
Phytyl Phenolipids: Structurally Modified Antioxidants with Superior Lipid Membrane Interaction

 , ,
, ,  ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
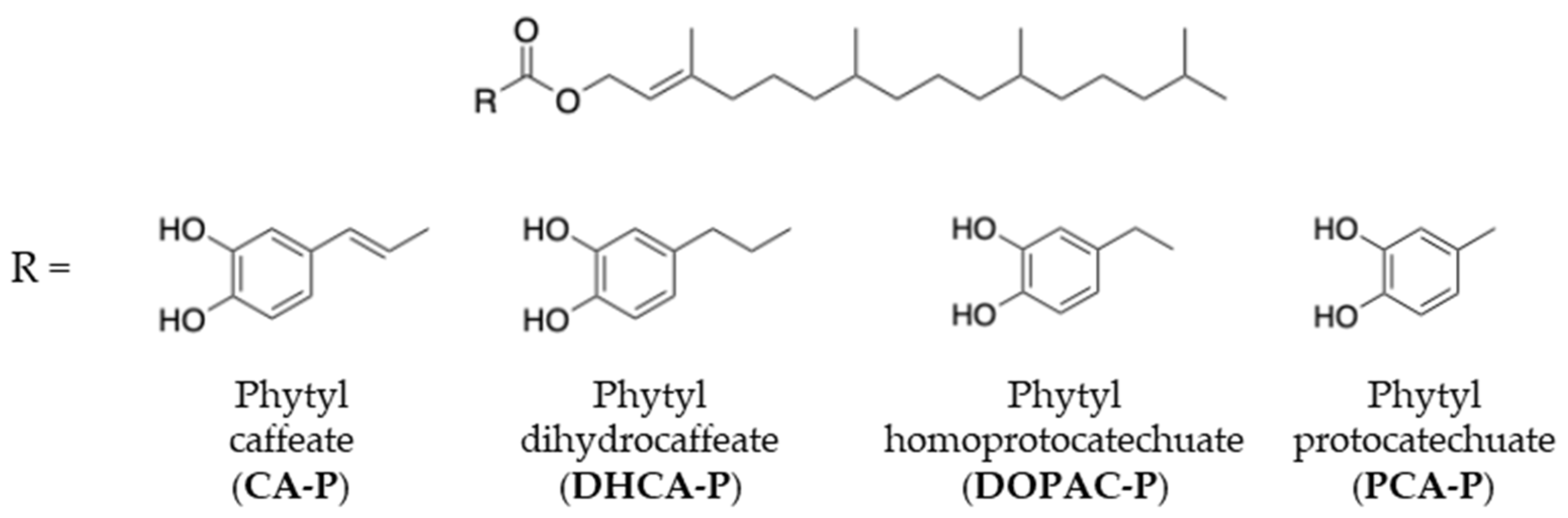
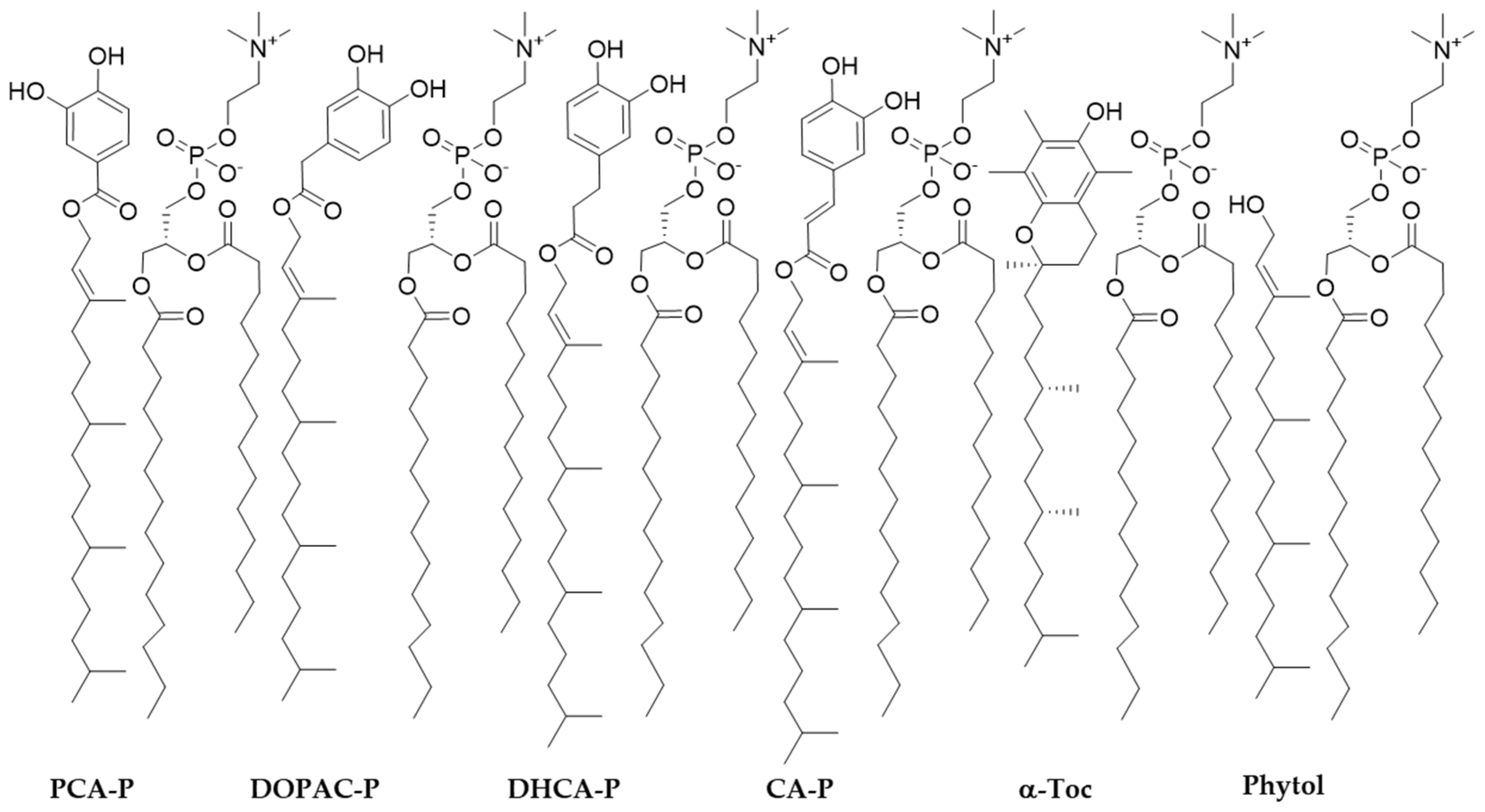
2.1. Synthesis of Phytyl Phenolipids
2.2. Evaluation of Free Radical Scavenging Capacity of Phytyl Phenolipids
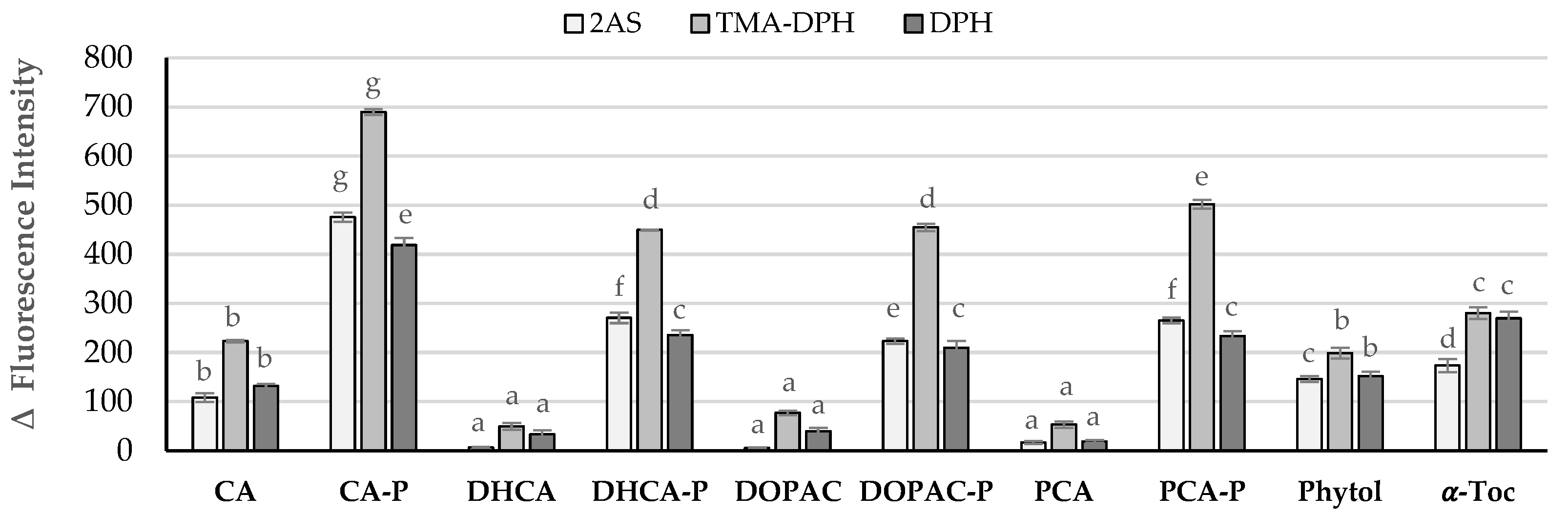
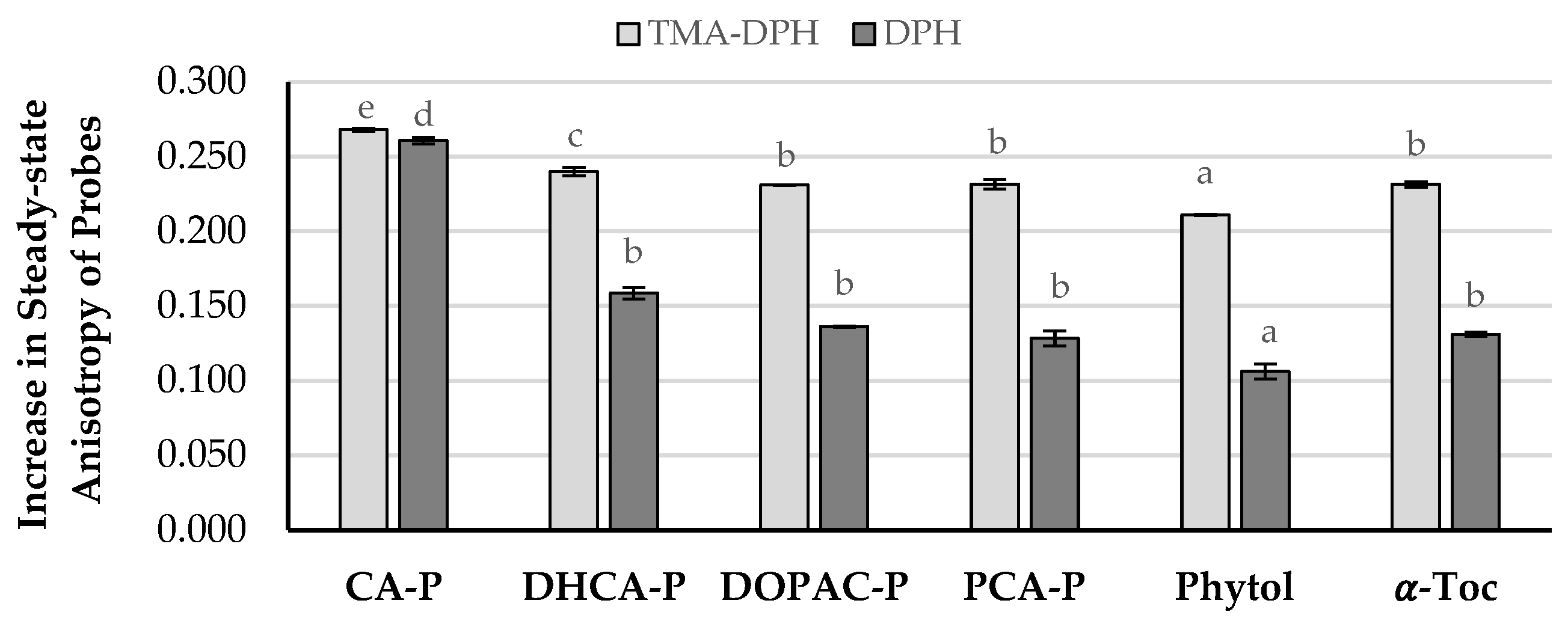
2.3. Interaction of Compounds with Liposomal Membranes
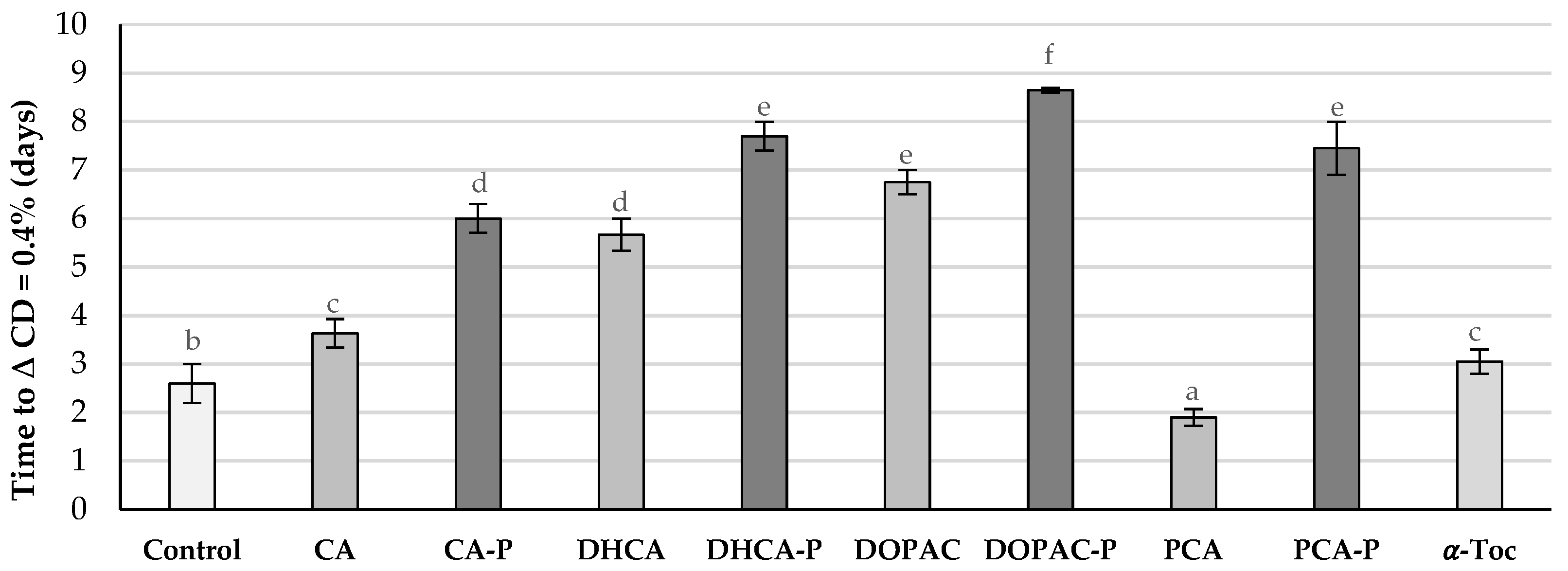
2.4. Antioxidant Capacity of Phytyl Phenolipids in Liposomal Systems
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Phytyl Esters of Polyphenolic Acids
3.2.1. Synthesis of CA-P
3.2.2. Synthesis of DHCA-P and DOPAC-P
3.2.3. Synthesis of PCA-P
3.3. Determination of miLog p Values
3.4. DPPH Radical Scavenging Capacity
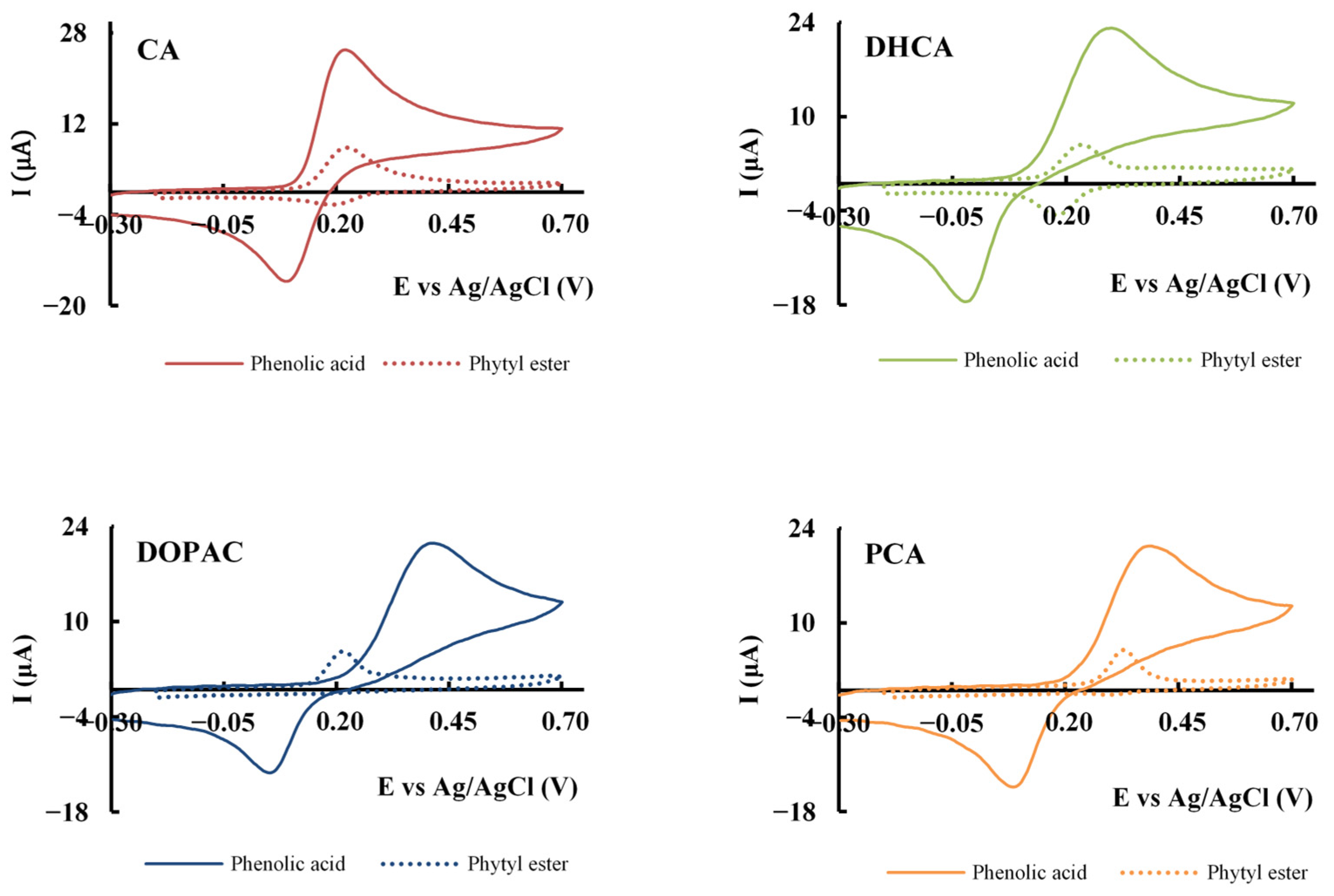
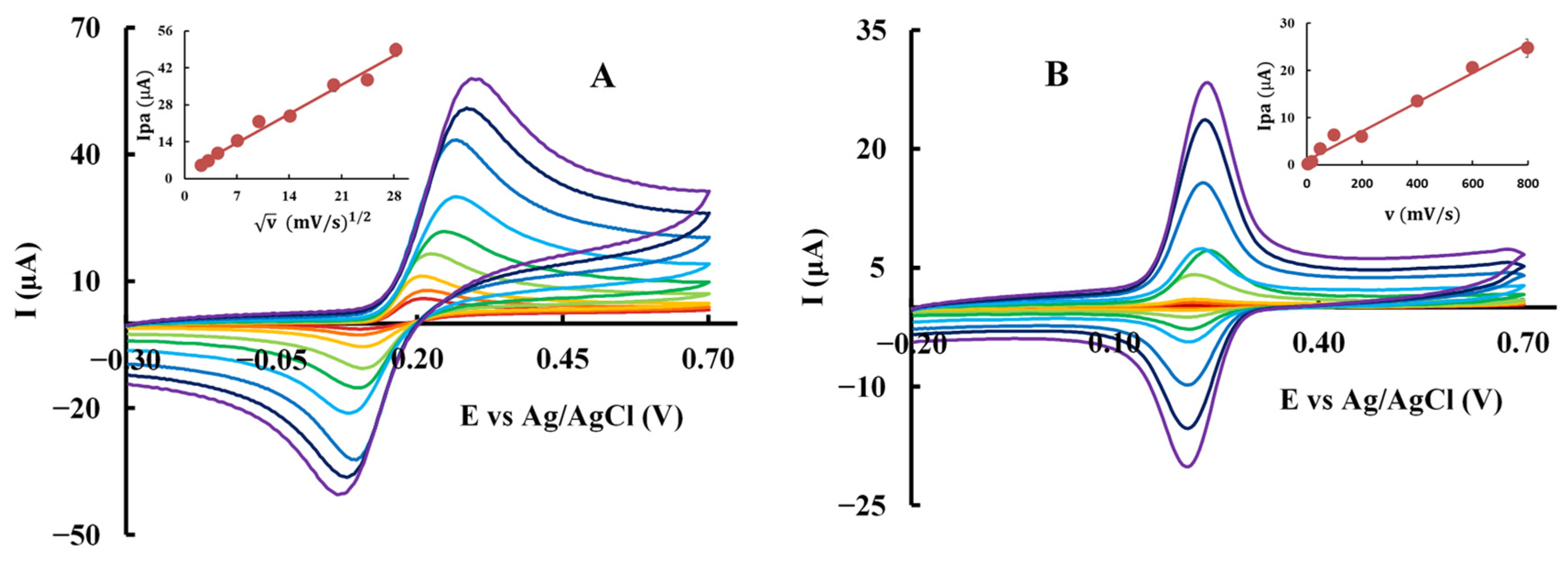
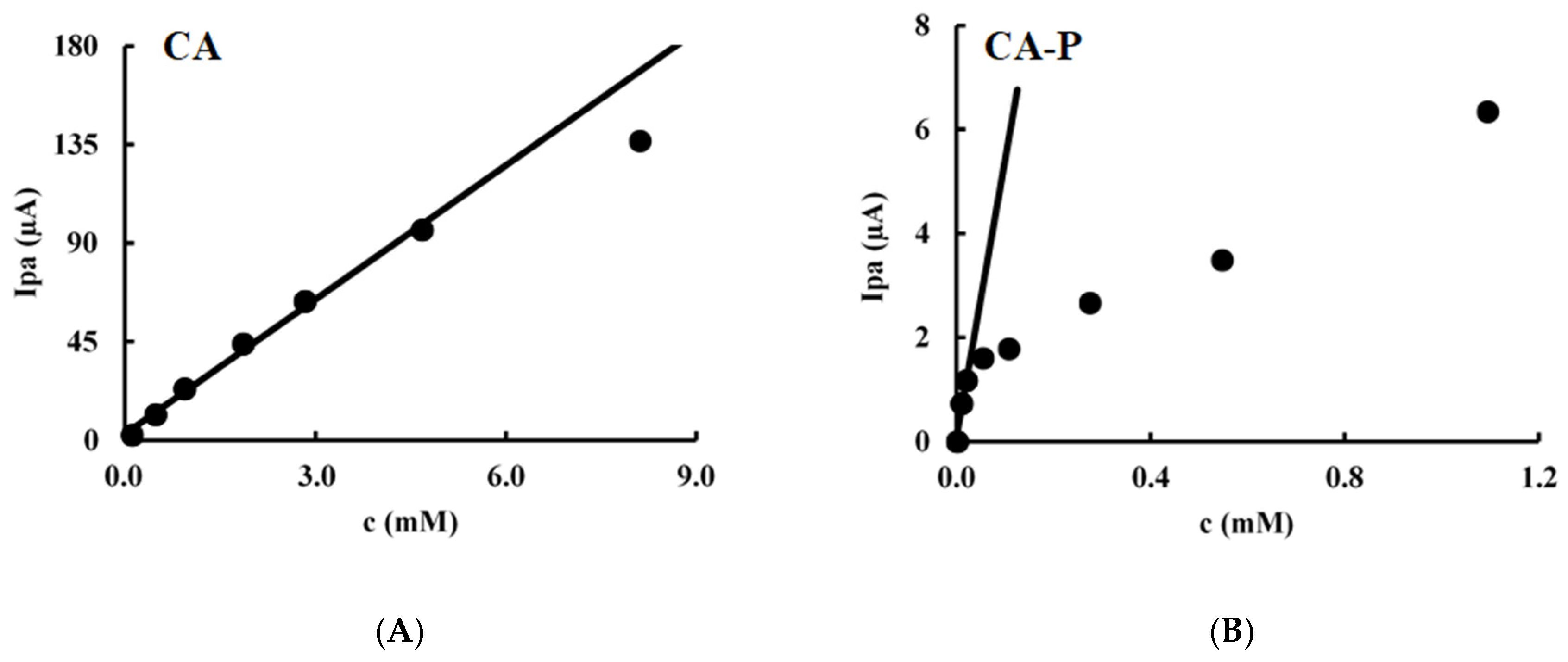
3.5. Cyclic Voltammetry
3.6. Preparation of Large Unilamellar Vesicles
3.7. Dynamic Light Scattering Measurements
3.8. Fluorescence Quenching Measurements
3.9. Effect of Compounds on the Fluorescence Polarization of Probes
3.10. Evaluation of the Antioxidant Activity of Compounds in PC Liposomes
3.11. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tomlinson, B.; Wu, Q.Y.; Zhong, Y.M.; Li, Y.H. Advances in Dyslipidaemia Treatments: Focusing on ApoC3 and ANGPTL3 Inhibitors. J. Lipid Atheroscler. 2024, 13, 2–20. [Google Scholar] [CrossRef]
- Bilson, J.; Mantovani, A.; Byrne, C.D.; Targher, G. Steatotic liver disease, MASLD and risk of chronic kidney disease. Diabetes Metab. 2024, 50, 101506. [Google Scholar] [CrossRef]
