva, D.; Campos, J.; Teixeira-Castro, A.; Sousa, R.A.; Fernandes-Platzgummer, A.; da Silva, C.L.; et al. Neurodifferentiation and Neuroprotection Potential of Mesenchymal Stromal Cell-Derived Secretome Produced in Different Dynamic Systems. Biomedicines 2023, 11, 1240. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.R.; Campos, J.; Sampaio-Marques, B.; Antunes, F.F.; dos Santos Cunha, R.M.; Silva, D.; Barata-Antunes, S.; Lima, R.; Fernandes-Platzgummer, A.; da Silva, C.L.; et al. Secretome of Bone Marrow Mesenchymal Stromal Cells Cultured in a Dynamic System Induces Neuroprotection and Modulates Microglial Responsiveness in an α-Synuclein Overexpression Rat Model. Cytotherapy 2024, 26, 700–713. [Google Scholar] [CrossRef]
- Narbute, K.; Piļipenko, V.; Pupure, J.; Dzirkale, Z.; Jonavičė, U.; Tunaitis, V.; Kriaučiūnaitė, K.; Jarmalavičiūtė, A.; Jansone, B.; Kluša, V.; et al. Intranasal Administration of Extracellular Vesicles Derived from Human Teeth Stem Cells Improves Motor Symptoms and Normalizes Tyrosine Hydroxylase Expression in the Substantia Nigra and Striatum of the 6-Hydroxydopamine-Treated Rats. Stem Cells Transl. Med. 2019, 8, 490–499. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Z.; Xing, H.; Wang, Y.; Guo, Y. Exosomes Derived from miR-188-3p-Modified Adipose-Derived Mesenchymal Stem Cells Protect Parkinson’s Disease. Mol. Ther. -Nucleic Acids 2021, 23, 1334–1344. [Google Scholar] [CrossRef]
- Ingram, D.K.; Zhu, M.; Mamczarz, J.; Zou, S.; Lane, M.A.; Roth, G.S.; DeCabo, R. Calorie Restriction Mimetics: An Emerging Research Field. Aging Cell 2006, 5, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Hofer, S.J.; Davinelli, S.; Bergmann, M.; Scapagnini, G.; Madeo, F. Caloric Restriction Mimetics in Nutrition and Clinical Trials. Front. Nutr. 2021, 8, 717343. [Google Scholar] [CrossRef]
- Nassar, K.; El-mekawey, D.; Elmasry, A.E.; Refaey, M.S.; El-Sayed Ghoneim, M.; Elshaier, Y.A.M.M. The Significance of Caloric Restriction Mimetics as Anti-Aging Drugs. Biochem. Biophys. Res. Commun. 2024, 692, 149354. [Google Scholar] [CrossRef]
- Goetz, C.G. The History of Parkinson’s Disease: Early Clinical Descriptions and Neurological Therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a008862. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy. JNP 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic Criteria for Parkinson Disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological Assessment of Parkinson’s Disease: Refining the Diagnostic Criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson Disease. Park. Relat. Disord. 2018, 46, S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s Disease: Substantia Nigra Regional Selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical Progression in Parkinson Disease and the Neurobiology of Axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Hernandez, L.F.; Obeso, I.; Costa, R.M.; Redgrave, P.; Obeso, J.A. Dopaminergic Vulnerability in Parkinson Disease: The Cost of Humans’ Habitual Performance. Trends Neurosci. 2019, 42, 375–383. [Google Scholar] [CrossRef]
- Blesa, J.; Foffani, G.; Dehay, B.; Bezard, E.; Obeso, J.A. Motor and Non-Motor Circuit Disturbances in Early Parkinson Disease: Which Happens First? Nat. Rev. Neurosci. 2022, 23, 115–128. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Klann, E.M.; Dissanayake, U.; Gurrala, A.; Farrer, M.; Shukla, A.W.; Ramirez-Zamora, A.; Mai, V.; Vedam-Mai, V. The Gut–Brain Axis and Its Relation to Parkinson’s Disease: A Review. Front. Aging Neurosci. 2022, 13, 782082. [Google Scholar] [CrossRef]
- Serratos, I.N.; Hernández-Pérez, E.; Campos, C.; Aschner, M.; Santamaría, A. An Update on the Critical Role of α-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Tissue to Cellular and Molecular Levels. Mol. Neurobiol. 2022, 59, 620–642. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s Disease: Separating the Wheat from the Chaff. J. Park. Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Luk, K.; Purtell, K.; Burke Nanni, S.; Stoessl, A.J.; Trudeau, L.-E.; Yue, Z.; Krainc, D.; Oertel, W.; Obeso, J.A.; et al. Neuronal Vulnerability in Parkinson Disease: Should the Focus Be on Axons and Synaptic Terminals? Mov. Disord. 2019, 34, 1406–1422. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xie, X.; Liu, R. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein Degradation and Protection against Misfolded or Damaged Proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-Synuclein and Protein Degradation Systems: A Reciprocal Relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, V. Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson’s Disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in Lysosomal and Proteasomal Markers in Parkinson’s Disease: Relationship to Alpha-Synuclein Inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.K.; Craig, E.L.; Chakrabarti, L. Mitochondrial Complex 1 Activity Measured by Spectrophotometry Is Reduced across All Brain Regions in Ageing and More Specifically in Neurodegeneration. PLoS ONE 2016, 11, e0157405. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Langston, J.W.; Langston, E.B.; Irwin, I. MPTP-Induced Parkinsonism in Human and Non-Human Primates--Clinical and Experimental Aspects. Acta Neurol. Scand. Suppl. 1984, 100, 49–54. [Google Scholar]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of Toxicity in Rotenone Models of Parkinson’s Disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s Disease α-Synuclein Transgenic Mice Develop Neuronal Mitochondrial Degeneration and Cell Death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Hwang, O. Role of Oxidative Stress in Parkinson’s Disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P.; Lerner, R.A.; Kelly, J.W. Elevated Levels of Oxidized Cholesterol Metabolites in Lewy Body Disease Brains Accelerate α-Synuclein Fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative Damage in Nucleic Acids and Parkinson’s Disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.V.; Halliday, G. Missing Pieces in the Parkinson’s Disease Puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Kahle, P.J.; Waak, J.; Gasser, T. DJ-1 and Prevention of Oxidative Stress in Parkinson’s Disease and Other Age-Related Disorders. Free. Radic. Biol. Med. 2009, 47, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9, 2045. [Google Scholar] [CrossRef]
