
Effects of Different Adduct Ions, Ionization Temperatures, and Solvents on the Ion Mobility of Glycans


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
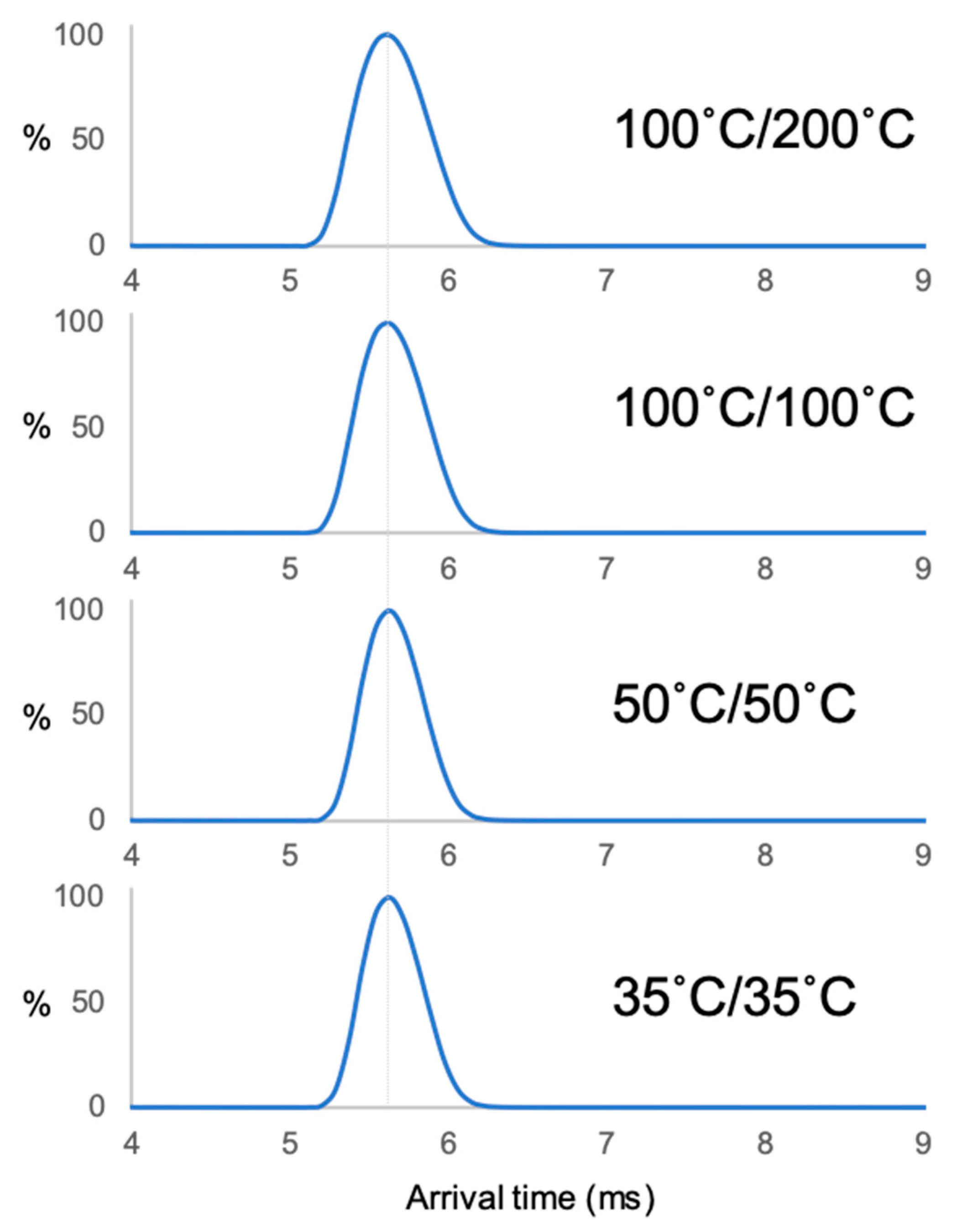
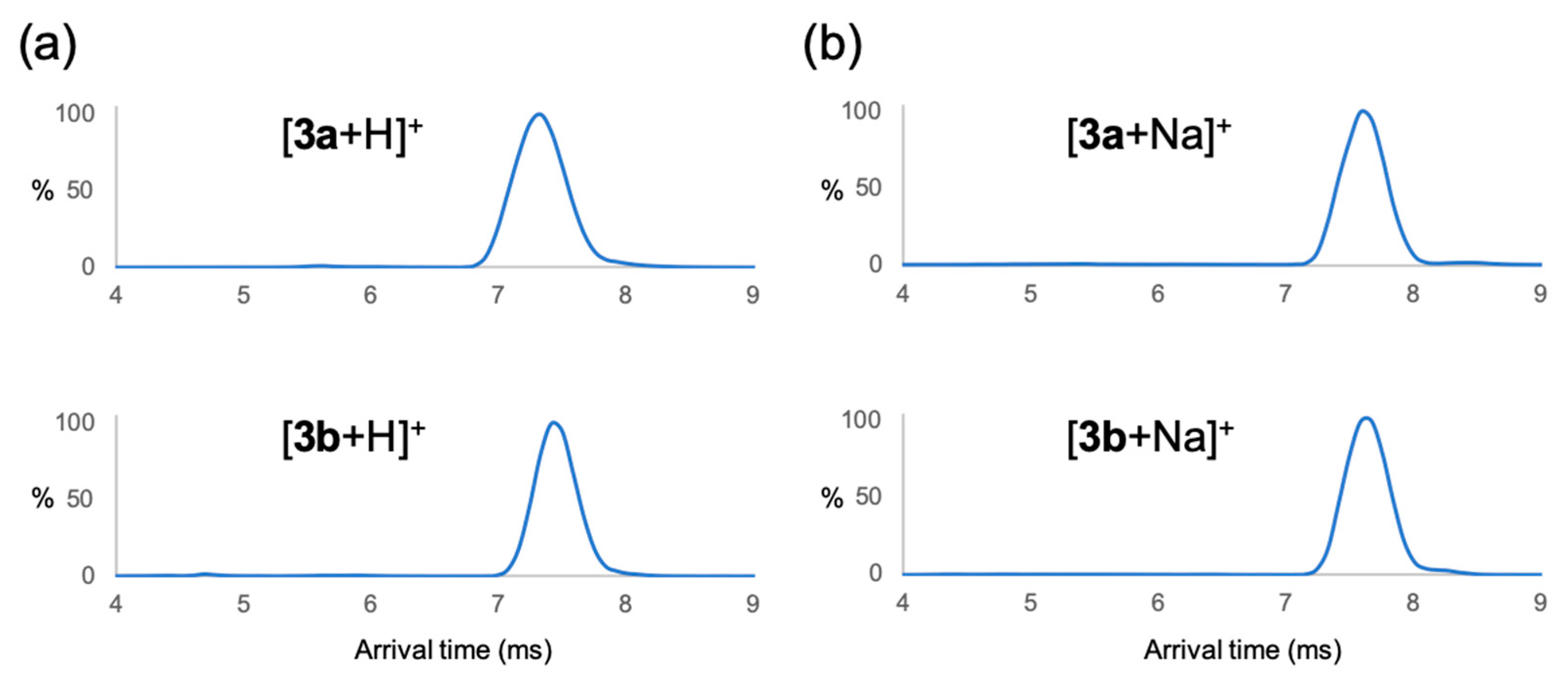
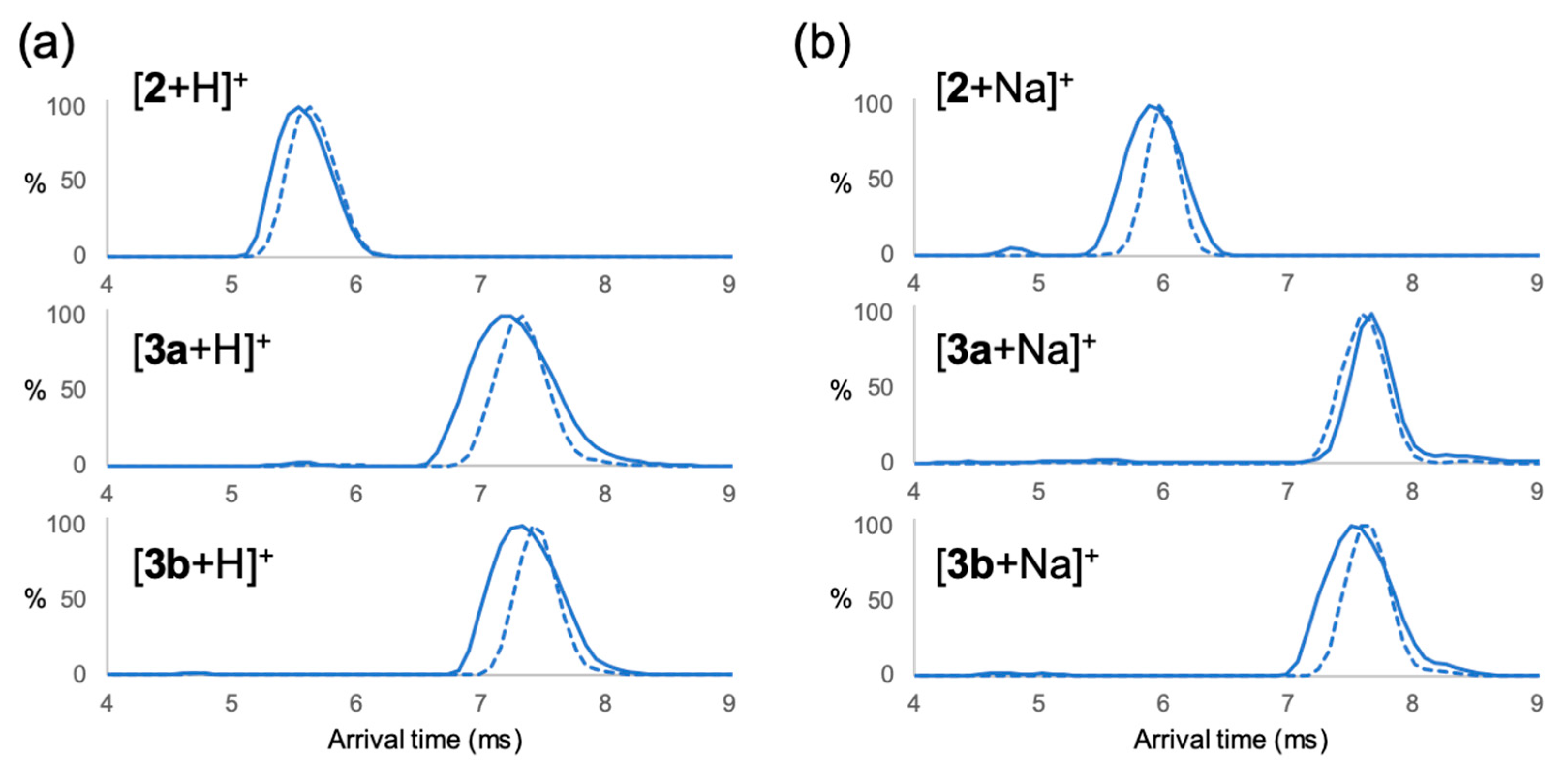
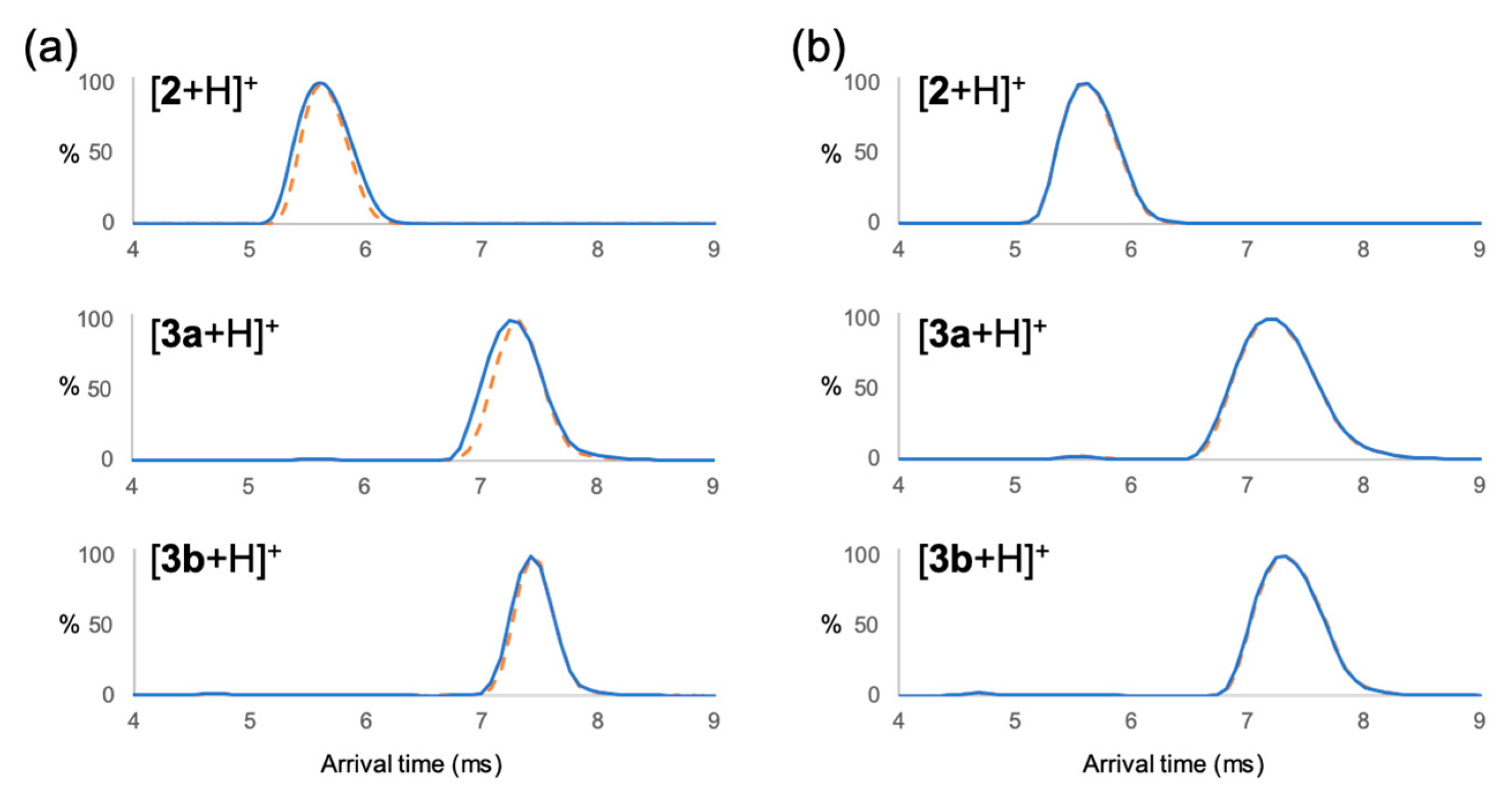
2. Results
3. Materials and Methods
3.1. Sample Preparation
3.2. ESI–IM–MS Measurement
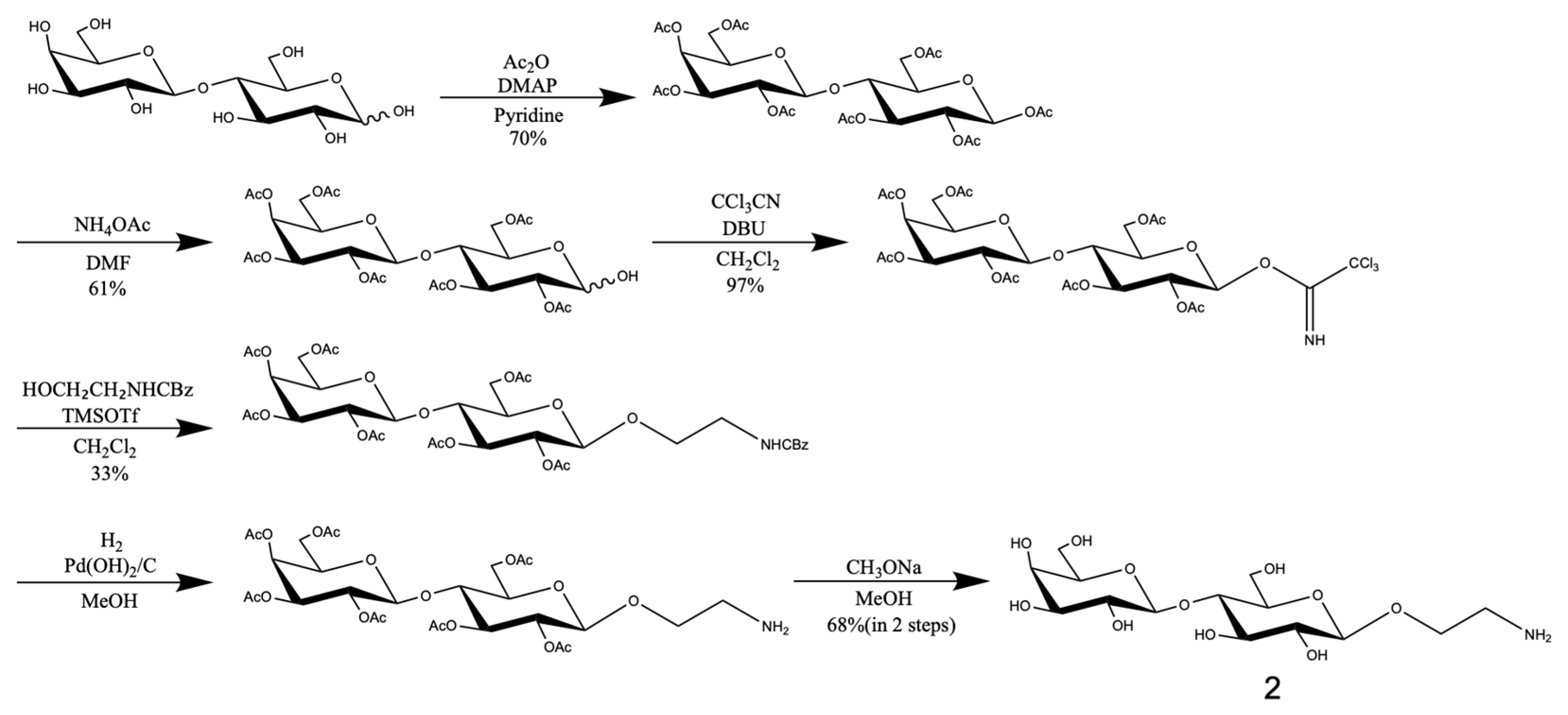
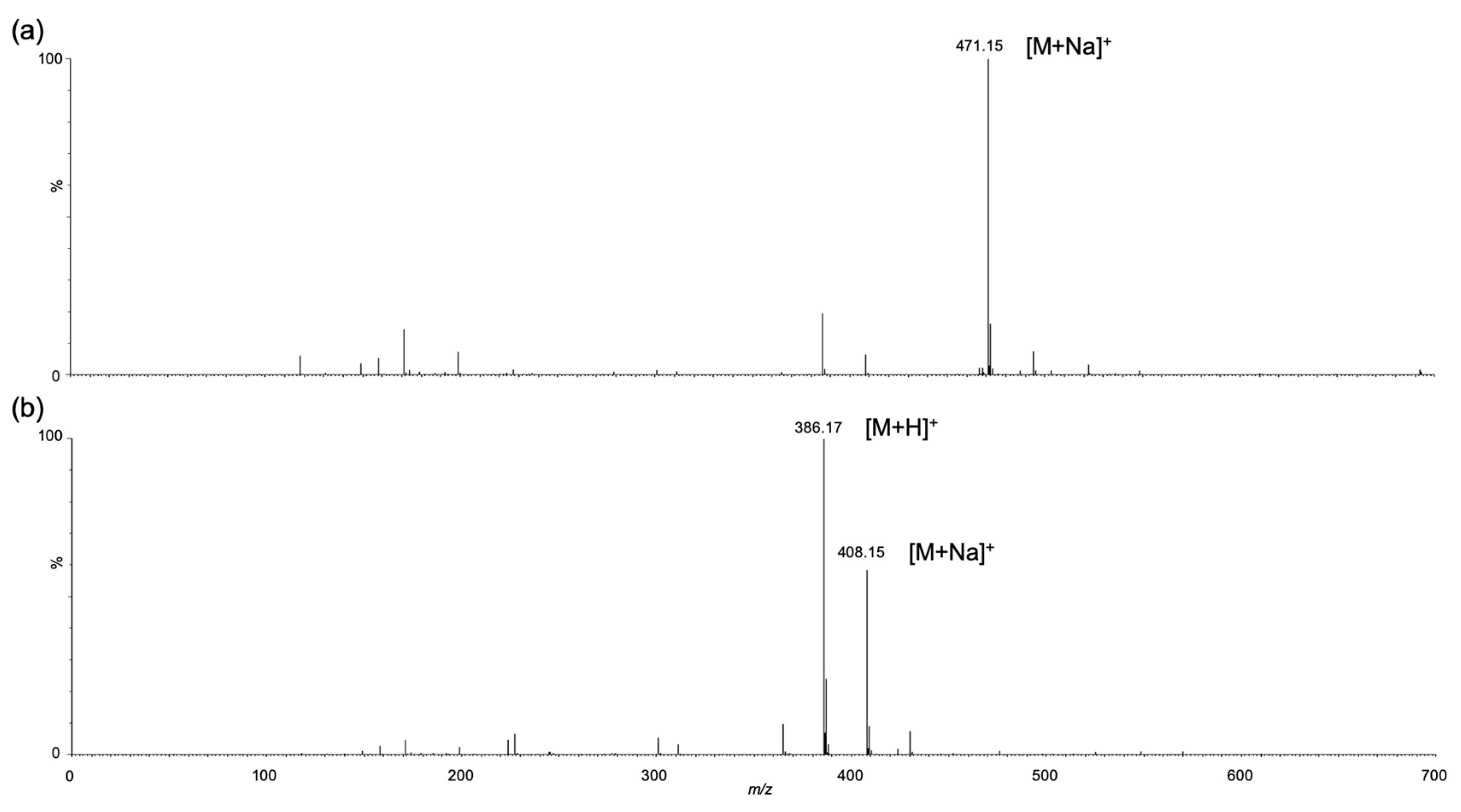
3.3. Synthesis of Trisaccharides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varki, A. Biological Roles of Glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.D. Stuck on Sugars—How Carbohydrates Regulate Cell Adhesion, Recognition, and Signaling. Glycoconj. J. 2019, 36, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.D.; Pierce, J.M. The Challenge and Promise of Glycomics. Chem. Biol. 2014, 21, 1–15. [Google Scholar] [CrossRef]
- Dwek, R.A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef]