- Habibullah, M.; Jemmieh, K.; Ouda, A.; Haider, M.Z.; Malki, M.I.; Elzouki, A.-N. Metabolic-associated fatty liver disease: A selective review of pathogenesis, diagnostic approaches, and therapeutic strategies. Front. Med. 2024, 11, 1291501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fleishman, J.S.; Li, T.; Li, Y.; Ren, Z.; Chen, J.; Ding, M. Pharmacological therapy of metabolic dysfunction-associated steatotic liver disease-driven hepatocellular carcinoma. Front. Pharmacol. 2024, 14, 1336216. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, B.; Marjanovic-Haljilji, M.; Mijac, D.; Lukic, S.; Kapor, S.; Kapor, S.; Starcevic, A.; Popovic, D.; Djokovic, A. Molecular Aspects of MAFLD—New Insights on Pathogenesis and Treatment. Curr. Issues Mol. Biol. 2023, 45, 9132–9148. [Google Scholar] [CrossRef] [PubMed]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef]
- Jia, G.; Bai, H.; Mather, B.; Hill, M.A.; Jia, G.; Sowers, J.R. Diabetic Vasculopathy: Molecular Mechanisms and Clinical Insights. Int. J. Mol. Sci. 2024, 25, 804. [Google Scholar] [CrossRef]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef]
- Toma, L.; Stancu, C.S.; Sima, A.V. Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies. Biomedicines 2021, 9, 18. [Google Scholar] [CrossRef]
- Steptoe, A.; Zaninotto, P. Lower socioeconomic status and the acceleration of aging: An outcome-wide analysis. Proc. Natl. Acad. Sci. USA 2020, 117, 14911–14917. [Google Scholar] [CrossRef]