- Shin, J.; Kovacheva, L.; Thomas, D.; Stojanovic, S.; Knowlton, C.J.; Mankel, J.; Boehm, J.; Farassat, N.; Paladini, C.; Striessnig, J.; et al. Cav1.3 Calcium Channels Are Full-Range Linear Amplifiers of Firing Frequencies in Lateral DA SN Neurons. Sci. Adv. 2022, 8, eabm4560. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. Calcium Homeostasis, Selective Vulnerability and Parkinson’s Disease. Trends Neurosci. 2009, 32, 249–256. [Google Scholar] [CrossRef]
- Virdi, G.S.; Choi, M.L.; Evans, J.R.; Yao, Z.; Athauda, D.; Strohbuecker, S.; Nirujogi, R.S.; Wernick, A.I.; Pelegrina-Hidalgo, N.; Leighton, C.; et al. Protein Aggregation and Calcium Dysregulation Are Hallmarks of Familial Parkinson’s Disease in Midbrain Dopaminergic Neurons. Npj. Park. Dis. 2022, 8, 162. [Google Scholar] [CrossRef]
- Harbauer, A.B.; Hees, J.T. Calcium Dysregulation and Mitochondrial Dysfunction Form a Vicious Cycle in Parkinson’s Disease. AJBSR 2019, 5, 246. [Google Scholar]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.H.; Riederer, P. The Relevance of Iron in the Pathogenesis of Parkinson’s Disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, J.; Rogers, J.; Xie, J. Brain Iron Metabolism Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 3078–3101. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B.H. Increased Iron (III) and Total Iron Content in Post Mortem Substantia Nigra of Parkinsonian Brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef]
- Lin, K.-J.; Chen, S.-D.; Lin, K.-L.; Liou, C.-W.; Lan, M.-Y.; Chuang, Y.-C.; Wang, P.-W.; Lee, J.-J.; Wang, F.-S.; Lin, H.-Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829. [Google Scholar] [CrossRef]
- Wang, H.; Wu, S.; Li, Q.; Sun, H.; Wang, Y. Targeting Ferroptosis: Acteoside as a Neuroprotective Agent in Salsolinol-Induced Parkinson’s Disease Models. FBL 2025, 30, 26679. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent Metal Transporter 1 (DMT1) Contributes to Neurodegeneration in Animal Models of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, Y.; Liu, X. The Mechanism of Cuproptosis in Parkinson’s Disease. Ageing Res. Rev. 2024, 95, 102214. [Google Scholar] [CrossRef]
- Telianidis, J.; Hung, Y.H.; Materia, S.; La Fontaine, S. Role of the P-Type ATPases, ATP7A and ATP7B in Brain Copper Homeostasis. Front. Aging Neurosci. 2013, 5, 44. [Google Scholar] [CrossRef]
- Liddell, J.R.; White, A.R. Nexus between Mitochondrial Function, Iron, Copper and Glutathione in Parkinson’s Disease. Neurochem. Int. 2018, 117, 126–138. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Moehle, M.S.; West, A.B. M1 and M2 Immune Activation in Parkinson’s Disease: Foe and Ally? Neuroscience 2015, 302, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Falcon, B.; Clavaguera, F.; Tolnay, M. Prion-like Mechanisms in the Pathogenesis of Tauopathies and Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2014, 14, 495. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ettle, B.; Bruno, A.; Kulinich, A.; Hoffmann, A.-C.; von Wittgenstein, J.; Winkler, J.; Xiang, W.; Schlachetzki, J.C.M. Alpha-Synuclein Activates BV2 Microglia Dependent on Its Aggregation State. Biochem. Biophys. Res. Commun. 2016, 479, 881–886. [Google Scholar] [CrossRef]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 Receptor Is Critical in α-Synuclein–Mediated Microglial NADPH Oxidase Activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein Activates Microglia in a Model of Parkinson’s Disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef]
- Gao, H.-M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuroinflammation and Oxidation/Nitration of α-Synuclein Linked to Dopaminergic Neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Ng, W.L.; Goh, S.Y.; Gulam, M.Y.; Wang, L.-F.; Tan, E.-K.; Ahn, M.; Chao, Y.-X. Targeting the Inflammasome in Parkinson’s Disease. Front. Aging Neurosci. 2022, 14, 957705. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Castillo-Rangel, C.; Marin, G.; Hernández-Contreras, K.A.; Vichi-Ramírez, M.M.; Zarate-Calderon, C.; Torres-Pineda, O.; Diaz-Chiguer, D.L.; De la Mora González, D.; Gómez Apo, E.; Teco-Cortes, J.A.; et al. Neuroinflammation in Parkinson’s Disease: From Gene to Clinic: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 5792. [Google Scholar] [CrossRef]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Deuschl, G.; Antonini, A.; Costa, J.; Śmiłowska, K.; Berg, D.; Corvol, J.-C.; Fabbrini, G.; Ferreira, J.; Foltynie, T.; Mir, P.; et al. European Academy of Neurology/Movement Disorder Society—European Section Guideline on the Treatment of Parkinson’s Disease: I. Invasive Therapies. Eur. J. Neurol. 2022, 29, 2580–2595. [Google Scholar] [CrossRef]
- Smilowska, K.; van Wamelen, D.J.; Pietrzykowski, T.; Calvano, A.; Rodriguez-Blazquez, C.; Martinez-Martin, P.; Odin, P.; Chaudhuri, K.R. Cost-Effectiveness of Device-Aided Therapies in Parkinson’s Disease: A Structured Review. J. Park. Dis. 2021, 11, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived with Disability of Parkinson’s Disease in 204 Countries/Territories From 1990 to 2019. Front. Public Health 2021, 9, 776874. [Google Scholar] [CrossRef]
- Currie, A.D.; Wong, J.K.; Okun, M.S. A Review of Temporal Interference, Nanoparticles, Ultrasound, Gene Therapy, and Designer Receptors for Parkinson Disease. NPJ. Park. Dis. 2024, 10, 195. [Google Scholar] [CrossRef]
- Pinjala, P.; Tryphena, K.P.; Prasad, R.; Khatri, D.K.; Sun, W.; Singh, S.B.; Gugulothu, D.; Srivastava, S.; Vora, L. CRISPR/Cas9 Assisted Stem Cell Therapy in Parkinson’s Disease. Biomater. Res. 2023, 27, 46. [Google Scholar] [CrossRef]
- Barker, R.A.; Drouin-Ouellet, J.; Parmar, M. Cell-Based Therapies for Parkinson Disease—Past Insights and Future Potential. Nat. Rev. Neurol. 2015, 11, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Fričová, D.; Korchak, J.A.; Zubair, A.C. Challenges and Translational Considerations of Mesenchymal Stem/Stromal Cell Therapy for Parkinson’s Disease. NPJ Regen. Med. 2020, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal Stem Cell Perspective: Cell Biology to Clinical Progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef]
