
Straightforward Access to Polyfunctionalized δ-Lactams via Domino Aza–Michael/Thia–Michael/Aldol Sequence
 , and
, and
Abstract

1. Introduction
2. Results and Discussion
2.1. The Optimization of the Thia–Michael/Aldol Domino Sequence
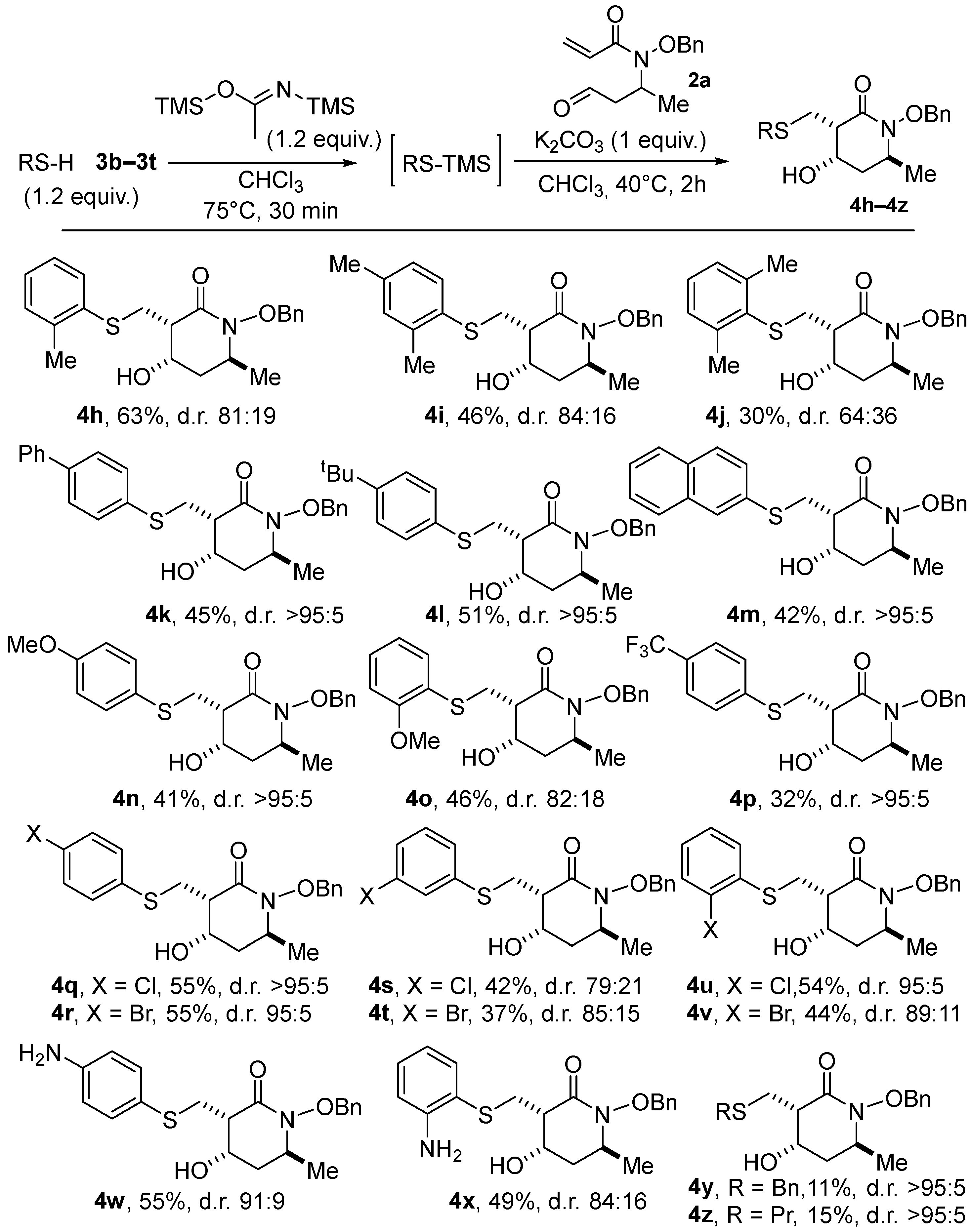
2.2. The Exemplification of the Thia–Michael/Aldol Domino Sequence
2.3. Sequential One-Pot Aza–Michael/Thia–Michael/Aldolization Sequence
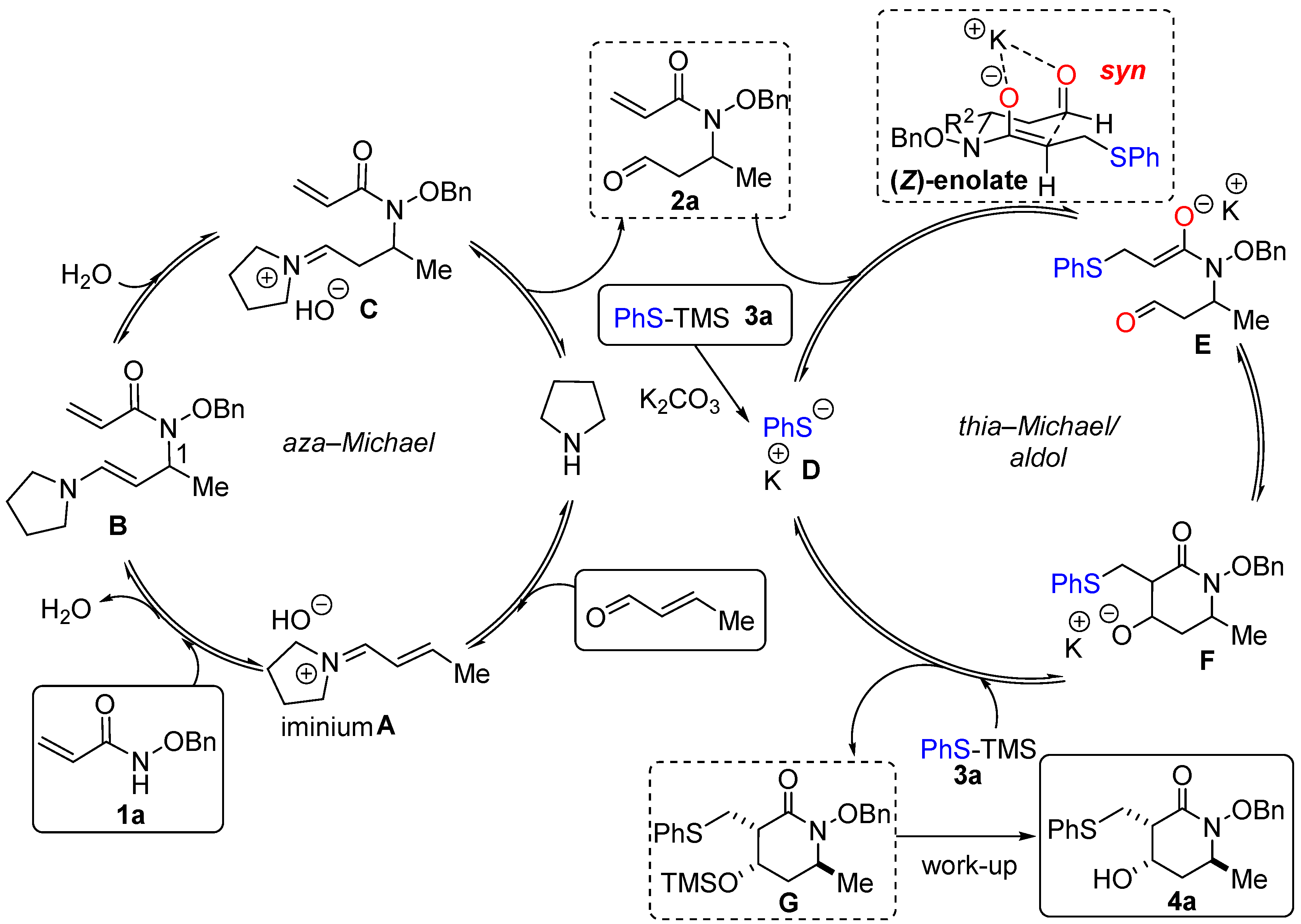
2.4. Proposed Mechanism
3. Materials and Methods
3.1. General Considerations
3.2. General Procedures
3.2.1. General Procedure A for the Thia–Michael/Aldolization Route
3.2.2. General Procedure B for the Silylation/Thia–Michael/Aldol Sequence
3.2.3. General Procedure C for the One-Pot Aza–Michael/Thia–Michael/Aldol Process
3.3. Experimental Data
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-6-methyl-3-((phenylthio)methyl)piperidin-2-one (4a). Prepared using general procedure A from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 2 h. Overall isolated yield: 68% (49 mg; d.r. = 88:12). Major diastereoisomer 4a1—White solid. Rf = 0.52 (EtOAc/Cyclohexane 4:6). mp: 112 °C. IR (cm−1): 3425, 1960, 1640. 1H-NMR (400 MHz, CDCl3, 20 °C) δ 7.48–7.17 (m, 10H), 5.04 (d syst AB, J = 9.5 Hz, 1H), 4.81 (d syst AB, J = 9.5 Hz, 1H), 4.43 (bs, 1H), 4.06–3.95 (m, 1H), 3.85 (dd, J = 14.0, 4.0 Hz, 1H), 3.11 (dd, J = 14.0, 11.3 Hz, 1H), 2.60 (dt, J = 11.3, 3.5 Hz, 1H), 2.28 (d, J = 4.1 Hz, 1H), 2.16 (dt, J = 14.0, 4.3 Hz, 1H), 1.65 (t, J = 12.7 Hz, 1H), 1.30 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 166.1, 135.7, 135.4, 129.5 (2C), 129.3 (2C), 129.1 (2C),128.7, 128.6 (2C), 126.3, 76.5, 63.8, 52.4, 47.0, 38.5, 30.1, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C20H23NO3S 358.1471, found 358.1467. Minor diastereoisomer 4a2—White solid. Rf = 0.36 (EtOAc/Cyclohexane 4:6). mp: 80–85 °C. 1H-NMR (300 MHz, CDCl3, 20 °C) δ 7.51–7.27 (m,), 7.23–7.18 (m, 1H17), 5.00 (d syst AB, J = 10 Hz, 1H), 4.89 (d syst AB, J = 10.0 Hz, 1H), 4.41 (m, 1H), 3.80 (dd, J = 13.8, 4.2 Hz, 1H), 3.75–3.65 (m, 1H), 3.18 (dd, J = 13.8, 10.7 Hz, 1H), 2.70 (dq, J = 10.6, 4.1 Hz, 1H), 2.01 (m, 3H), 1.45 (d, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3, 20 °C) δ (ppm) 167.1, 135.5, 129.5 (2C), 129.3 (5C), 128.8, 128.6 (2C), 126.5, 76.4, 65.1, 54.5, 47.2, 35.7, 30.7, 20.8.