- Brewer, M.S. Natural Antioxidants: Sources, Compounds, Mechanisms of Action, and Potential Applications. Compr. Rev. Food Sci. Food Saf. 2011, 10, 221–247. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Costa, M.; Arques, F.; Ferreira, M.; Gameiro, P.; Geraldo, D.; Monteiro, L.S.; Paiva-Martins, F. Cholesteryl Phenolipids as Potential Biomembrane Antioxidants. Molecules 2024, 29, 4959. [Google Scholar] [CrossRef]
- Chalas, J.; Claise, C.; Edeas, M.; Messaoudi, C.; Vergnes, L.; Abella, A.; Lindenbaum, A. Effect of ethyl esterification of phenolic acids on low-density lipoprotein oxidation. Biomed. Pharmacother. 2001, 55, 54–60. [Google Scholar] [CrossRef]
- Katsoura, M.H.; Polydera, A.C.; Tsironis, L.D.; Petraki, M.P.; Rajacić, S.K.; Tselepis, A.D.; Stamatis, H. Efficient enzymatic preparation of hydroxycinnamates in ionic liquids enhances their antioxidant effect on lipoproteins oxidative modification. New Biotechnol. 2009, 26, 83–91. [Google Scholar] [CrossRef]
- Lopes, R.; Costa, M.; Ferreira, M.; Gameiro, P.; Fernandes, S.; Catarino, C.; Santos-Silva, A.; Paiva-Martins, F. Caffeic acid phenolipids in the protection of cell membranes from oxidative injuries. Interaction with the membrane phospholipid bilayer. Biochim. Biophys. Acta (BBA)—Biomembr. 2021, 1863, 183727. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Silva, T.C.; Losada-Barreiro, S.; Valentão, P.; Paiva-Martins, F.; Andrade, P.B. Toxicity of phenolipids: Protocatechuic acid alkyl esters trigger disruption of mitochondrial membrane potential and caspase activation in macrophages. Chem. Phys. Lipids 2017, 206, 16–27. [Google Scholar] [CrossRef]
- Rabiej-Kozioł, D.; Roszek, K.; Krzemiński, M.P.; Szydłowska-Czerniak, A. Phenolipids as new food additives: From synthesis to cell-based biological activities. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2022, 39, 1365–1379. [Google Scholar] [CrossRef]