- Gray, C.J.; Migas, L.G.; Barran, P.E.; Pagel, K.; Seeberger, P.H.; Eyers, C.E.; Boons, G.J.; Pohl, N.L.B.; Compagnon, I.; Widmalm, G.; et al. Advancing Solutions to the Carbohydrate Sequencing Challenge. J. Am. Chem. Soc. 2019, 141, 14463–14479. [Google Scholar] [CrossRef]
- Wuhrer, M. Glycomics Using Mass Spectrometry. Glycoconj. J. 2013, 30, 11–22. [Google Scholar] [CrossRef]
- Duus, J.Ø.; Gotfredsen, C.H.; Bock, K. Carbohydrate Structural Determination by NMR Spectroscopy: Modern Methods and Limitations. Chem. Rev. 2000, 100, 4589–4614. [Google Scholar] [CrossRef]
- Widmalm, G. Glycan Shape, Motions, and Interactions Explored by NMR Spectroscopy. JACS Au 2024, 4, 20–39. [Google Scholar] [CrossRef]
- Angulo, J. NMR Investigations of Glycan Conformation, Dynamics, and Interactions. Prog. Nucl. Magn. Reson. Spectrosc. 2024, 144–145, 97–152. [Google Scholar] [CrossRef]
- Yamaguchi, T. Delineating the Dynamic Conformations of Oligosaccharides by Combining Molecular Simulation and NMR Spectroscopy. Trends Glycosci. Glycotechnol. 2020, 32, E93–E98. [Google Scholar] [CrossRef]