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-6-ethyl-4-hydroxy-3-((phenylthio)methyl)piperidin-2-one (4b). Prepared using general procedure A from (N-(benzyloxy)-N-(1-oxopentan-3-yl)acrylamide 2b (0.2 mmol, 53 mg, 1 equiv). Reaction time: 2 h. White solid. Overall isolated yield: 45% (33 mg; d.r. = 86:14). Rf = 0.46 (EtOAc/Cyclohexane 4:6). mp: 94 °C. IR (cm−1): 3397, 2932, 1724, 1656. 1H-NMR (400 MHz, CDCl3, 20 °C) 7.54–7.10 (m, 10H), 5.01 (d, J = 9.5 Hz, 1H), 4.84 (d, J = 9.6 Hz, 1H), 4.47 (m, 1H), 3.88–3.82 (m, 2H), 3.12 (dd, J = 14.0, 11.2 Hz, 1H), 2.59 (dt, J = 11.1, 3.5 Hz, 1H), 2.25–2.07 (m, 2H), 1.93 (d, J = 24.3 Hz, 1H), 1.63 (d, J = 38.3 Hz, 2H), 0.87 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.8, 135.7, 135.4, 129.5 (2C), 129.3 (2C), 129.1 (2C), 128.7, 128.6 (2C), 126.3, 76.0, 63.8, 57.1, 47.0, 34.8, 30.2, 25.5, 8.8. HRMS (ESI+) m/z: [M + H]+ calculated for C21H26NO3S 372.1627, found 372.1628.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-6-ethyl-4-hydroxy-3-((phenylthio)methyl)piperidin-2-one (4c). Prepared using general procedure A from N-(benzyloxy)-N-(1-oxopentan-3-yl)acrylamide 2c (0.2 mmol, 56 mg, 1 equiv). Reaction time: 2 h. White solid. Overall isolated yield: 50% (39 mg; d.r. = 83:17). Rf = 0.46 (EtOAc/Cyclohexane 4:6). mp: 90 °C. IR (cm−1): 3325, 2943, 2867, 1629, 1582. 1H-NMR (400 MHz, CDCl3, 20 °C) 7.55–7.14 (m, 10H), 5.02 (d, J = 9.6 Hz, 1H), 4.83 (d, J = 9.6 Hz, 1H), 4.46 (m, 1H), 3.93–3.81 (m, 2H), 3.11 (dd, J = 14.0, 11.2 Hz, 1H), 2.59 (dt, J = 11.2, 3.5 Hz, 1H), 2.20 (dt, J = 14.1, 4.7 Hz, 1H), 1.91 (ddt, J = 13.8, 5.7, 3.3 Hz, 1H), 1.83 (d, J = 4.5 Hz, 1H), 1.71–1.44 (m, 2H), 1.43–1.17 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.7, 135.7, 135.4, 129.6 (2C), 129.3 (2C), 129.2 (2C), 128.8, 128.6 (2C), 126.4, 76.2, 63.9, 56.2, 47.0, 35.5, 35.1, 30.3, 17.9, 14.3. HRMS (ESI+) m/z: [M + H]+ calculated for C22H28NO3S: 373.1660, found 373.1661.