- Islam, M.T.; Ali, E.S.; Uddin, S.J.; Shaw, S.; Islam, M.A.; Ahmed, M.I.; Chandra Shill, M.; Karmakar, U.K.; Yarla, N.S.; Khan, I.N.; et al. Phytol: A review of biomedical activities. Food Chem. Toxicol. 2018, 121, 82–94. [Google Scholar] [CrossRef]
- Singh, S.P.; Sashidhara, K.V. Lipid lowering agents of natural origin: An account of some promising chemotypes. Eur. J. Med. Chem. 2017, 140, 331–348. [Google Scholar] [CrossRef]
- Condo, A.M.; Baker, D.C.; Moreau, R.A.; Hicks, K.B. Improved Method for the Synthesis of trans-Feruloyl-β-sitostanol. J. Agric. Food Chem. 2001, 49, 4961–4964. [Google Scholar] [CrossRef] [PubMed]
- Winkler-Moser, J.K.; Hwang, H.-S.; Bakota, E.L.; Palmquist, D.A. Synthesis of steryl ferulates with various sterol structures and comparison of their antioxidant activity. Food Chem. 2015, 169, 92–101. [Google Scholar] [CrossRef]
- Tan, Z.; Shahidi, F. Chemoenzymatic Synthesis of Phytosteryl Ferulates and Evaluation of Their Antioxidant Activity. J. Agric. Food Chem. 2011, 59, 12375–12383. [Google Scholar] [CrossRef]
- Pletcher, D.; Greff, R.; Peat, R.; Peter, L.M.; Robinson, J. Introduction to the fundamental concepts of electrochemistry. In Instrumental Methods in Electrochemistry; Pletcher, D., Greff, R., Peat, R., Peter, L.M., Robinson, J., Eds.; Woodhead Publishing: Cambridge, UK, 2010; pp. 15–41. [Google Scholar] [CrossRef]
- Terao, J.; Piskula, M.; Yao, Q. Protective Effect of Epicatechin, Epicatechin Gallate, and Quercetin on Lipid Peroxidation in Phospholipid Bilayers. Arch. Biochem. Biophys. 1994, 308, 278–284. [Google Scholar] [CrossRef]