- Lukomska, B.; Stanaszek, L.; Zuba-Surma, E.; Legosz, P.; Sarzynska, S.; Drela, K. Challenges and Controversies in Human Mesenchymal Stem Cell Therapy. Stem Cells Int. 2019, 2019, 9628536. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ramos, J.; Song, S.; Cardozo-Pelaez, F.; Hazzi, C.; Stedeford, T.; Willing, A.; Freeman, T.B.; Saporta, S.; Janssen, W.; Patel, N.; et al. Adult Bone Marrow Stromal Cells Differentiate into Neural Cells In Vitro. Exp. Neurol. 2000, 164, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Woodbury, D.; Schwarz, E.J.; Prockop, D.J.; Black, I.B. Adult Rat and Human Bone Marrow Stromal Cells Differentiate into Neurons. J. Neurosci. Res. 2000, 61, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Choong, P.F.; Mok, P.L.; Cheong, S.K.; Leong, C.F.; Then, K.Y. Generating Neuron-like Cells from BM-Derived Mesenchymal Stromal Cells In Vitro. Cytotherapy 2007, 9, 170–183. [Google Scholar] [CrossRef]
- Lu, P.; Blesch, A.; Tuszynski, M.H. Induction of bone marrow stromal cells to neurons: Differentiation, transdifferentiation, or artifact? J. Neurosci. Res. 2004, 77, 174–191. [Google Scholar] [CrossRef]
- Neirinckx, V.; Coste, C.; Rogister, B.; Wislet-Gendebien, S. Concise Review: Adult Mesenchymal Stem Cells, Adult Neural Crest Stem Cells, and Therapy of Neurological Pathologies: A State of Play. STEM CELLS Transl. Med. 2013, 2, 284–296. [Google Scholar] [CrossRef]
- Liu, J.; Song, L.; Jiang, C.; Liu, Y.; George, J.; Ye, H.; Cui, Z. Electrophysiological Properties and Synaptic Function of Mesenchymal Stem Cells during Neurogenic Differentiation—A Mini-Review. Int. J. Artif. Organs. 2012, 35, 323–337. [Google Scholar] [CrossRef]
- Tondreau, T.; Lagneaux, L.; Dejeneffe, M.; Massy, M.; Mortier, C.; Delforge, A.; Bron, D. Bone Marrow–Derived Mesenchymal Stem Cells Already Express Specific Neural Proteins before Any Differentiation. Differentiation 2004, 72, 319–326. [Google Scholar] [CrossRef]
- Montzka, K.; Lassonczyk, N.; Tschöke, B.; Neuss, S.; Führmann, T.; Franzen, R.; Smeets, R.; Brook, G.A.; Wöltje, M. Neural Differentiation Potential of Human Bone Marrow-Derived Mesenchymal Stromal Cells: Misleading Marker Gene Expression. BMC Neurosci. 2009, 10, 16. [Google Scholar] [CrossRef]
- Kemp, K.; Gordon, D.; Wraith, D.C.; Mallam, E.; Hartfield, E.; Uney, J.; Wilkins, A.; Scolding, N. Fusion between Human Mesenchymal Stem Cells and Rodent Cerebellar Purkinje Cells. Neuropathol. Appl. Neurobiol. 2011, 37, 166–178. [Google Scholar] [CrossRef]
- Terada, N.; Hamazaki, T.; Oka, M.; Hoki, M.; Mastalerz, D.M.; Nakano, Y.; Meyer, E.M.; Morel, L.; Petersen, B.E.; Scott, E.W. Bone Marrow Cells Adopt the Phenotype of Other Cells by Spontaneous Cell Fusion. Nature 2002, 416, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yang, J.; Fang, J.; Zhou, Y.; Candi, E.; Wang, J.; Hua, D.; Shao, C.; Shi, Y. The Secretion Profile of Mesenchymal Stem Cells and Potential Applications in Treating Human Diseases. Sig. Transduct. Target Ther. 2022, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, S.H.; Levy, O.; Inamdar, M.S.; Karp, J.M. Harnessing the Mesenchymal Stem Cell Secretome for the Treatment of Cardiovascular Disease. Cell Stem Cell 2012, 10, 244–258. [Google Scholar] [CrossRef]
- Drago, D.; Cossetti, C.; Iraci, N.; Gaude, E.; Musco, G.; Bachi, A.; Pluchino, S. The Stem Cell Secretome and Its Role in Brain Repair. Biochimie 2013, 95, 2271–2285. [Google Scholar] [CrossRef]
- Bari, E.; Ferrarotti, I.; Torre, M.L.; Corsico, A.G.; Perteghella, S. Mesenchymal Stem/Stromal Cell Secretome for Lung Regeneration: The Long Way through “Pharmaceuticalization” for the Best Formulation. J. Control. Release 2019, 309, 11–24. [Google Scholar] [CrossRef]
- Damayanti, R.H.; Rusdiana, T.; Wathoni, N. Mesenchymal Stem Cell Secretome for Dermatology Application: A Review. CCID 2021, 14, 1401–1412. [Google Scholar] [CrossRef]
- Műzes, G.; Sipos, F. Mesenchymal Stem Cell-Derived Secretome: A Potential Therapeutic Option for Autoimmune and Immune-Mediated Inflammatory Diseases. Cells 2022, 11, 2300. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, H.J.; Oh, J.-H.; Park, H.-G.; Ra, J.C.; Chang, K.-A.; Suh, Y.-H. Therapeutic Potentials of Human Adipose-Derived Stem Cells on the Mouse Model of Parkinson’s Disease. Neurobiol. Aging 2015, 36, 2885–2892. [Google Scholar] [CrossRef]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Wrage, P.C.; Keefer, E.W.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Autologous Transplants of Adipose-Derived Adult Stromal (ADAS) Cells Afford Dopaminergic Neuroprotection in a Model of Parkinson’s Disease. Exp. Neurol. 2008, 210, 14–29. [Google Scholar] [CrossRef]
- Sadan, O.; Bahat-Stromza, M.; Barhum, Y.; Levy, Y.S.; Pisnevsky, A.; Peretz, H.; Ilan, A.B.; Bulvik, S.; Shemesh, N.; Krepel, D.; et al. Protective Effects of Neurotrophic Factor–Secreting Cells in a 6-OHDA Rat Model of Parkinson Disease. Stem Cells Dev. 2009, 18, 1179–1190. [Google Scholar] [CrossRef]
- Wang, F.; Yasuhara, T.; Shingo, T.; Kameda, M.; Tajiri, N.; Yuan, W.J.; Kondo, A.; Kadota, T.; Baba, T.; Tayra, J.T.; et al. Intravenous Administration of Mesenchymal Stem Cells Exerts Therapeutic Effects on Parkinsonian Model of Rats: Focusing on Neuroprotective Effects of Stromal Cell-Derived Factor-1α. BMC Neurosci. 2010, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.L.; Medicetty, S.; Bledsoe, A.R.; Rachakatla, R.S.; Choi, M.; Merchav, S.; Luo, Y.; Rao, M.S.; Velagaleti, G.; Troyer, D. Human Umbilical Cord Matrix Stem Cells: Preliminary Characterization and Effect of Transplantation in a Rodent Model of Parkinson’s Disease. Stem Cells 2006, 24, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.F.; Costa, R.O.; Pedro, J.R.; Aguiar, P.; Serra, S.C.; Teixeira, F.G.; Sousa, N.; Salgado, A.J.; Almeida, R.D. Mesenchymal Stem Cells Secretome-Induced Axonal Outgrowth Is Mediated by BDNF. Sci. Rep. 2017, 7, 4153. [Google Scholar] [CrossRef] [PubMed]
- Vilaça-Faria, H.; Salgado, A.J.; Teixeira, F.G. Mesenchymal Stem Cells-Derived Exosomes: A New Possible Therapeutic Strategy for Parkinson’s Disease? Cells 2019, 8, 118. [Google Scholar] [CrossRef]
- Belkozhayev, A.M.; Al-Yozbaki, M.; George, A.; Ye Niyazova, R.; Sharipov, K.O.; Byrne, L.J.; Wilson, C.M. Extracellular Vesicles, Stem Cells and the Role of miRNAs in Neurodegeneration. Curr. Neuropharmacol. 2022, 20, 1450–1478. [Google Scholar] [CrossRef]
- Turovsky, E.A.; Golovicheva, V.V.; Varlamova, E.G.; Danilina, T.I.; Goryunov, K.V.; Shevtsova, Y.A.; Pevzner, I.B.; Zorova, L.D.; Babenko, V.A.; Evtushenko, E.A.; et al. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Afford Neuroprotection by Modulating PI3K/AKT Pathway and Calcium Oscillations. Int. J. Biol. Sci. 2022, 18, 5345–5368. [Google Scholar] [CrossRef]