- (±)-(3S,4S,6S)-4-Hydroxy-1-methoxy-6-methyl-3-((phenylthio)methyl)piperidin-2-one (4d). Prepared using general procedure A from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2d (0.2 mmol, 50 mg, 1 equiv). Reaction time: 2 h. White solid. Yield: 37% (21 mg; d.r. > 95:5). Rf = 0.50 (EtOAc/Cyclohexane 4:6). mp.: 96 °C. IR (cm−1): 3396, 2932, 2245, 1639. 1H-NMR (400 MHz, CDCl3, 20 °C) δ 7.41–7.14 (m, 5H), 4.46–4.40 (m, 1H), 4.09–4.01 (m, 1H), 3.81 (dd, J = 14.1, 4.0 Hz, 1H), 3.74 (d, J = 1.3 Hz, 3H), 3.08 (dd, J = 14.0, 11.2 Hz, 1H), 2.56 (dt, J = 11.3, 3.3 Hz, 1H), 2.25–2.14 (m, 2H), 1.75–1.60 (m, 1H), 1.31 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.1, 135.6, 129.3 (3C), 126.4 (2C), 63.8, 62.3, 51.9, 46.8, 38.5, 30.1, 19.9. HRMS (ESI+) m/z: [M + H]+ calculated for C14H20NO3S 282.1158, found 282.1158.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-4,6-dimethyl-3-((phenylthio)methyl) piperidin-2-one (4e). Prepared using general procedure A from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2e (0.2 mmol, 52 mg, 1 equiv). Reaction time: 24 h. White solid. Yield: 51% (38 mg; d.r. > 95:5). Rf = 0.52 (EtOAc/Cyclohexane 4:6). mp: 109 °C. IR (cm−1): 3399, 2931, 2860, 1652. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.50–7.21 (m, 10H), 5.03 (d, J = 9.7 Hz, 1H), 4.78 (d, J = 9.7 Hz, 1H), 3.57 (m, 1H), 3.48–3.29 (m, 2H), 2.76 (m, 1H), 2.23 (m, 1H), 2.04 (dd, J = 13.5, 10.1 Hz, 1H), 1.89 (dd, J = 13.6, 5.4 Hz, 1H), 1.34 (d, J = 6.2 Hz, 3H), 1.30 (s, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.6, 135.9, 135.3, 129.7 (2C), 129.5 (2C), 129.3 (2C), 128.9, 128.6 (2C), 126.5, 76.7, 70.0, 54.4, 53.7, 41.5, 33.3, 28.9, 20.3. HRMS (ESI+) m/z: [M + H]+ calculated for C21H26NO3S 372.1620, found 372.1628.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-6-methyl-3-((o-tolylthio)methyl)piperidin-2-one (4f). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 63% (47 mg; d.r. = 81:19). White solid. Rf = 0.54 (EtOAc/Cyclohexane 4:6). mp: 79 °C. IR (cm−1): 3406, 2925, 2244, 1639. 1HNMR(400 MHz, CDCl3, 20 °C) δ 7.50–7.45 (m, 2H), 7.41–7.32 (m, 4H), 7.22–7.07 (m, 3H), 5.06 (d, J = 9.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.45 (m, 1H), 4.04–4.01 (m, 1H), 3.82 (dd, J = 13.8, 3.9 Hz, 1H), 3.11 (dd, J = 13.8, 11.2 Hz, 1H), 2.65 (dt, J = 11.2, 3.4 Hz, 1H), 2.38 (s, 3H), 2.19 (dt, J = 14.2, 4.5 Hz, 1H), 2.04 (d, J = 4.3 Hz, 1H), 1.69 (ddd, J = 13.7, 11.4, 1.9 Hz, 1H), 1.32 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.0, 137.6, 135.4, 134.9, 130.5, 129.6 (2C), 128.8, 128.6 (2C), 127.7, 126.8, 126.0, 76.58, 64.05, 52.4, 46.9, 38.6, 29.3, 20.5, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C21H25NO3S 373.1660, found 373.1661.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((2,4-dimethylphenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4g). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 3 h. Overall isolated yield: 51% (46 mg; d.r. = 84:16). White solid. Rf = 0.59 (EtOAc/Cyclohexane 4:6). mp: 90 °C. IR (cm−1): 3391, 2988, 2923, 2288, 1980, 1886, 1722, 1638. 1H NMR(400 MHz, CDCl3, 20 °C) 7.51–6.95 (m, 8H), 5.04 (d, J = 9.5 Hz, 1H), 4.81 (d, J = 9.5 Hz, 1H), 4.45 (m, 1H), 4.06–3.97 (m, 1H), 3.76 (dd, J = 13.9, 4.0 Hz, 1H), 3.06 (dd, J = 13.9, 11.3 Hz, 1H), 2.61 (dt, J = 11.2, 3.5 Hz, 1H), 2.37 (s, 3H), 2.29 (s, 3H), 2.18 (m, 1H), 2.08 (d, J = 4.2 Hz, 1H), 1.68 (ddd, J = 13.7, 11.3, 1.9 Hz, 1H), 1.31 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.1, 138.2, 136.3, 135.4, 131.5, 131.0, 129.5 (2C), 129.1, 128.7, 128.6 (2C), 127.5, 76.5, 64.0, 52.4, 47.0, 38.5, 30.0, 21.0, 20.5, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C22H27NO3S 386.1740, found m/z 386.1780.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((2,6-dimethylphenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4h). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 6 h. Overall isolated yield: 30% (46 mg; d.r. = 64:36). White solid. Rf = 0.56 (EtOAc/Cyclohexane 4:6). mp: 92 °C. IR (cm−1): 3403, 2985, 2922, 1978, 1734, 1638. 1HNMR(400 MHz, CDCl3, 20 °C) δ 7.49–7.04 (m, 8H), 5.02 (d, J = 9.5 Hz, 1H), 4.78 (d, J = 9.5 Hz, 1H), 4.49 (m, 1H), 4.04–3.96 (m, 1H), 3.43 (dd, J = 13.2, 4.1 Hz, 1H), 3.00–2.85 (m, 1H), 2.56 (s, 6H), 2.53 (m, 1H), 2.21 (dt, J = 14.1, 4.5 Hz, 1H), 1.97 (d, J = 4.2 Hz, 1H), 1.71 (ddd, J = 13.7, 11.3, 1.9 Hz, 1H), 1.32 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.0, 142.9, 135.4, 133.2 (2C), 129.5 (2C), 128.7, 128.6 (2C), 128.5, 128.4 (2C), 76.6, 64.6, 52.5, 48.0, 38.7, 32.2, 22.2 (2C), 20.1. HRMS (ESI+) m/z: [M + H]+ calculated for C22H27NO3S 386.1784, found m/z 386.1784.