- Freiría-Gandara, J.; Losada-Barreiro, S.; Paiva-Martins, F.; Bravo-Díaz, C. Differential partitioning of bioantioxidants in edible oil−water and octanol−water systems: Linear Free Energy Relationships. J. Chem. Eng. Data 2018, 63, 2999–3007. [Google Scholar] [CrossRef]
- Paiva-Martins, F.; Gordon, M.H. Interactions of ferric ions with olive oil phenolic compounds. J. Agric. Food Chem. 2005, 53, 2704–2709. [Google Scholar] [CrossRef]
- Leth, T.; Sondergaard, H. Biological activity of vitamin E compounds and natural materials by the resorption-gestation test, and chemical determination of the vitamin E activity in foods and feeds. J. Nutr. 1977, 107, 2236–2243. [Google Scholar] [CrossRef]
- Islam, M.T.; Ayatollahi, S.A.; Zihad, S.; Sifat, N.; Khan, M.R.; Paul, A.; Salehi, B.; Islam, T.; Mubarak, M.S.; Martins, N.; et al. Phytol anti-inflammatory activity: Pre-clinical assessment and possible mechanism of action elucidation. Cell. Mol. Biol. 2020, 66, 264–269. [Google Scholar] [CrossRef] [PubMed]
- de Alencar, M.V.O.B.; Islam, M.T.; da Mata, A.M.O.F.; dos Reis, A.C.; de Lima, R.M.T.; de Oliveira Ferreira, J.R.; de Castro e Sousa, J.M.; Ferreira, P.M.P.; de Carvalho Melo-Cavalcante, A.A.; Rauf, A.; et al. Anticancer effects of phytol against Sarcoma (S-180) and Human Leukemic (HL-60) cancer cells. Environ. Sci. Pollut. Res. 2023, 30, 80996–81007. [Google Scholar] [CrossRef]
- Bobe, G.; Zhang, Z.; Kopp, R.; Garzotto, M.; Shannon, J.; Takata, Y. Phytol and its metabolites phytanic and pristanic acids for risk of cancer: Current evidence and future directions. Eur. J. Cancer Prev. 2020, 29, 191–200. [Google Scholar] [CrossRef]