- Oh, S.H.; Kim, H.N.; Park, H.J.; Shin, J.Y.; Kim, D.Y.; Lee, P.H. The Cleavage Effect of Mesenchymal Stem Cell and Its Derived Matrix Metalloproteinase-2 on Extracellular α-Synuclein Aggregates in Parkinsonian Models. Stem Cells Transl. Med. 2017, 6, 949–961. [Google Scholar] [CrossRef]
- Oh, S.H.; Kim, H.N.; Park, H.J.; Shin, J.Y.; Bae, E.-J.; Sunwoo, M.K.; Lee, S.-J.; Lee, P.H. Mesenchymal Stem Cells Inhibit Transmission of α-Synuclein by Modulating Clathrin-Mediated Endocytosis in a Parkinsonian Model. Cell Rep. 2016, 14, 835–849. [Google Scholar] [CrossRef]
- Park, H.J.; Shin, J.Y.; Kim, H.N.; Oh, S.H.; Lee, P.H. Neuroprotective Effects of Mesenchymal Stem Cells through Autophagy Modulation in a Parkinsonian Model. Neurobiol. Aging 2014, 35, 1920–1928. [Google Scholar] [CrossRef]
- Shin, J.Y.; Lee, P.H. Mesenchymal Stem Cells Modulate Misfolded α-Synuclein in Parkinsonian Disorders: A Multitarget Disease-Modifying Strategy. Stem Cell Res. 2020, 47, 101908. [Google Scholar] [CrossRef]
- Ceccariglia, S.; Cargnoni, A.; Silini, A.R.; Parolini, O. Autophagy: A Potential Key Contributor to the Therapeutic Action of Mesenchymal Stem Cells. Autophagy 2020, 16, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling Integrators for Homeostasis Maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Shanware, N.P.; Bray, K.; Abraham, R.T. The PI3K, Metabolic, and Autophagy Networks: Interactive Partners in Cellular Health and Disease. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 89–106. [Google Scholar] [CrossRef]
- Domingues, A.V.; Pereira, I.M.; Vilaça-Faria, H.; Salgado, A.J.; Rodrigues, A.J.; Teixeira, F.G. Glial Cells in Parkinson´s Disease: Protective or Deleterious? Cell. Mol. Life Sci. 2020, 77, 5171–5188. [Google Scholar] [CrossRef]
- Fan, X.-L.; Zhang, Y.; Li, X.; Fu, Q.-L. Mechanisms Underlying the Protective Effects of Mesenchymal Stem Cell-Based Therapy. Cell. Mol. Life Sci. 2020, 77, 2771–2794. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Y.; Liu, S.; Xiao, J.; He, Y.; Shao, Z.; Zhang, Y.; Cai, X.; Xiong, L. Tunneling Nanotube-Mediated Mitochondrial Transfer Rescues Nucleus Pulposus Cells from Mitochondrial Dysfunction and Apoptosis. Oxidative Med. Cell. Longev. 2022, 2022, 3613319. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.A.; Fahey, M.J.; Pugliese, B.R.; Irwin, R.M.; Antonyak, M.A.; Delco, M.L. Human Mesenchymal Stromal Cells Release Functional Mitochondria in Extracellular Vesicles. Front. Bioeng. Biotechnol. 2022, 10, 870193. [Google Scholar] [CrossRef]
- Norris, R.P. Transfer of Mitochondria and Endosomes between Cells by Gap Junction Internalization. Traffic 2021, 22, 174–179. [Google Scholar] [CrossRef]
- Wada, K.-I.; Hosokawa, K.; Ito, Y.; Maeda, M. Quantitatively Controlled Intercellular Mitochondrial Transfer by Cell Fusion-Based Method Using a Microfluidic Device. In Mitochondrial Medicine: Volume 3: Manipulating Mitochondria and Disease—Specific Approaches; Weissig, V., Edeas, M., Eds.; Springer: New York, NY, USA, 2021; pp. 39–47. ISBN 978-1-0716-1270-5. [Google Scholar]
- Gomzikova, M.O.; James, V.; Rizvanov, A.A. Mitochondria Donation by Mesenchymal Stem Cells: Current Understanding and Mitochondria Transplantation Strategies. Front. Cell Dev. Biol. 2021, 9, 653322. [Google Scholar] [CrossRef]
- Qin, Y.; Jiang, X.; Yang, Q.; Zhao, J.; Zhou, Q.; Zhou, Y. The Functions, Methods, and Mobility of Mitochondrial Transfer Between Cells. Front. Oncol. 2021, 11, 672781. [Google Scholar] [CrossRef]
- Sanz-Ros, J.; Mas-Bargues, C.; Romero-García, N.; Huete-Acevedo, J.; Dromant, M.; Borrás, C. The Potential Use of Mitochondrial Extracellular Vesicles as Biomarkers or Therapeutical Tools. Int. J. Mol. Sci. 2023, 24, 7005. [Google Scholar] [CrossRef]
- Huang, P.-J.; Kuo, C.-C.; Lee, H.-C.; Shen, C.-I.; Cheng, F.-C.; Wu, S.-F.; Chang, J.-C.; Pan, H.-C.; Lin, S.-Z.; Liu, C.-S.; et al. Transferring Xenogenic Mitochondria Provides Neural Protection against Ischemic Stress in Ischemic Rat Brains. Cell Transpl. 2016, 25, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Marzookian, K.; Aliakbari, F.; Hourfar, H.; Sabouni, F.; Otzen, D.E.; Morshedi, D. The Neuroprotective Effect of Human Umbilical Cord MSCs-Derived Secretome against α-Synuclein Aggregates on the Blood-Brain Barrier. Int. J. Biol. Macromol. 2025, 304, 140387. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rodríguez, P.; Cuenca-Martagón, A.; Castillo-González, J.; Serrano-Martínez, I.; Luque, R.M.; Delgado, M.; González-Rey, E. Novel Therapeutic Opportunities for Neurodegenerative Diseases with Mesenchymal Stem Cells: The Focus on Modulating the Blood-Brain Barrier. Int. J. Mol. Sci. 2023, 24, 14117. [Google Scholar] [CrossRef]
- Tambe, P.; Undale, V.; Sanap, A.; Bhonde, R.; Mante, N. The Prospective Role of Mesenchymal Stem Cells in Parkinson’s Disease. Park. Relat. Disord. 2024, 127, 107087. [Google Scholar] [CrossRef]
- Lo, B.; Parham, L. Ethical Issues in Stem Cell Research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Markovic, B.S.; Gazdic, M.; Volarevic, A.; Jovicic, N.; Arsenijevic, N.; Armstrong, L.; Djonov, V.; Lako, M.; Stojkovic, M. Ethical and Safety Issues of Stem Cell-Based Therapy. Int. J. Med. Sci. 2018, 15, 36–45. [Google Scholar] [CrossRef]
- Gwam, C.; Mohammed, N.; Ma, X. Stem Cell Secretome, Regeneration, and Clinical Translation: A Narrative Review. Ann. Transl. Med. 2021, 9, 70. [Google Scholar] [CrossRef]
- Sajjad, U.; Ahmed, M.; Iqbal, M.Z.; Riaz, M.; Mustafa, M.; Biedermann, T.; Klar, A.S. Exploring Mesenchymal Stem Cells Homing Mechanisms and Improvement Strategies. Stem Cells Transl. Med. 2024, 13, 1161–1177. [Google Scholar] [CrossRef]
- Kumar, P.; Kandoi, S.; Misra, R.; Vijayalakshmi, S.; Rajagopal, K.; Verma, R.S. The Mesenchymal Stem Cell Secretome: A New Paradigm towards Cell-Free Therapeutic Mode in Regenerative Medicine. Cytokine Growth Factor Rev. 2019, 46, 1–9. [Google Scholar] [CrossRef]
- Hezam, K.; Fu, E.; Zhang, J.; Li, Z. Therapeutic Trends of Priming Mesenchymal Stem Cells: A Bibliometric Analysis. Biochem. Biophys. Rep. 2024, 38, 101708. [Google Scholar] [CrossRef] [PubMed]
- Robb, K.P.; Galipeau, J.; Shi, Y.; Schuster, M.; Martin, I.; Viswanathan, S. Failure to Launch Commercially-Approved Mesenchymal Stromal Cell Therapies: What’s the Path Forward? Proceedings of the International Society for Cell & Gene Therapy (ISCT) Annual Meeting Roundtable Held in May 2023, Palais Des Congrès de Paris, Organized by the ISCT MSC Scientific Committee. Cytotherapy 2024, 26, 413–417. [Google Scholar] [CrossRef]