- (±)-(3S,4S,6S)-3-(([1,1′-Biphenyl]-4-ylthio)methyl)-1-(benzyloxy)-4-hydroxy-6-methylpiperidin-2-one (4i). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 3 h. Yield: 45% yield (87 mg; d.r. > 95:5). White solid. Rf = 0.73 (EtOAc/Cyclohexane 4:6). mp: 146 °C. IR (cm−1): 3378, 2960, 2935, 2162, 1963, 1632. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.61–7.29 (m, 14H), 5.06 (d, J = 9.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.47 (m, 1H), 4.07–3.98 (m, 1H), 3.91 (dd, J = 14.0, 4.0 Hz, 1H), 3.15 (dd, J = 14.0, 11.2 Hz, 1H), 2.66 (dt, J = 11.1, 3.5 Hz, 1H), 2.19 (dt, J = 14.1, 4.5 Hz, 1H), 2.02 (d, J = 4.1 Hz, 1H), 1.70 (ddd, J = 13.8, 11.4, 1.9 Hz, 1H), 1.32 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.0, 140.4, 139.4, 135.4, 134.7, 129.6 (2C), 129.5 (2C), 129.0 (2C), 128.8, 128.6 (2C), 127.9 (2C), 127.6, 127.0 (2C), 76.6, 63.9, 52.4, 47.0, 38.6, 30.2, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C26H27NO3S 434.1742, found 434.1782.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((4-(tert-butyl)phenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4j). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 3 h. Yield: 51% (43 mg; d.r. > 95:5). White solid. Rf = 0.55 (EtOAc/Cyclohexane 4:6). mp: 98 °C. IR (cm−1): 3395, 2961, 2869, 2287, 1642, 1498. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.49–7.31 (m, 9H), 5.05 (d, J = 9.5 Hz, 1H), 4.81 (d, J = 9.5 Hz, 1H), 4.47 (m, 1H), 4.05–3.97 (m, 1H), 3.82 (dd, J = 14.0, 4.1 Hz, 1H), 3.08 (dd, J = 14.0, 11.3 Hz, 1H), 2.62 (dt, J = 11.3, 3.6 Hz, 1H), 2.18 (m, 1H), 1.85 (d, J = 4.3 Hz, 1H), 1.69 (ddd, J = 13.8, 11.4, 1.9 Hz, 1H), 1.34–1.27 (m, 12H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.1, 149.9, 135.4, 131.9, 129.6 (2C), 129.5 (2C), 128.8, 128.6 (2C), 126.4 (2C), 76.6, 63.9, 52.4, 47.0, 38.5, 34.6, 31.4, 30.7 (3C), 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C24H31NO3S 414.2097, found 414.2091.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-6-methyl-3-((naphthalen-2-ylthio)methyl) piperidin-2-one (4k). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 4 h. Yield: 42% (35 mg; d.r. > 95:5). White solid. Rf = 0.68 (EtOAc/Cyclohexane 4:6). mp: 140 °C. IR (cm−1): 3376, 3054, 2965, 2853, 1745, 1642, 1593. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.86–7.72 (m, 4H), 7.53–7.30 (m, 8H), 5.06 (d, J = 9.5 Hz, 1H), 4.81 (d, J = 9.5 Hz, 1H), 4.43 (m, 1H), 4.06–3.89 (m, 2H), 3.20 (dd, J = 14.0, 11.3 Hz, 1H), 2.67 (dt, J = 11.3, 3.5 Hz, 1H), 2.17 ((dt, J = 14.2, 4.5 Hz, 1H), 1.91 (d, J = 4.3 Hz, 1H), 1.67 (ddd, J = 13.7, 11.5, 1.7 Hz, 1H), 1.31 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.1, 135.4, 133.9, 133.0, 132.0, 129.6, 128.9, 128.8, 128.6, 127.9, 127.3, 127.1 (2C), 126.9 (3C), 126.0, 76.6, 63.9, 52.4, 46.9, 38.6, 30.0, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C24H25NO3S 408.1628, found m/z 408.1624.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-3-(((4-methoxyphenyl)thio)methyl)-6-methylpiperidin-2-one (4l). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Yield: 41% (32 mg; d.r. > 95:5). White solid. Rf = 0.35 (EtOAc/Cyclohexane 4:6). mp: 85 °C. IR (cm−1): 3377, 1638, 1493, 1242, 823. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.87–7.84 (m, 2H), 7.47–7.45 (m, 2H), 7.39–7.33 (m, 5H), 5.03 (d, J = 9.5 Hz, 1H), 4.79 (d, J = 9.5 Hz, 1H), 4.47 (m, 1H), 4.04–3.96 (m, 1H), 3.80 (s, 3H), 3.72 (dd, J = 14.0, 4.1 Hz, 1H), 3.03 (dd, J = 14.1, 11.2 Hz, 1H), 2.54 (dt, J = 11.2, 3.6 Hz, 1H), 2.18 (dt, J = 14.1, 4.5 Hz, 1H), 1.90 (d, J = 4.3 Hz, 1H), 1.73–1.63 (m, 1H), 1.31 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.2, 159.2, 135.4, 132.9 (2C), 129.55 (2C), 128.7, 128.6 (2C), 125.6, 115.0 (2C), 76.6, 63.8, 55.5, 52.4, 47.0, 38.5, 32.4, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C21H25NO4S 388.1577, found 388.1575.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-3-(((2-methoxyphenyl)thio)methyl)-6-methylpiperidin-2-one (4m). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 46% (36 mg; d.r. = 82:18). White solid. Rf = 0.37 (EtOAc/Cyclohexane 4:6). mp: 108 °C. IR (cm−1): 3380, 2968, 2901, 2833, 1637. 