- Lopes, R.; Costa, M.; Ferreira, M.; Gameiro, P.; Paiva-Martins, F. A new family of hydroxytyrosol phenolipids for the antioxidant protection of liposomal systems. Biochim. Biophys. Acta (BBA) Biomembr. 2021, 1863, 183505. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.O.; Sousa, F.B.; Damasceno, S.R.; Carvalho, N.S.; Silva, V.G.; Oliveira, F.R.; Sousa, D.P.; Aragão, K.S.; Barbosa, A.L.; Freitas, R.M.; et al. Phytol, a diterpene alcohol, inhibits the inflammatory response by reducing cytokine production and oxidative stress. Fundam. Clin. Pharmacol. 2014, 28, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Jéssica, P.C.; Md, T.I.; Pauline, S.S.; Paula, B.F.; George, L.S.O.; Marcus, V.O.B.A.; Marcia, F.C.J.P.; Éverton, L.F.F.; Chistiane, M.F.; Antonia, M.G.L.C.; et al. Evaluation of Antioxidant Activity of Phytol Using Non- and Pre-Clinical Models. Curr. Pharm. Biotechnol. 2016, 17, 1278–1284. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.; Ai, W.; Zhang, F.; Yang, K.; Wang, L.; Zhu, X.; Gao, P.; Shu, G.; Jiang, Q.; et al. Phytol increases adipocyte number and glucose tolerance through activation of PI3K/Akt signaling pathway in mice fed high-fat and high-fructose diet. Biochem. Biophys. Res. Commun. 2017, 489, 432–438. [Google Scholar] [CrossRef]
- Papaccio, F.; D’Arino, A.; Caputo, S.; Bellei, B. Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants 2022, 11, 1121. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Acid Catalysis | Enzymatic Catalysis | ||||
|---|---|---|---|---|---|---|
| Toluene | THF | Toluene | Dioxane | DCM | THF | |
| DCA-P | NR | 17% | 95% | 50% | NR | NR |
| DOPAC-P | NR | 56% | 46% | 83% | NR | NR |
| PCA-P | NR | NR | 18% | 0% | NR | NR |