- de Noronha, N.C.; Mizukami, A.; Caliári-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C.R. Priming Approaches to Improve the Efficacy of Mesenchymal Stromal Cell-Based Therapies. Stem Cell Res. Ther. 2019, 10, 131. [Google Scholar] [CrossRef]
- Beegle, J.; Lakatos, K.; Kalomoiris, S.; Stewart, H.; Isseroff, R.R.; Nolta, J.A.; Fierro, F.A. Hypoxic Preconditioning of Mesenchymal Stromal Cells Induces Metabolic Changes, Enhances Survival, and Promotes Cell Retention In Vivo. Stem Cells 2015, 33, 1818–1828. [Google Scholar] [CrossRef]
- Fuentes, P.; Torres, M.J.; Arancibia, R.; Aulestia, F.; Vergara, M.; Carrión, F.; Osses, N.; Altamirano, C. Dynamic Culture of Mesenchymal Stromal/Stem Cell Spheroids and Secretion of Paracrine Factors. Front. Bioeng. Biotechnol. 2022, 10, 916229. [Google Scholar] [CrossRef]
- Bianconi, S.; Oliveira, K.M.C.; Klein, K.-L.; Wolf, J.; Schaible, A.; Schröder, K.; Barker, J.; Marzi, I.; Leppik, L.; Henrich, D. Pretreatment of Mesenchymal Stem Cells with Electrical Stimulation as a Strategy to Improve Bone Tissue Engineering Outcomes. Cells 2023, 12, 2151. [Google Scholar] [CrossRef]
- Al-Azab, M.; Idiiatullina, E.; Safi, M.; Hezam, K. Enhancers of Mesenchymal Stem Cell Stemness and Therapeutic Potency. Biomed. Pharmacother. 2023, 162, 114356. [Google Scholar] [CrossRef] [PubMed]
- Al-Azab, M.; Safi, M.; Idiiatullina, E.; Al-Shaebi, F.; Zaky, M.Y. Aging of Mesenchymal Stem Cell: Machinery, Markers, and Strategies of Fighting. Cell. Mol. Biol. Lett. 2022, 27, 69. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Sasso, J.M.; Wang, X.; Zhou, Q.A. Antiaging Strategies and Remedies: A Landscape of Research Progress and Promise. ACS Chem. Neurosci. 2024, 15, 408–446. [Google Scholar] [CrossRef]
- Flanagan, E.W.; Most, J.; Mey, J.T.; Redman, L.M. Calorie Restriction and Aging in Humans. Annu. Rev. Nutr. 2020, 40, 105–133. [Google Scholar] [CrossRef]
- Trisal, A.; Singh, A.K. Clinical Insights on Caloric Restriction Mimetics for Mitigating Brain Aging and Related Neurodegeneration. Cell Mol. Neurobiol. 2024, 44, 67. [Google Scholar] [CrossRef] [PubMed]
- Trisal, A.; Singh, A.K. Mechanisms and Early Efficacy Data of Caloric Restriction and Caloric Restriction Mimetics in Neurodegenerative Disease. Neuroscience 2025, 567, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.; Chang, S.-H.; Wu, C.-Y.; Peng, H.-H.; Hwang, T.-L.; Ko, Y.-F.; Young, J.D.; Ojcius, D.M. Recent Advances in the Field of Caloric Restriction Mimetics and Anti-Aging Molecules. Ageing Res. Rev. 2021, 66, 101240. [Google Scholar] [CrossRef] [PubMed]
- Marote, A.; Santos, D.; Mendes-Pinheiro, B.; Serre-Miranda, C.; Anjo, S.I.; Vieira, J.; Ferreira-Antunes, F.; Correia, J.S.; Borges-Pereira, C.; Pinho, A.G.; et al. Cellular Aging Secretes: A Comparison of Bone-Marrow-Derived and Induced Mesenchymal Stem Cells and Their Secretome Over Long-Term Culture. Stem Cell Rev. Rep. 2023, 19, 248–263. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Salgado, A.J. Mesenchymal Stem Cells Secretome: Current Trends and Future Challenges. Neural. Regen. Res. 2019, 15, 75–77. [Google Scholar] [CrossRef]
- Barata-Antunes, S.; Sousa, R.A.; Salgado, A.J.; Silva, B.F.B. Whole Secretome of Mesenchymal Stem Cells Is Fully Incorporated in Lipid Bicontinuous Cubic Phases. Front. Med. Eng. 2025, 3, 1397406. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, G.; Alimujiang, A.; Xie, H.; Yang, W.; Yin, F.; Huang, D. Protective Effects of Querectin against MPP+-Induced Dopaminergic Neurons Injury via the Nrf2 Signaling Pathway. FBL 2023, 28, 42. [Google Scholar] [CrossRef]
- Wang, W.-W.; Han, R.; He, H.-J.; Li, J.; Chen, S.-Y.; Gu, Y.; Xie, C. Administration of Quercetin Improves Mitochondria Quality Control and Protects the Neurons in 6-OHDA-Lesioned Parkinson’s Disease Models. Aging 2021, 13, 11738–11751. [Google Scholar] [CrossRef]
- Bao, D.; Wang, J.; Pang, X.; Liu, H. Protective Effect of Quercetin against Oxidative Stress-Induced Cytotoxicity in Rat Pheochromocytoma (PC-12) Cells. Molecules 2017, 22, 1122. [Google Scholar] [CrossRef]
- Ay, M.; Luo, J.; Langley, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Molecular Mechanisms Underlying Protective Effects of Quercetin against Mitochondrial Dysfunction and Progressive Dopaminergic Neurodegeneration in Cell Culture and MitoPark Transgenic Mouse Models of Parkinson’s Disease. J. Neurochem. 2017, 141, 766–782. [Google Scholar] [CrossRef]
- Sharma, S.; Raj, K.; Singh, S. Neuroprotective Effect of Quercetin in Combination with Piperine Against Rotenone- and Iron Supplement–Induced Parkinson’s Disease in Experimental Rats. Neurotox Res. 2020, 37, 198–209. [Google Scholar] [CrossRef]
- Büttner, S.; Broeskamp, F.; Sommer, C.; Markaki, M.; Habernig, L.; Alavian-Ghavanini, A.; Carmona-Gutierrez, D.; Eisenberg, T.; Michael, E.; Kroemer, G.; et al. Spermidine Protects against α-Synuclein Neurotoxicity. Cell Cycle 2014, 13, 3903–3908. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, M.; Dai, Y.; Sun, Y.; Aman, Y.; Xu, Y.; Yu, P.; Zheng, Y.; Yang, J.; Zhu, X. Spermidine Inhibits Neurodegeneration and Delays Aging via the PINK1-PDR1-Dependent Mitophagy Pathway in C. Elegans. Aging 2020, 12, 16852–16866. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Jiao, Y.; Wei, W.; Yan, A. Spermidine Inhibits M1 Microglia Polarization in a Mouse Model of Parkinson’s Disease and BV2 Cells via NF-κB/STAT-1 Pathway. Brain Behav. 2025, 15, e70410. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kumar, P.; Deshmukh, R. Neuroprotective Potential of Spermidine against Rotenone Induced Parkinson’s Disease in Rats. Neurochem. Int. 2018, 116, 104–111. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of Resveratrol on Mitochondrial Function: Implications in Parkin-Associated Familiar Parkinson’s Disease. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, T.; Dong, S.-Y.; Guo, Y.-J.; Jankovic, J.; Xu, H.; Wu, Y.-C. Rotenone Affects P53 Transcriptional Activity and Apoptosis via Targeting SIRT1 and H3K9 Acetylation in SH-SY5Y Cells. J. Neurochem. 2015, 134, 668–676. [Google Scholar] [CrossRef]