1HNMR(400 MHz, CDCl3, 20 °C) δ 7.50–7.19 (m, 7H), 6.94 (td, J = 7.5, 1.2 Hz, 1H), 6.87 (dd, J = 8.2, 1.1 Hz, 1H), 5.04 (d, J = 9.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.50 (m, 1H), 4.07–3.99 (m, 1H), 3.90 (s, 3H), 3.77 (dd, J = 13.5, 4.0 Hz, 1H), 3.07 (dd, J = 13.5, 11.4 Hz, 1H), 2.63 (dt, J = 11.4, 3.5 Hz, 1H), 2.44 (d, J = 3.7 Hz, 1H), 2.20 (dt, J = 14.1, 4.4 Hz, 1H), 1.77–1.61 (m, 1H), 1.32 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.1, 157.8, 135.4, 130.8, 129.5 (2C), 128.7, 128.6 (2C), 128.1, 123.4, 121.5, 110.9, 76.5, 64.0, 56.0, 52.4, 46.8, 38.4, 29.5, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C21H25NO4S 388.1577, found 388.1571.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-6-methyl-3-(((4-(trifluoromethyl)phenyl)thio) methyl)piperidin-2-one (4n). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. White oil. Yield: 32% yield (28 mg; d.r. > 95:5). Rf = 0.53 (EtOAc/Cyclohexane 4:6). IR (cm−1): 3379, 2928, 2894, 1745, 1639, 1604, 1328. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.53–7.32 (m, 9H), 5.06 (d, J = 9.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.39 (m, 1H), 4.06–3.97 (m, 1H), 3.91 (dd, J = 14.0, 3.8 Hz, 1H), 3.17 (dd, J = 14.0, 11.1 Hz, 1H), 2.63 (dt, J = 11.0, 3.4 Hz, 1H), 2.20 (m, 1H), 2.16 (t, J = 4.5 Hz, 1H), 1.76–1.62 (m, 1H), 1.32 (d, J = 6.3 Hz, 3H) 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.8, 141.4, 135.2, 129.6 (2C), 128.8, 128.6 (2C), 127.8 (q, J = 33 Hz), 127.4 (2C), 126.0 (q, J = 4Hz, 2C), 124.2 (q, J = 270 Hz), 76.7, 63.9, 52.5, 46.9, 38.6, 28.8, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C21H22F3NO3S 426.1345, found 426.1343.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((4-chlorophenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4o). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. White solid. Yield: 55% (44 mg; d.r. > 95:5). Rf = 0.55 (EtOAc/Cyclohexane 4:6). mp: 136 °C. IR (cm−1): 3406, 3059, 2899, 1639, 1589, 1453. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.47 (dd, J = 7.4, 2.1 Hz, 2H), 7.40–7.24 (m, 7H), 5.05 (d, J = 9.5 Hz, 1H), 4.81 (d, J = 9.5 Hz, 1H), 4.42 (m, 1H), 4.05–3.97 (m, 1H), 3.82 (dd, J = 14.0, 4.0 Hz, 1H), 3.10 (dd, J = 14.0, 11.1 Hz, 1H), 2.59 (dt, J = 11.0, 3.5 Hz, 1H), 2.18 (dt, J = 14.1, 4.5 Hz, 1H), 1.90 (d, J = 4.5 Hz, 1H), 1.69 (ddd, J = 13.8, 11.4, 1.8 Hz, 1H), 1.32 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.8, 138.0, 135.4, 135.0, 130.3, 129.6 (2C), 128.8, 128.6 (2C), 128.5, 126.7, 126.4, 76.6, 64.0, 52.4, 46.9, 38.6, 30.0, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C20H22ClNO3S 392.1082, found 392.1081.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((4-bromophenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4p). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Yield: 55% (48 mg; d.r. = 95:5). Orange solid. Rf = 0.52 (EtOAc/Cyclohexane 4:6). mp: 145 °C. IR (cm−1): 3379, 2925, 2854, 2444, 1923, 1787, 1649, 1595, 1507. 1H NMR(400 MHz, CDCl3, 20 °C) δ 7.55–7.11 (m, 9H), 5.06 (d, J = 9.5 Hz, 1H), 4.84 (d, J = 9.5 Hz, 1H), 4.48 (m, 1H), 4.03 (m, 1H), 3.86 (dd, J = 13.7, 3.7 Hz, 1H), 3.15 (dd, J = 13.7, 11.3 Hz, 1H), 2.67 (dt, J = 11.3, 3.3 Hz, 1H), 2.20 (dt, J = 14.1, 4.4 Hz, 1H), 1.95 (d, J = 4.4 Hz, 1H), 1.71 (ddd, J = 13.8, 11.4, 1.9 Hz, 1H), 1.33 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.7, 135.4, 135.3, 133.5, 129.9, 129.6 (2C), 128.8, 128.6 (2C), 128.3, 127.6, 126.8, 76.6, 64.0, 52.4, 46.7, 38.6, 29.2, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C20H23BrNO3S 436.0571, found 436.0577.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((3-chlorophenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4q). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 42% (33 mg; d.r. = 79:21). White solid. Rf = 0.51 (EtOAc/Cyclohexane 4:6). mp: 130 °C. IR (cm−1): 3377, 2967, 2927, 2050, 1639, 1578. 1H NMR (300 MHz, CDCl3, 20 °C) δ 7.51–7.11 (m, 9H), 5.06 (d, J = 9.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.42 (m, 1H), 4.05–3.97 (m, 1H), 3.84 (dd, J = 13.9, 4.0 Hz, 1H), 3.12 (dd, J = 13.9, 11.1 Hz, 1H), 2.62 (dt, J = 11.3, 3.5 Hz, 1H), 2.19 (dt, J = 14.2, 4.5 Hz, 1H), 1.84 (d, J = 4.3 Hz, 1H), 1.71 (dd, J = 14.2, 11.7 Hz, 1H), 1.33 (d, J = 6.2 Hz, 3H). 