| Compound | miLog p | EC50 ** | Compound | miLog p | EC50 ** | ||
|---|---|---|---|---|---|---|---|
| 5 min | 30 min | 5 min | 30 min | ||||
| Phytol | 6.76 | - | - | α-Toc | 9.04 | 0.33 | 0.29 |
| PCA | 0.86 | 0.22 | 0.19 | PCA-P | 8.20 | 0.19 | 0.14 |
| DOPAC | 0.39 | 0.13 | 0.13 | DOPAC-P | 8.09 | 0.21 | 0.18 |
| DHCA | 0.91 | 0.19 | 0.13 | DHCA-P | 8.26 | 0.29 | 0.29 |
| CA | 0.94 | 0.23 | 0.21 | CA-P | 8.49 | 0.23 | 0.22 |
| Compound | Epa (V) * | Epc (V) * | Ipa (µA) * | −Ipc (µA) * | Compound | Epa (V) * | Epc (V) * | Ipa (µA) * | −Ipc (µA) * |
|---|---|---|---|---|---|---|---|---|---|
| CA | 0.223 | 0.091 | 21.67 | 17.79 | CA-P | 0.235 | 0.198 | 6.71 | 2.25 |
| DHCA | 0.297 | −0.026 | 17.31 | 14.63 | DHCA-P | 0.250 | 0.171 | 4.49 | 4.22 |
| DOPAC | 0.403 | 0.053 | 14.67 | 12.72 | DOPAC-P | 0.219 | - | 4.56 | - |
| PCA | 0.387 | 0.082 | 15.18 | 10.35 | PCA-P | 0.327 | 0.303 | 4.55 | 0.35 |
| α-Toc | 0.219 | - | 4.10 | - |
| Compound | Anodic Process | Cathodic Process | Controlled by | ||
|---|---|---|---|---|---|
| Linear Regression Equation | r | Linear Regression Equation | r | ||
| CA | Ip = (1.57 ± 0.08) v1/2 + (2.6 ± 1.2) | 0.991 | −Ip = (1.63 ± 0.07) v1/2 + (−0.6 ± 1.2) | 0.993 | Diffusion |
| DHCA | Ip = (1.54 ± 0.04) v1/2 + (2.0 ± 0.6) | 0.998 | −Ip = (1.23 ± 0.09) v1/2 + (1.0 ± 1.4) | 0.980 | Diffusion |
| PCA | Ip = (1.43 ± 0.01) v1/2 + (1.0 ± 0.2) | 0.999 | −Ip = (1.25 ± 0.03) v1/2 + (−0.9 ± 0.5) | 0.998 | Diffusion |
| DOPAC | Ip = (1.30 ± 0.02) v1/2 + (2.1 ± 0.3) | 0.999 | −Ip = (1.20 ± 0.04) v1/2 + (−1.5 ± 0.7) | 0.995 | Diffusion |
| CA-P | Ip = (0.031 ± 0.002) v + (0.9 ± 0.6) | 0.991 | −Ip = (0.0231 ± 0.0004) v + (−0.2 ± 0.1) | 0.999 | Adsorption |