- Abolaji, A.O.; Adedara, A.O.; Adie, M.A.; Vicente-Crespo, M.; Farombi, E.O. Resveratrol Prolongs Lifespan and Improves 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Oxidative Damage and Behavioural Deficits in Drosophila Melanogaster. Biochem. Biophys. Res. Commun. 2018, 503, 1042–1048. [Google Scholar] [CrossRef]
- Huang, N.; Zhang, Y.; Chen, M.; Jin, H.; Nie, J.; Luo, Y.; Zhou, S.; Shi, J.; Jin, F. Resveratrol Delays 6-Hydroxydopamine-Induced Apoptosis by Activating the PI3K/Akt Signaling Pathway. Exp. Gerontol. 2019, 124, 110653. [Google Scholar] [CrossRef]
- Katila, N.; Bhurtel, S.; Park, P.-H.; Choi, D.-Y. Metformin Attenuates Rotenone-Induced Oxidative Stress and Mitochondrial Damage via the AKT/Nrf2 Pathway. Neurochem. Int. 2021, 148, 105120. [Google Scholar] [CrossRef]
- Ay, M.; Charli, A.; Langley, M.; Jang, A.; Padhi, P.; Jin, H.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.; Kanthasamy, A.G. Mito-Metformin Protects against Mitochondrial Dysfunction and Dopaminergic Neuronal Degeneration by Activating Upstream PKD1 Signaling in Cell Culture and MitoPark Animal Models of Parkinson’s Disease. Front. Neurosci. 2024, 18, 1356703. [Google Scholar] [CrossRef]
- Saewanee, N.; Praputpittaya, T.; Malaiwong, N.; Chalorak, P.; Meemon, K. Neuroprotective Effect of Metformin on Dopaminergic Neurodegeneration and α-Synuclein Aggregation in C. Elegans Model of Parkinson’s Disease. Neurosci. Res. 2021, 162, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin Rescues Parkinson’s Disease Phenotypes Caused by Hyperactive Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.-S.; Choi, D.-Y. Metformin Lowers α-Synuclein Phosphorylation and Upregulates Neurotrophic Factor in the MPTP Mouse Model of Parkinson’s Disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Ivan, A.; Lukinich-Gruia, A.T.; Cristea, I.-M.; Pricop, M.-A.; Calma, C.L.; Simina, A.-G.; Tatu, C.A.; Galuscan, A.; Păunescu, V. Quercetin and Mesenchymal Stem Cell Metabolism: A Comparative Analysis of Young and Senescent States. Molecules 2024, 29, 5755. [Google Scholar] [CrossRef]
- Chen, G.; Ye, Y.; Cheng, M.; Tao, Y.; Zhang, K.; Huang, Q.; Deng, J.; Yao, D.; Lu, C.; Huang, Y. Quercetin Combined with Human Umbilical Cord Mesenchymal Stem Cells Regulated Tumour Necrosis Factor-α/Interferon-γ-Stimulated Peripheral Blood Mononuclear Cells via Activation of Toll-Like Receptor 3 Signalling. Front. Pharmacol. 2020, 11, 499. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, W.; Su, J.; Zhou, B.; Han, Q. Spermidine Retarded the Senescence of Multipotent Mesenchymal Stromal Cells In Vitro and In Vivo through SIRT3-Mediated Antioxidation. Stem Cells Int. 2023, 2023, 9672658. [Google Scholar] [CrossRef]
- Yoon, D.S.; Choi, Y.; Choi, S.M.; Park, K.H.; Lee, J.W. Different Effects of Resveratrol on Early and Late Passage Mesenchymal Stem Cells through β-Catenin Regulation. Biochem. Biophys. Res. Commun. 2015, 467, 1026–1032. [Google Scholar] [CrossRef]
- Wang, X.; Ma, S.; Meng, N.; Yao, N.; Zhang, K.; Li, Q.; Zhang, Y.; Xing, Q.; Han, K.; Song, J.; et al. Resveratrol Exerts Dosage-Dependent Effects on the Self-Renewal and Neural Differentiation of hUC-MSCs. Mol. Cells 2016, 39, 418–425. [Google Scholar] [CrossRef]
- Geng, Y.-W.; Zhang, Z.; Liu, M.-Y.; Hu, W.-P. Differentiation of Human Dental Pulp Stem Cells into Neuronal by Resveratrol. Cell Biol. Int. 2017, 41, 1391–1398. [Google Scholar] [CrossRef]
- Acar, M.B.; Ayaz-Güner, Ş.; Gunaydin, Z.; Karakukcu, M.; Peluso, G.; Di Bernardo, G.; Özcan, S.; Galderisi, U. Proteomic and Biological Analysis of the Effects of Metformin Senomorphics on the Mesenchymal Stromal Cells. Front. Bioeng. Biotechnol. 2021, 9, 730813. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Li, S.; Lu, S.; Liu, H.; Li, G.; Ma, L.; Luo, R.; Ke, W.; Wang, B.; Xiang, Q.; et al. Metformin Facilitates Mesenchymal Stem Cell-Derived Extracellular Nanovesicles Release and Optimizes Therapeutic Efficacy in Intervertebral Disc Degeneration. Biomaterials 2021, 274, 120850. [Google Scholar] [CrossRef]
- Ahn, M.-J.; Cho, G.-W. Metformin Promotes Neuronal Differentiation and Neurite Outgrowth through AMPK Activation in Human Bone Marrow–Mesenchymal Stem Cells. Biotechnol. Appl. Biochem. 2017, 64, 836–842. [Google Scholar] [CrossRef]
- Mlcek, J.; Jurikova, T.; Skrovankova, S.; Sochor, J. Quercetin and Its Anti-Allergic Immune Response. Molecules 2016, 21, 623. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef]
- Hu, Y.; Gui, Z.; Zhou, Y.; Xia, L.; Lin, K.; Xu, Y. Quercetin Alleviates Rat Osteoarthritis by Inhibiting Inflammation and Apoptosis of Chondrocytes, Modulating Synovial Macrophages Polarization to M2 Macrophages. Free Radic. Biol. Med. 2019, 145, 146–160. [Google Scholar] [CrossRef]
- Grewal, A.K.; Singh, T.G.; Sharma, D.; Sharma, V.; Singh, M.; Rahman, M.d.H.; Najda, A.; Walasek-Janusz, M.; Kamel, M.; Albadrani, G.M.; et al. Mechanistic Insights and Perspectives Involved in Neuroprotective Action of Quercetin. Biomed. Pharmacother. 2021, 140, 111729. [Google Scholar] [CrossRef]
- Cui, Z.; Zhao, X.; Amevor, F.K.; Du, X.; Wang, Y.; Li, D.; Shu, G.; Tian, Y.; Zhao, X. Therapeutic Application of Quercetin in Aging-Related Diseases: SIRT1 as a Potential Mechanism. Front. Immunol. 2022, 13, 943321. [Google Scholar] [CrossRef]
- Islam, R.; Al-Imran, I.K.; Zehravi, M.; Sweilam, S.H.; Mortuza, M.R.; Gupta, J.K.; Shanmugarajan, T.S.; Devi, K.; Tummala, T.; Alshehri, M.A.; et al. Targeting Signaling Pathways in Neurodegenerative Diseases: Quercetin’s Cellular and Molecular Mechanisms for Neuroprotection. Anim. Models Exp. Med. 2025. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The Pro- and Anti-Inflammatory Properties of the Cytokine Interleukin-6. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in Health and Disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Wilson, J.; Kolathur, K.K.; Mudgal, J.; Lewis, S.A.; Arora, D.; Nampoothiri, M. Spermidine as an Epigenetic Regulator of Autophagy in Neurodegenerative Disorders. Eur. J. Pharmacol. 2024, 979, 176823. [Google Scholar] [CrossRef]
- Jiang, D.; Guo, Y.; Niu, C.; Long, S.; Jiang, Y.; Wang, Z.; Wang, X.; Sun, Q.; Ling, W.; An, X.; et al. Exploration of the Antioxidant Effect of Spermidine on the Ovary and Screening and Identification of Differentially Expressed Proteins. Int. J. Mol. Sci. 2023, 24, 5793. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Park, H.Y. Anti-Inflammatory Effects of Spermidine in Lipopolysaccharide-Stimulated BV2 Microglial Cells. J. Biomed. Sci. 2012, 19, 31. [Google Scholar] [CrossRef]