- Ruda, A.; Aytenfisu, A.H.; Angles d’Ortoli, T.; MacKerell, A.D.; Widmalm, G. Glycosidic α-Linked Mannopyranose Disaccharides: An NMR Spectroscopy and Molecular Dynamics Simulation Study Employing Additive and Drude Polarizable Force Fields. Phys. Chem. Chem. Phys. 2023, 25, 3042–3060. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.J.; Thomas, B.; Upton, R.; Migas, L.G.; Eyers, C.E.; Barran, P.E.; Flitsch, S.L. Applications of Ion Mobility Mass Spectrometry for High Throughput, High Resolution Glycan Analysis. Biochim. Biophys. Acta 2016, 1860, 1688–1709. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Harvey, D.J.; Struwe, W.B.; Karlsson, N.G. Separation of Isomeric O-Glycans by Ion Mobility and Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2019, 91, 10604–10613. [Google Scholar] [CrossRef]
- May, J.C.; McLean, J.A. Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem. 2015, 87, 1422–1436. [Google Scholar] [CrossRef]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The Power of Ion Mobility-Mass Spectrometry for Structural Characterization and the Study of Conformational Dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef]
- Harvey, D.J.; Scarff, C.A.; Crispin, M.; Scanlan, C.N.; Bonomelli, C.; Scrivens, J.H. Confident Identification of Isomeric N-Glycan Structures by Combined Ion Mobility Mass Spectrometry and Hydrophilic Interaction Liquid Chromatography. Rapid Commun. Mass Spectrom. 2012, 26, 175–180. [Google Scholar]
- Hofmann, J.; Hahm, H.; Seeberger, P.H.; Pagel, K. Identification of Carbohydrate Anomers Using Ion Mobility–Mass Spectrometry. Nature 2015, 526, 241–244. [Google Scholar] [CrossRef]
- Gray, C.J.; Thomas, B.; Upton, R.; Migas, L.G.; Eyers, C.E.; Barran, P.E.; Flitsch, S.L. Bottom-Up Elucidation of Glycosidic Bond Stereochemistry. Anal. Chem. 2017, 89, 4540–4549. [Google Scholar] [CrossRef]
- Manabe, N.; Ohno, S.; Matsumoto, K.; Kawase, T.; Hirose, K.; Masuda, K.; Yamaguchi, Y. A Data Set of Ion Mobility Collision Cross Sections and Liquid Chromatography Retention Times from 71 Pyridylaminated N-Linked Oligosaccharides. J. Am. Soc. Mass Spectrom. 2022, 33, 1772–1783. [Google Scholar] [CrossRef]
- Struwe, W.B.; Pagel, K.; Benesch, J.L.P.; Harvey, D.J.; Campbell, M.P. GlycoMob: An Ion Mobility-Mass Spectrometry Collision Cross Section Database for Glycomics. Glycoconj. J. 2016, 33, 399–404. [Google Scholar] [CrossRef]