| DHCA-P | Ip = (0.0388 ± 0.0006) v + (0.8 ± 0.1) | 0.999 | −Ip = (0.054 ± 0.001) v + (−0.8 ± 0.4) | 0.997 | Adsorption |
| PCA-P | Ip = (0.030 ± 0.001) v + (0.7 ± 0.4) | 0.994 | −Ip = (0.0125 ± 0.0006) v + (−0.5 ± 0.2) | 0.993 | Adsorption |
| DOPAC-P | Ip = (0.0251 ± 0.0009) v + (1.0 ± 0.3) | 0.996 | - | - | Adsorption |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, V.; Costa, M.; Rebelo, R.; Arques, F.; Ferreira, M.; Gameiro, P.; Barros, T.; Geraldo, D.; Monteiro, L.S.; Paiva-Martins, F. Phytyl Phenolipids: Structurally Modified Antioxidants with Superior Lipid Membrane Interaction. Molecules 2025, 30, 2193. https://doi.org/10.3390/molecules30102193
Costa V, Costa M, Rebelo R, Arques F, Ferreira M, Gameiro P, Barros T, Geraldo D, Monteiro LS, Paiva-Martins F. Phytyl Phenolipids: Structurally Modified Antioxidants with Superior Lipid Membrane Interaction. Molecules. 2025; 30(10):2193. https://doi.org/10.3390/molecules30102193
Chicago/Turabian StyleCosta, Vânia, Marlene Costa, Rute Rebelo, Francisca Arques, Mariana Ferreira, Paula Gameiro, Tomás Barros, Dulce Geraldo, Luís S. Monteiro, and Fátima Paiva-Martins. 2025. "Phytyl Phenolipids: Structurally Modified Antioxidants with Superior Lipid Membrane Interaction" Molecules 30, no. 10: 2193. https://doi.org/10.3390/molecules30102193
APA StyleCosta, V., Costa, M., Rebelo, R., Arques, F., Ferreira, M., Gameiro, P., Barros, T., Geraldo, D., Monteiro, L. S., & Paiva-Martins, F. (2025). Phytyl Phenolipids: Structurally Modified Antioxidants with Superior Lipid Membrane Interaction. Molecules, 30(10), 2193. https://doi.org/10.3390/molecules30102193