- Ni, Y.-Q.; Liu, Y.-S. New Insights into the Roles and Mechanisms of Spermidine in Aging and Age-Related Diseases. Aging Dis. 2021, 12, 1948–1963. [Google Scholar] [CrossRef]
- Saiki, S.; Sasazawa, Y.; Fujimaki, M.; Kamagata, K.; Kaga, N.; Taka, H.; Li, Y.; Souma, S.; Hatano, T.; Imamichi, Y.; et al. A Metabolic Profile of Polyamines in Parkinson Disease: A Promising Biomarker. Ann. Neurol. 2019, 86, 251–263. [Google Scholar] [CrossRef]
- Cressman, A.; Morales, D.; Zhang, Z.; Le, B.; Foley, J.; Murray-Stewart, T.; Genetos, D.C.; Fierro, F.A. Effects of Spermine Synthase Deficiency in Mesenchymal Stromal Cells Are Rescued by Upstream Inhibition of Ornithine Decarboxylase. Int. J. Mol. Sci. 2024, 25, 2463. [Google Scholar] [CrossRef]
- Takaoka, M. Resveratrol, a New Phenolic Compound, from Veratrum Grandiflorum. Nippon Kagaku Kaishi 1939, 60, 1090–1100. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef]
- Campos, J.; Silva, N.A.; Salgado, A.J. Nutritional Interventions for Spinal Cord Injury: Preclinical Efficacy and Molecular Mechanisms. Nutr. Rev. 2022, 80, 1206–1221. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.-D.; Cheng, J.; Li, J.; Wu, S.-X.; Xiong, R.-G.; Huang, S.-Y.; Cheung, P.C.-K.; Li, H.-B. Resveratrol and Its Analogues: Anti-Ageing Effects and Underlying Mechanisms. In Biochemistry and Cell Biology of Ageing: Part V, Anti-Ageing Interventions; Korolchuk, V.I., Harris, J.R., Eds.; Subcellular Biochemistry; Springer: Cham, Switzerland, 2024; pp. 183–203. ISBN 978-3-031-66768-8. [Google Scholar]
- Handschin, C. Caloric Restriction and Exercise “mimetics’’: Ready for Prime Time? Pharmacol. Res. 2016, 103, 158–166. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small Molecule Activators of Sirtuins Extend Saccharomyces Cerevisiae Lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.; Kim, S.K.; Berdichevsky, A.; Guarente, L. A Role for SIR-2.1 Regulation of ER Stress Response Genes in Determining C. Elegans Life Span. Dev. Cell 2005, 9, 605–615. [Google Scholar] [CrossRef]
- Bauer, J.H.; Goupil, S.; Garber, G.B.; Helfand, S.L. An Accelerated Assay for the Identification of Lifespan-Extending Interventions in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 2004, 101, 12980–12985. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, D.R.; Terzibasi, E.; Genade, T.; Cattaneo, A.; Domenici, L.; Cellerino, A. Resveratrol Prolongs Lifespan and Retards the Onset of Age-Related Markers in a Short-Lived Vertebrate. Curr. Biol. 2006, 16, 296–300. [Google Scholar] [CrossRef]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Lifespan. Cell Metab. 2008, 8, 157–168. [Google Scholar] [CrossRef]
- Bitterman, J.L.; Chung, J.H. Metabolic Effects of Resveratrol: Addressing the Controversies. Cell. Mol. Life Sci. 2015, 72, 1473–1488. [Google Scholar] [CrossRef]
- Hu, C.; Li, L. The Application of Resveratrol to Mesenchymal Stromal Cell-Based Regenerative Medicine. Stem Cell Res. Ther. 2019, 10, 307. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The Mechanisms of Action of Metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Mohammed, I.; Hollenberg, M.D.; Ding, H.; Triggle, C.R. A Critical Review of the Evidence That Metformin Is a Putative Anti-Aging Drug That Enhances Healthspan and Extends Lifespan. Front. Endocrinol. 2021, 12, 718942. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gan, D.; Lin, S.; Zhong, Y.; Chen, M.; Zou, X.; Shao, Z.; Xiao, G. Metformin in Aging and Aging-Related Diseases: Clinical Applications and Relevant Mechanisms. Theranostics 2022, 12, 2722–2740. [Google Scholar] [CrossRef]
- Chen, J.; Ou, Y.; Li, Y.; Hu, S.; Shao, L.-W.; Liu, Y. Metformin Extends C. Elegans Lifespan through Lysosomal Pathway. eLife 2017, 6, e31268. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin Improves Healthspan and Lifespan in Mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Agostini, F.; Masato, A.; Bubacco, L.; Bisaglia, M. Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges. Int. J. Mol. Sci. 2021, 23, 398. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Zimprich, A.; Carvajal Berrio, D.A.; Schindler, K.M.; Maurer, B.; Schulte, C.; Bus, C.; Hauser, A.-K.; Kübler, M.; Lewin, R.; et al. Metformin Reverses TRAP1 Mutation-Associated Alterations in Mitochondrial Function in Parkinson’s Disease. Brain 2017, 140, 2444–2459. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tian, T.; Zhou, H.; Jiang, S.-Y.; Jiao, Y.-Y.; Zhu, Z.; Xia, J.; Ma, J.-H.; Du, R.-H. Metformin Normalizes Mitochondrial Function to Delay Astrocyte Senescence in a Mouse Model of Parkinson’s Disease through Mfn2-cGAS Signaling. J. Neuroinflammation 2024, 21, 81. [Google Scholar] [CrossRef]
- Campos, J.; Sampaio-Marques, B.; Santos, D.; Barata-Antunes, S.; Ribeiro, M.; Serra, S.C.; Pinho, T.S.; Canto-Gomes, J.; Marote, A.; Cortez, M.; et al. Lipid Priming of Adipose Mesenchymal Stromal Cells with Docosahexaenoic Acid: Impact on Cell Differentiation, Senescence and the Secretome Neuroregulatory Profile. Tissue Eng. Regen. Med. 2025, 22, 113–128. [Google Scholar] [CrossRef]
- Casado-Díaz, A.; Anter, J.; Dorado, G.; Quesada-Gómez, J.M. Effects of Quercetin, a Natural Phenolic Compound, in the Differentiation of Human Mesenchymal Stem Cells (MSC) into Adipocytes and Osteoblasts. J. Nutr. Biochem. 2016, 32, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Z.; Liu, L.; Zhang, P.; Wang, Y.; Ding, G. Metformin-Mediated Effects on Mesenchymal Stem Cells and Mechanisms: Proliferation, Differentiation and Aging. Front. Pharmacol. 2024, 15, 1465697. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.A.; Tatarskiy, V.; Kalinina, E. Synthetic Pathways and the Therapeutic Potential of Quercetin and Curcumin. Int. J. Mol. Sci. 2022, 23, 14413. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| CRM | Type of Study | Model System | PD Model | Effects |
|---|---|---|---|---|
| Quercetin | In vitro | SH-SY5Y | MPTP | Reduced apoptosis, MDA, NCOA4; Upregulated GPX4, Nrf2 and SLC7A11 [158]. |
| PC12 | 6-OHDA | Enhanced PINK1/Parkin expression; Prevented neuronal loss [159]. | ||
| H2O2 | Downregulated Bax and caspase-3; Upregulated Bcl-2; Reduced apoptosis [160]. | |||
| MN9D | - | Activated PGC-1α, PKD1, Akt, and CREB; Upregulated BDNF; Increased basal OCR and ATP-linked respiration [161]. | ||
| 6-OHDA | Toxin resistance [161]. | |||