- Christofi, E.; Barran, P.E. Ion Mobility Mass Spectrometry (IM-MS) for Structural Biology: Insights Gained by Measuring Mass, Charge, and Collision Cross Section. Chem. Rev. 2023, 123, 2902–2949. [Google Scholar] [CrossRef] [PubMed]
- Karch, K.R.; Denizio, J.E.; Black, B.E.; Garcia, B.A. High-Resolution Native Mass Spectrometry. Chem. Rev. 2021, 121, 806–841. [Google Scholar]
- Re, S.; Watabe, S.; Nishima, W.; Sugita, Y. Characterization of Conformational Ensembles of Protonated N-Glycans in the Gas-Phase. Sci. Rep. 2018, 8, 1644. [Google Scholar] [CrossRef] [PubMed]
- Hinneburg, H.; Hofmann, J.; Struwe, W.B.; Thader, A.; Altmann, F.; Varon Silva, D.; Seeberger, P.H.; Pagel, K. Identification of Isomeric N-Glycans by Conformer Distribution Fingerprinting Using Ion Mobility Mass Spectrometry. Chem. Eur. J. 2020, 26, 12213–12220. [Google Scholar]
- Harvey, D.J.; Scarff, C.A.; Crispin, M.; Scanlan, C.N.; Bonomelli, C.; Scrivens, J.H. Travelling-Wave Ion Mobility and Negative Ion Fragmentation of High Mannose N-Glycans. Rapid Commun. Mass Spectrom. 2016, 30, 235–244. [Google Scholar] [CrossRef]