13C NMR (75 MHz, CDCl3, 20 °C) δ (ppm) 168.8, 138.0, 135.3, 135.0, 130.3, 129.6 (2C), 128.8, 128.6 (2C), 128.3, 126.6, 126.3, 76.6, 63.9, 52.5, 46.9, 38.6, 29.9, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C20H22ClNO3S 392.1082, found 392.1080.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((3-bromophenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4r). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 42% (37 mg; d.r. = 79:21). Orange solid. Rf = 0.48 (EtOAc/Cyclohexane 4:6). IR (cm−1): 3355, 2934, 2865, 1925, 1756, 1652. 1H NMR (400 MHz, CDCl3, 20 °C) δ 7.51–7.14 (m, 9H), 5.06 (d, J = 9.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.42 (m, 1H), 4.05–3.97 (m, 1H), 3.84 (dd, J = 13.9, 4.0 Hz, 1H), 3.12 (dd, J = 13.9, 11.2 Hz, 1H), 2.63 (dt, J = 11.0, 3.3 Hz, 1H), 2.24–2.13 (m, 1H), 1.88 (d, J = 4.2 Hz, 1H), 1.69 (d, J = 13.4 Hz, 1H), 1.33 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.8, 138.3, 135.4, 131.3, 130.6, 129.6 (2C), 129.3, 128.8, 128.7, 128.6 (2C), 127.8, 127.1, 76.6, 64.0, 52.4, 46.9, 38.7, 30.0, 20.0. HRMS (ESI+) C20H23BrNO3S 436.0573, found 436.0576.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((2-chlorophenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4s). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 54% (43 mg; d.r. = 95:5). Pink solid. Rf = 0.53 (EtOAc/Cyclohexane 4:6). mp: 126 °C. IR (cm−1): 3376, 2991, 2928, 2162, 1980, 1824, 1637. 1H NMR (400 MHz, CDCl3, 20 °C) δ 7.53–7.30 (m, 7H), 7.25 (td, J = 7.7, 1.4 Hz, 1H), 7.13 (td, J = 7.7, 1.5 Hz, 1H), 5.06 (d, J = 9.5 Hz, 1H), 4.84 (d, J = 9.5 Hz, 1H), 4.48 (m, 1H), 4.07–3.99 (m, 1H), 3.86 (dd, J = 13.7, 3.7 Hz, 1H), 3.16 (dd, J = 13.7, 11.3 Hz, 1H), 2.66 (dt, J = 11.4, 3.4 Hz, 1H), 2.20 (dt, J = 14.2, 4.5 Hz, 1H), 2.02 (d, J = 4.4 Hz, 1H), 1.71 (ddd, J = 13.8, 11.4, 1.9 Hz, 1H), 1.33 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.7, 135.4, 135.2, 133.5, 129.9, 129.6 (2C), 128.8, 128.6 (2C), 128.4, 127.6, 126.8, 76.6, 64.0, 52.4, 46.5, 38.6, 29.2, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C20H22ClNO3S 392.1082, found 392.1081.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-(((2-bromophenyl)thio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4t). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 54% (47 mg; d.r. = 89:11). White solid. Rf = 0.52 (EtOAc/Cyclohexane 4:6). mp: 143 °C. IR (cm−1): 3377, 2961, 2869, 2287, 1641, 1498. 1H NMR (400 MHz, CDCl3, 20 °C) δ 7.53–7.30 (m, 7H), 7.25 (td, J = 7.7, 1.4 Hz, 1H), 7.13 (td, J = 7.7, 1.5 Hz, 1H), 5.06 (d, J = 9.5 Hz, 1H), 4.84 (d, J = 9.5 Hz, 1H), 4.48 (m, 1H), 4.08–3.99 (m, 1H), 3.86 (dd, J = 13.7, 3.7 Hz, 1H), 3.16 (dd, J = 13.7, 11.3 Hz, 1H), 2.66 (dt, J = 11.4, 3.4 Hz, 1H), 2.20 (dt, J = 14.2, 4.5 Hz, 1H), 2.02 (d, J = 4.4 Hz, 1H), 1.71 (ddd, J = 13.8, 11.4, 1.9 Hz, 1H), 1.33 (d, J = 6.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 168.7, 137.3, 135.3, 133.2, 129.6 (2C), 128.8, 128.6 (2C), 128.2, 128.1, 126.9, 123.5, 76.6, 64.0, 52.4, 46.7, 38.6, 29.6, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C20H22BrNO3S 436.0573, found 436.0579.
- (±)-(3S,4S,6S)-3-(((4-Aminophenyl)thio)methyl)-1-(benzyloxy)-4-hydroxy-6-methylpiperidin-2-one (4u). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 3a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 55% (41 mg; d.r. = 91:9). Yellow solid. Rf = 0.15 (EtOAc/Cyclohexane 4:6). mp: 48 °C. IR (cm−1): 3340, 2937, 2244, 1626, 1615. 1H NMR (400 MHz, CDCl3, 20 °C) δ 7.46–7.21 (m, 7H), 7.07 (td, J = 7.7, 1.6 Hz, 1H), 5.01 (d, J = 9.5 Hz, 1H), 4.78 (d, J = 9.5 Hz, 1H), 4.47 (m, 1H), 4.03–3.94 (m, 1H), 3.78 (s, 2H), 3.65 (dd, J = 14.0, 4.1 Hz, 1H), 2.97 (dd, J = 14.0, 11.2 Hz, 1H), 2.58–2.48 (m, 1H), 2.16 (dt, J = 14.1, 4.4 Hz, 1H), 1.77 (bs, 1H), 1.65 (ddd, J = 13.8, 11.4, 2.0 Hz, 1H), 1.30 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.3, 147.1, 135.3, 133.9, 129.4 (2C), 128.6, 128.5 (2C), 125.7 (2C), 115.4 (2C), 76.5, 63.6, 52.3, 46.9, 38.4, 32.8, 19.9. HRMS (ESI+) m/z: [M + H]+ calculated for C20H24N2O3S 373.1573, found 373.1569.