| In vivo | C. elegans | Transgenic neuronal mt-Rosella | Induced mitophagy; Reduced oxidative stress, mitochondrial damage, and asyn accumulation [159]. | |
| Rat | 6-OHDA | Enhanced PINK1/Parkin expression; Decreased neuronal loss and behavioral deficits [159]. | ||
| Rotenone | Reduced TNF-α, IL-1β, and IL-6; Attenuated motor deficits; Improved biochemical and neurotransmitter alterations [162]. | |||
| Spermidine | In vivo | C. elegans | Transgenic asyn expression | Decreased neuronal degeneration. (UA44 strain) [163]. |
| Increased mean lifespan, locomotor capacity, and chemotaxis-based cognitive ability; Reduced asyn; Upregulated bec-1; Downregulated sqst-1. (NL5901 strain) [164]. | ||||
| D. melanogaster | Transgenic asyn expression exposed to manganese | Increased mean lifespan and Atg8a-II levels; Decreased motor deficits [163]. | ||
| Mouse | MPTP | Reduced IL-1β, IL-6, TNF-α, and M1 microglial markers (CD16, CD32, CD86); Increased M2 microglial markers (Arg-1, CD206, Ym1), STAT6 activation, behavioral scores, TH-positive neurons, and TH expression in SN; Decreased activation of NF-κB p65, STAT1, and p38 MAPK [165]. | ||
| Rat | Rotenone | Decreased weight loss, motor dysfunction, and MDA, nitrite, TNF-α, IL-1β, IL-6, and glutamate levels; Increased GSH, GABA, and norepinephrine, dopamine, serotonin, and respective metabolites [166]. | ||
| Resveratrol | In vitro | Fibroblasts (early-onset patients) | PARK2 heterozygous mutations | Increased OCR, ATP production, complex I and citrate synthase activity, relative mitochondrial DNA content, AMPK activation, NAD+/NADH ratio, PGC-1α, mitochondrial transcriptional factor A, cytochrome c, cyclooxygenase 1, SOD2, CAT, SIRT1, and LC3-independent macroautophagy; Decreased mitochondrial ROS and acetylated-H3 [167]. |
| SH-SY5Y | Rotenone | Decreased cell death, Bax, apoptotic cells, P53, cells in G0/G1 phase and acetylated H3K9; Increased Bcl-2, AMPK activation, SIRT1, cells in G2/M phase and tri-methylated H3K9 [168]. | ||
| In vivo | D. melanogaster | MPTP | Increased climbing rate, acetylcholinesterase, CAT and GSH activity, emergence of flies, and cell viability; Reduced H2O2 and NO [169]. | |
| Rat | 6-OHDA | Improved motor function and body weight; Increased Bcl-2, PI3K-110α, p-Akt Ser473, and TH-positive cells in SN; Decreased Bax and active caspase-3; Delayed apoptosis [170]. | ||
| Metformin | In vitro | SH-SY5Y | Rotenone | Improved cell viability; Inhibited caspase-3 activation; Reduced intracellular and mitochondrial ROS; Increased GSH activity, cytosolic and mitochondrial SOD, PGC-1α, and Nrf2 levels [171]. |
| N27 | MPTP | Increased mitochondrial bioenergetics capacity, TFAM, and mitochondrial DNA content. Reduced mitochondrial fragmentation and dopaminergic neuronal degeneration [172]. | ||
| In vivo | C. elegans | 6-OHDA | Reduced neurodegeneration and asyn aggregation; Restored food-sensing behavior; Upregulated cat-2 and sod-3 gene expression [173]. | |
| b-cat1 knockdown | Reduced mitochondrial respiration to control levels. Improved motor function and neuronal viability [174]. | |||
| Mouse | MPTP | Improved motor function; Increased TH-positive neurons, striatal dopamine, methylated PP2A levels, and BDNF expression; Reduced microglia activation, asyn accumulation, and mTOR signaling; Activated AMPK, Akt, and ERK [175]. |
| CRM | MSCs Cell Source | Condition | Effects |
|---|---|---|---|
| Quercetin | SHEDs | Early Passages (Passage 5) | Increased metabolic activity, mitochondrial respiration, and levels of lauric and myristic acids; reduced levels of oleic acid [176]. |
| Later Passages (Passage 16) | Preserved mitochondrial function; increased levels of stearic acid; modulated expression of oxidative stress genes and sirtuins [176]. | ||
| hUC-MSCs | - (Passage 3–5) | Reduced activation of Akt and IκB; increased expression of TLR-3; enhanced production of NO, IDO, and IL-6 [177]. | |
| Spermidine | hUC-MSCs | Later Passages (Passage 26) | Increased proliferation, Ki67, SIRT3; Reduced SA-β-gal, p-P53, P53, P21 and ROS; Improved mitochondrial function; Maintained adipogenic/osteogenic potential; SIRT3 knockout abolished these benefits—indicating SIRT3-dependency [178]. |
| Resveratrol | hBM-MSCs | Early Passages (Passage 1–3) | Reduced ERK activation [179]. |
| Late Passages (Passage 9–10)/SIRT1 knockdown | Increased ERK, β-catenin, ROS, and senescence; Indicates SIRT1-dependent dual effect [179]. | ||
| hUC-MSCs | - (Passage 4) | Increased SIRT1, βIII-tubulin, NSE, Ngn2 and Mash1; Decreased P53, P16, Nestin and Ngn1; Induced morphological changes [180]. | |
| DPSCs | - (Passage 3–5) | Increased Nestin, Musashi, and NF-M [181]. | |
| Metformin | ASCs | - (Passage 3) | Supported long-term viability; Reduced senescence, apoptosis, and β-gal; Increased DNA synthesis, SOD1/2, CAT, GLRX, GST, and secretion of molecules involved in α-adrenergic signaling, detox, and aspartate degradation [182]. |
| hBM-MSCs | - (Passage 2–3) | Increased EV production via autophagy-related pathways and secretome functional relevance [183]. | |
| - (Passage 7) | Increased βIII-tubulin, MAP2, and key neurogenic signaling [184]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carneiro-Pereira, B.; Ferreira-Antunes, F.; Campos, J.; Salgado, A.J.; Sampaio-Marques, B. Caloric Restriction Mimetics as Priming Agents of Mesenchymal Stem Cells Secretome to Enhance Regenerative Responses to Parkinson’s Disease. Molecules 2025, 30, 2260. https://doi.org/10.3390/molecules30112260
Carneiro-Pereira B, Ferreira-Antunes F, Campos J, Salgado AJ, Sampaio-Marques B. Caloric Restriction Mimetics as Priming Agents of Mesenchymal Stem Cells Secretome to Enhance Regenerative Responses to Parkinson’s Disease. Molecules. 2025; 30(11):2260. https://doi.org/10.3390/molecules30112260
Chicago/Turabian StyleCarneiro-Pereira, Bárbara, Filipa Ferreira-Antunes, Jonas Campos, António J. Salgado, and Belém Sampaio-Marques. 2025. "Caloric Restriction Mimetics as Priming Agents of Mesenchymal Stem Cells Secretome to Enhance Regenerative Responses to Parkinson’s Disease" Molecules 30, no. 11: 2260. https://doi.org/10.3390/molecules30112260
APA StyleCarneiro-Pereira, B., Ferreira-Antunes, F., Campos, J., Salgado, A. J., & Sampaio-Marques, B. (2025). Caloric Restriction Mimetics as Priming Agents of Mesenchymal Stem Cells Secretome to Enhance Regenerative Responses to Parkinson’s Disease. Molecules, 30(11), 2260. https://doi.org/10.3390/molecules30112260