- Gabelica, V.; De Pauw, E.; Karas, M. Influence of the capillary temperature and the source pressure on the internal energy distribution of electrosprayed ions. Int. J. Mass Spectrom. 2004, 231, 189–195. [Google Scholar] [CrossRef]
- Smith, D.P.; Knapman, T.W.; Campuzano, I.; Malham, R.W.; Berryman, J.T.; Radford, S.H.; Ashcroft, A.E. Deciphering Drift Time Measurements from Travelling Wave Ion Mobility Spectrometry-Mass Spectrometry Studies. Eur. J. Mass Spectrom. 2009, 15, 113–130. [Google Scholar] [CrossRef]
- Morsa, D.; Gabelica, V.; De Pauw, E. Effective Temperature of Ions in Traveling Wave Ion Mobility Spectrometry. Anal. Chem. 2011, 83, 5775–5782. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.; Wang, J.; Zhang, X.; Wang, X.; Wang, Y.; Zhang, Y.; Zhang, Y.; Zhang, Y.; Zhang, Y. Role of Globotriaosylceramide in Physiology and Pathology. Front. Mol. Biosci. 2022, 9, 813637. [Google Scholar]
- Warnke, S.; Seo, J.; Boschmans, J.; Sobott, F.; Scrivens, J.H.; Bleiholder, C.; Bowers, M.T.; Gewinner, S.; Schöllkopf, W.; Pagel, K. Protomers of Benzocaine: Solvent and Permittivity Dependence. J. Am. Chem. Soc. 2015, 137, 4236–4242. [Google Scholar] [CrossRef]