- (±)-(3S,4S,6S)-3-(((2-Aminophenyl)thio)methyl)-1-(benzyloxy)-4-hydroxy-6-methylpiperidin-2-one (4v). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 2 h. Overall isolated yield: 49% (37 mg; d.r. = 84:16). Yellow solid. Rf = 0.18 (EtOAc/Cyclohexane 4:6). mp: 51 °C. IR (cm−1): 3346, 2928, 2245, 1635, 1606. 1H NMR (400 MHz, CDCl3, 20 °C) δ 7.46–7.16 (m, 6H), 7.07 (td, J = 7.7, 1.6 Hz, 1H), 6.71–6.60 (m, 2H), 5.02 (d, J = 9.5 Hz, 1H), 4.78 (d, J = 9.6 Hz, 1H), 4.48 (m, 1H), 4.38 (s, 2H), 4.03–3.94 (m, 1H), 3.64 (dd, J = 13.7, 4.3 Hz, 1H), 2.94 (dd, J = 13.7, 10.7 Hz, 1H), 2.55 (dt, J = 10.6, 3.5 Hz, 1H), 2.18 (dt, J = 14.1, 4.5 Hz, 1H), 2.00 (bs, 1H), 1.70 (ddd, J = 13.8, 11.4, 1.9 Hz, 1H), 1.31 (d, J = 6.1 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.4, 148.2, 135.5 (2C), 130.0, 129.5 (2C), 128.7, 128.6 (2C), 118.9, 117.3, 115.5, 76.6, 64.2, 52.5, 47.6, 38.5, 31.4, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C20H24N2O3S 373.1573, found m/z 373.1580.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-3-((benzylthio)methyl)-4-hydroxy-6-methylpiperidin-2-one (4w). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 3 h. White solid. Yield: 11% (8 mg; d.r. > 95:5). Rf = 0.63 (EtOAc/Cyclohexane 4:6). mp: 84 °C IR (cm−1): 3405, 2988, 2923, 2288, 1980, 1886, 1722, 1638, 1495. 1H NMR (400 MHz, CDCl3, 20 °C) δ 7.49–7.03 (m, 10H), 4.94 (d, J = 9.5 Hz, 1H), 4.72 (d, J = 9.5 Hz, 1H), 4.20 (m, 1H), 3.93–3.84 (m, 1H), 3.68 (s, 2H), 3.19 (dd, J = 13.3, 4.3 Hz, 1H), 2.66 (dd, J = 13.2, 10.9 Hz, 1H), 2.42 (dt, J = 10.7, 3.5 Hz, 1H), 2.07 (dt, J = 14.0, 4.4 Hz, 1H), 1.93 (m, 1H), 1.54–1.51 (m, 1H), 1.22 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.1, 138.5, 135.4, 129.5 (2C), 129.0 (2C), 128.8 (2C), 128.7, 128.6 (2C), 127.4, 76.5, 64.4, 52.5, 47.1, 38.4, 37.3, 28.8, 20.0. HRMS (ESI+) m/z: [M + H]+ calculated for C21H25NO3S 372.1628, found 372.1626.
- (±)-(3S,4S,6S)-1-(Benzyloxy)-4-hydroxy-6-methyl-3-((propylthio)methyl)piperidin-2-one (4x). Prepared using general procedure B from N-(benzyloxy)-N-(4-oxobutan-2-yl)acrylamide 2a (0.2 mmol, 50 mg, 1 equiv). Reaction time: 30 min + 3 h. White solid. Yield: 15% (10 mg; d.r. > 95:5). Rf = 0.51 (EtOAc/Cyclohexane 4:6). mp: 75 °C. IR (cm−1): 3398, 2966, 2935, 2901, 2876, 1815, 1650, 1636. 1H NMR (400 MHz, CDCl3, 20 °C) δ 7.49–7.46 (m, 2H), 7.38–7.33 (m,3H), 5.04 (d, J = 9.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.39 (m, 1H), 4.07–3.98 (m, 1H), 3.25 (dd, J = 13.3, 4.4 Hz, 1H), 2.86 (dd, J = 13.2, 10.7 Hz, 1H), 2.64 (dq, J = 10.7, 4.1, 1H), 2.58–2.48 (m, 2H), 2.31 (d, J = 3.7 Hz, 1H), 2.21 (dt, J = 14.1, 4.5 Hz, 1H), 1.81–1.54 (m, 3H), 1.34 (d, J = 6.3 Hz, 3H), 0.99 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3, 20 °C) δ (ppm) 169.1, 135.4, 129.6 (2C), 128.7, 128.6 (2C), 76.6, 64.5, 52.5, 47.2, 38.6, 34.9, 29.2, 23.0, 20.1, 13.6. HRMS (ESI+) m/z: [M + H]+ calculated for C17H25NO3S 324.1628, found 324.1620.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferro, C.T.B.; Santos, B.F.; Silva, C.D.G.; Brand, G.; Silva, B.A.L.; Domingues, N.L.C. Review of the Syntheses and Activities of Some Sulfur-Containing Drugs. Curr. Org. Synth. 2020, 17, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Ahangarpour, M.; Kavianinia, I.; Brimble, M.A. Thia-Michael addition: The route to promising opportunities for fast and cysteine-specific modification. Org. Biomol. Chem. 2023, 21, 3057–3072. [Google Scholar] [CrossRef] [PubMed]
- Berne, D.; Ladmiral, V.; Leclerc, E.; Caillol, S. Thia-Michael Reaction: The Route to Promising Covalent Adaptable Networks. Polymers 2022, 14, 4457. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.; Kharbanda, A.; Sharma, A. Thia-Michael Addition: An Emerging Strategy in Organic Synthesis. Asian J. Org. Chem. 2018, 7, 634–661. [Google Scholar] [CrossRef]
- Niu, C.; Du, D.-M. Recent Advances in Organocatalyzed Asymmetric Sulfa-Michael Addition Triggered Cascade Reactions. Chem. Rec. 2023, 23, e202200258. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, Y.; Zhao, J.; Luo, S. CPA-catalyzed asymmetric domino thia-Michael/aldol reactions for simultaneous chiral center and axial chirality formation. Org. Biomol. Chem. 2023, 21, 6697–6701. [Google Scholar] [CrossRef] [PubMed]
- Alahyen, I.; Benhamou, L.; Dalla, V.; Taillier, C.; Comesse, S. 20 Years of Forging N-Heterocycles from Acrylamides Through Domino/Cascade Reactions. Synthesis 2021, 53, 3409–3439. [Google Scholar]
- Kamimura, A.; Omata, Y.; Mitsudera, H.; Kakehi, A. A simple preparation of syn-NH-amide aldols and amide-Baylis-Hillman adducts via a Michael-aldol tandem process. J. Chem. Soc. Perkin Trans. 1 2000, 4499–4504. [Google Scholar] [CrossRef]
- Zu, L.; Wang, J.; Li, H.; Xie, H.; Jiang, W.; Wang, W. Cascade Michael-Aldol Reactions Promoted by Hydrogen Bonding Mediated Catalysis. J. Am. Chem. Soc. 2007, 129, 1036–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-P.; You, Y.; Zhao, J.-Q.; Zhang, X.-M.; Xu, X.-Y.; Yuan, W.-C. Chiral Bifunctional Amine-Squaramide-Catalyzed Highly Diastereoand Enantioselective Michael/Aldol Cascade Reaction of 2-Mercaptobenzaldehyde and α,β-Unsaturated 7-Azaindoline Amides. J. Org. Chem. 2019, 84, 7984–7994. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Miyazaki, T.; Ogawa, H.; Masu, H. PyBidine-Ni(OAc)2-Catalyzed Michael/Aldol Reaction of Methyleneindolinones and Thiosalicylaldehydes for Stereochemically Divergent Thiochromanyl-spirooxindoles. Org. Lett. 2016, 18, 5824–5827. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Li, Z.-H.; Lian, P.-F.; Li, Q.-Z.; She, Y.; Ma, Z.-G.; Zhang, S.-Y. Stereoselective Sulfa-Michael/Aldol Reaction Promoted by an Axially Chiral Styrene-Based Organocatalyst. Org. Lett. 2023, 25, 6913–6918. [Google Scholar] [CrossRef] [PubMed]
- Champetter, P.; Castillo-Aguilera, O.; Taillier, C.; Brière, J.-F.; Dalla, V.; Oudeyer, S.; Comesse, S. N-Alkoxyacrylamides in Domino Reactions: Catalytic and Stereoselective Access to δ-Lactams. Eur. J. Org. Chem. 2019, 2019, 7703–7710. [Google Scholar] [CrossRef]
- Alahyen, I.; Taillier, C.; Lhoste, J.; Dalla, V.; Comesse, S. N-Benzyloxyacrylamides as bis-Nucleophiles in an Organocatalyzed Domino aza-Michael/Morita-Baylis-Hillman Sequence. Org. Lett. 2024, 26, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Champetter, P.; Alahyen, I.; Taillier, C.; Brière, J.-F.; Dalla, V.; Oudeyer, S.; Comesse, S. Probing N-Alkoxy Effects in Domino Reactions of α-Bromoacetamide Derivatives Towards Functionalized γ-Lactams. ChemistrySelect 2022, 7, e202203305. [Google Scholar] [CrossRef]
- Khodair, A.I.; Al-Masoudi, N.A.; Gesson, J.-P. A new approach to the synthesis of benzothiazole, benzoxazole, and pyridine nucleosides as potential antitumor agents. Nucleosides Nucleotides Nucleic Acids 2003, 22, 2061–2076. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. “SHELXL-2014”, Program for Crystal Structure Determination; Universität of Göttingen: Göttingen, Germany, 2014. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Additive (equiv.) | Solvent | T °C | Overall Yield 4a (%) 2 | Ratio 3 4a1:4a2:4a3 | Yield 5a (%) 2 |
| 1 | - | CHCl3 | 20 | 0 | - | 8 |
| 2 | TBAF (0.2) | CHCl3 | 20 | 55 | 44:0:56 | 11 |
| 3 | TBAT (0.2) | CHCl3 | 20 | 0 | - | 8 |
| 4 | tBuOK (0.2) | CHCl3 | 20 | 45 | 84:16:0 | 2 |
| 5 | Na2CO3 (0.2) | CHCl3 | 20 | 0 | - | - |
| 6 | Cs2CO3 (0.2) | CHCl3 | 20 | 29 | 93:7:0 | 2 |
| 7 | K2CO3 (0.2) | CHCl3 | 20 | 37 | 95:0:5 | 3 |
| 8 | K2CO3 (0.2) | CHCl3 | 40 | 46 | 87:0:13 | 10 |
| 9 | K2CO3 (1.0) | CHCl3 | 40 | 62 | 87:13:0 | 5 |
| 10 | K2CO3 (1.0) | CH2Cl2 | 40 | n.d. 4 | - | n.o. 5 |
| 11 | K2CO3 (1.0) | DCE | 40 | n.d. 4 | - | n.o. 5 |
| 12 | K2CO3 (1.0) | MeCN | 40 | 24 | 100:0:0 | 20 |
| 13 | K2CO3 (1.0) | toluene | 40 | 36 | 100:0:0 | 6 |
| 14 | K2CO3 (1.0) | DMF | 40 | 44 | 67:11:22 | 6 |
| 15 | K2CO3 (1.0) | Et2O | 40 | 44 | 77:23:0 | 6 |
| 16 | K2CO3 (1.0) | THF | 40 | 68 | 88:12:0 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genty, A.; Alahyen, I.; Tranchant, M.-J.; Lhoste, J.; Dalla, V.; Taillier, C.; Comesse, S. Straightforward Access to Polyfunctionalized δ-Lactams via Domino Aza–Michael/Thia–Michael/Aldol Sequence. Molecules 2025, 30, 2154. https://doi.org/10.3390/molecules30102154
Genty A, Alahyen I, Tranchant M-J, Lhoste J, Dalla V, Taillier C, Comesse S. Straightforward Access to Polyfunctionalized δ-Lactams via Domino Aza–Michael/Thia–Michael/Aldol Sequence. Molecules. 2025; 30(10):2154. https://doi.org/10.3390/molecules30102154
Chicago/Turabian StyleGenty, Axelle, Ismail Alahyen, Marie-José Tranchant, Jérôme Lhoste, Vincent Dalla, Catherine Taillier, and Sébastien Comesse. 2025. "Straightforward Access to Polyfunctionalized δ-Lactams via Domino Aza–Michael/Thia–Michael/Aldol Sequence" Molecules 30, no. 10: 2154. https://doi.org/10.3390/molecules30102154
APA StyleGenty, A., Alahyen, I., Tranchant, M.-J., Lhoste, J., Dalla, V., Taillier, C., & Comesse, S. (2025). Straightforward Access to Polyfunctionalized δ-Lactams via Domino Aza–Michael/Thia–Michael/Aldol Sequence. Molecules, 30(10), 2154. https://doi.org/10.3390/molecules30102154