- Chung, N.A.; May, J.C.; Robinson, R.A.S.; McLean, J.A. Solvent Composition Can Have a Measurable Influence on the Ion Mobility-Derived Collision Cross Section of Small Molecules. J. Am. Soc. Mass Spectrom. 2023, 34, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Taraszka, J.A.; Counterman, A.E.; Clemmer, D.E. Influence of Solvent Composition and Capillary Temperature on the Conformations of Electrosprayed Ions: Unfolding of Compact Ubiquitin Conformers from Pseudonative and Denatured Solutions. Int. J. Mass Spectrom. 1999, 185–187, 37–47. [Google Scholar] [CrossRef]
- Vékey, K. Internal energy effects in mass spectrometry. J. Mass Spectrom. 1996, 31, 445–463. [Google Scholar] [CrossRef]
- Pendrill, R.; Engström, O.; Volpato, A.; Zerbetto, M.; Polimeno, A.; Widmalm, G. Flexibility at a Glycosidic Linkage Revealed by Molecular Dynamics, Stochastic Modeling, and 13C NMR Spin Relaxation: Conformational Preferences of α-L-Rhap-α-(1→2)-α-L-Rhap-OMe in Water and Dimethyl Sulfoxide Solutions. Phys. Chem. Chem. Phys. 2016, 18, 3086–3098. [Google Scholar] [CrossRef]
- Shvartsburg, A.A.; Smith, R.D. Fundamentals of Traveling Wave Ion Mobility Spectrometry. Anal. Chem. 2008, 80, 9689–9699. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, H.; Yamaguchi, T. Effects of Different Adduct Ions, Ionization Temperatures, and Solvents on the Ion Mobility of Glycans. Molecules 2025, 30, 2177. https://doi.org/10.3390/molecules30102177
Feng H, Yamaguchi T. Effects of Different Adduct Ions, Ionization Temperatures, and Solvents on the Ion Mobility of Glycans. Molecules. 2025; 30(10):2177. https://doi.org/10.3390/molecules30102177
Chicago/Turabian StyleFeng, Hao, and Takumi Yamaguchi. 2025. "Effects of Different Adduct Ions, Ionization Temperatures, and Solvents on the Ion Mobility of Glycans" Molecules 30, no. 10: 2177. https://doi.org/10.3390/molecules30102177
APA StyleFeng, H., & Yamaguchi, T. (2025). Effects of Different Adduct Ions, Ionization Temperatures, and Solvents on the Ion Mobility of Glycans. Molecules, 30(10), 2177. https://doi.org/10.3390/molecules30102177