
Diastereoselective Transfer Hydrogenation of Cyclic and Bicyclic Ketones over Selected Metal Oxides as Catalysts
 , and
, and
Abstract

1. Introduction
- -
- Aliphatic ketones with increasing steric hindrance in close proximity to the carbonyl group caused by the introduction of an increasing number of methyl substituents at both α-carbons [14];
- -
- Straight-chained aliphatic ketones with 11 carbon atoms in the chain, in which the carbonyl group was present in different positions along the chain (positions 2–6) [14];
- -
- 1-phenyl-1-alkanones substituted with alkyl groups in the ring and in the side chain [15];
- -
- Diaryl ketones with different substituents present in the aromatic rings [16];
- -
- -
- 4-t-butylcyclohexanone [19];
- -
- 1- and 2-acetylnaphthalenes, and diacylbenzenes [20].
2. Results and Discussion
2.1. Characterization of Catalysts
2.1.1. Scanning Electron Microscopy Coupled with Energy-Dispersive X-Ray Spectroscopy (SEM-EDX)
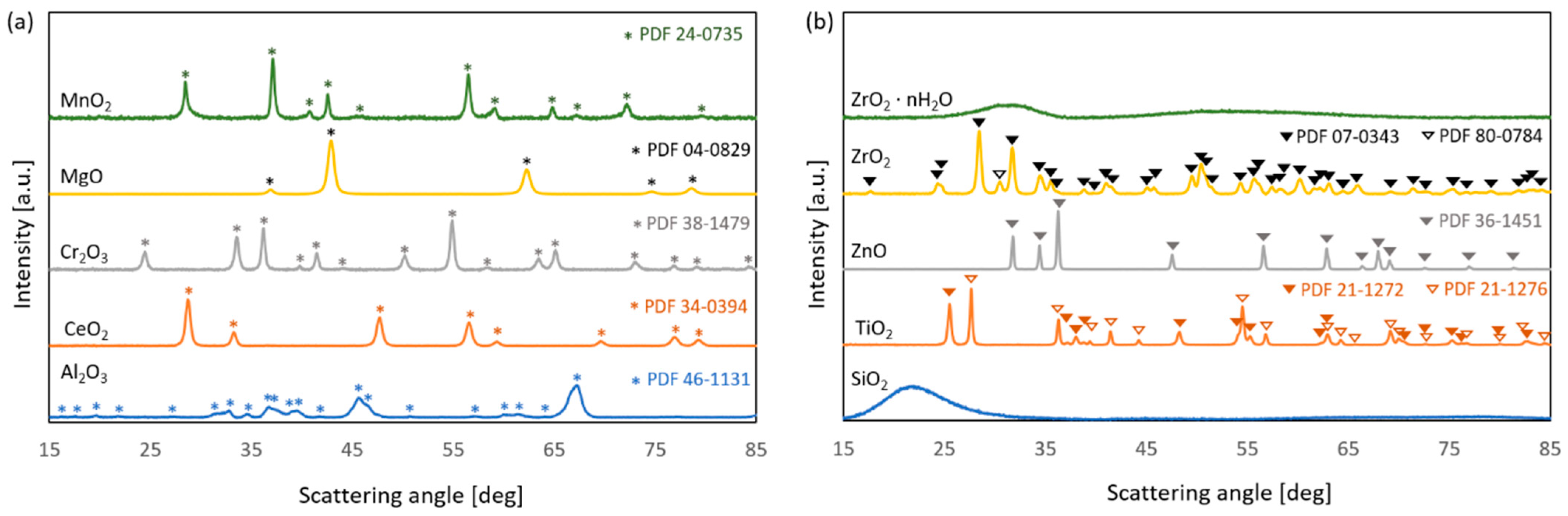
2.1.2. Powder X-Ray Diffraction (PXRD) and Nitrogen Physisorption
2.1.3. Acid/Base Properties
2.2. Activity Measurements
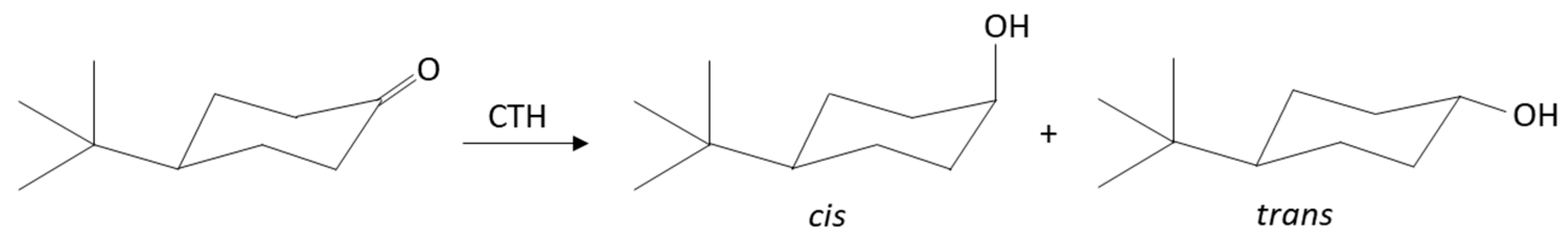
2.2.1. Liquid- and Vapor-Phase Transfer Hydrogenation of 4-t-Butylcyclohexanone
2.2.2. Liquid- and Vapor-Phase Transfer Hydrogenation of x-Methylcyclohexanones (x = 2, 3 or 4)
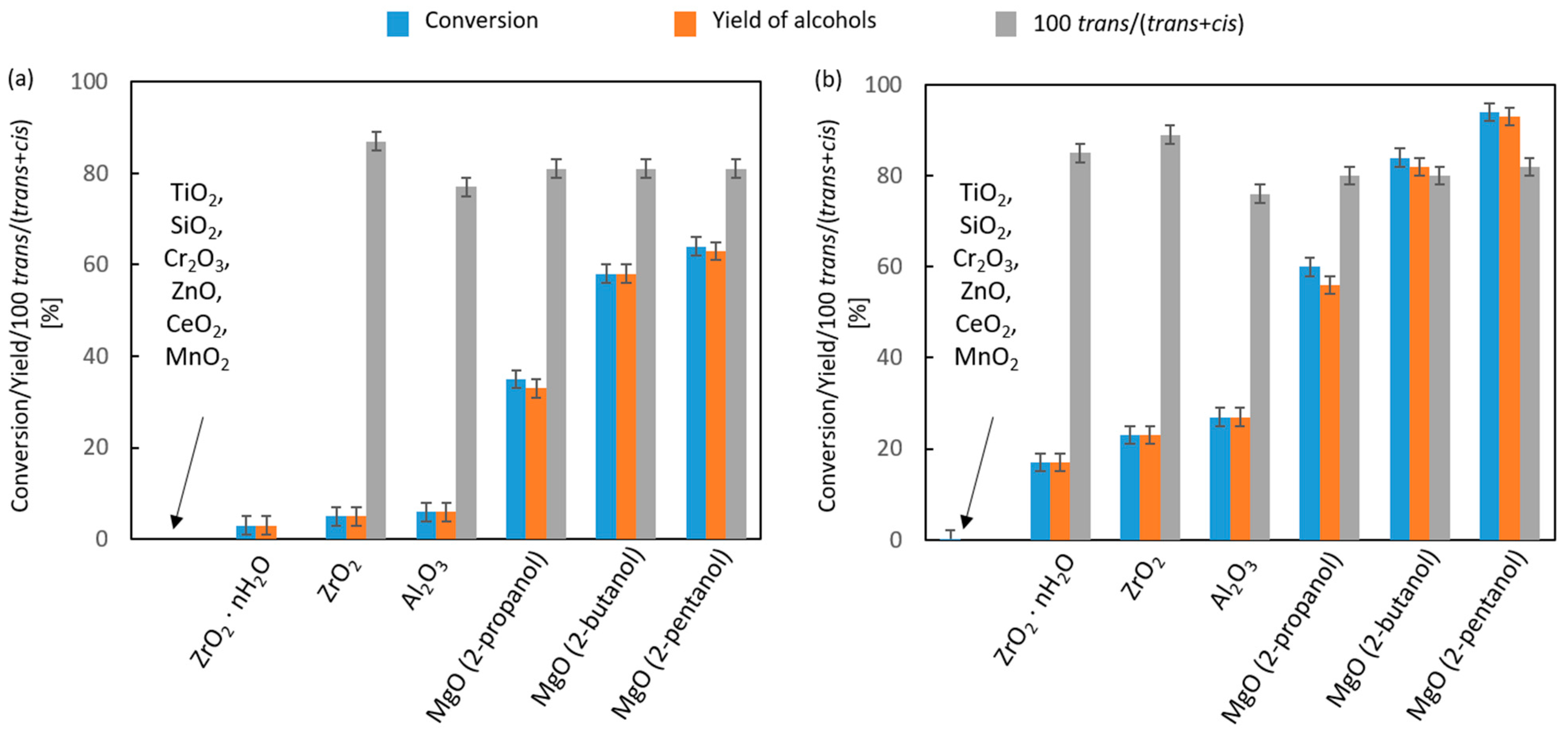
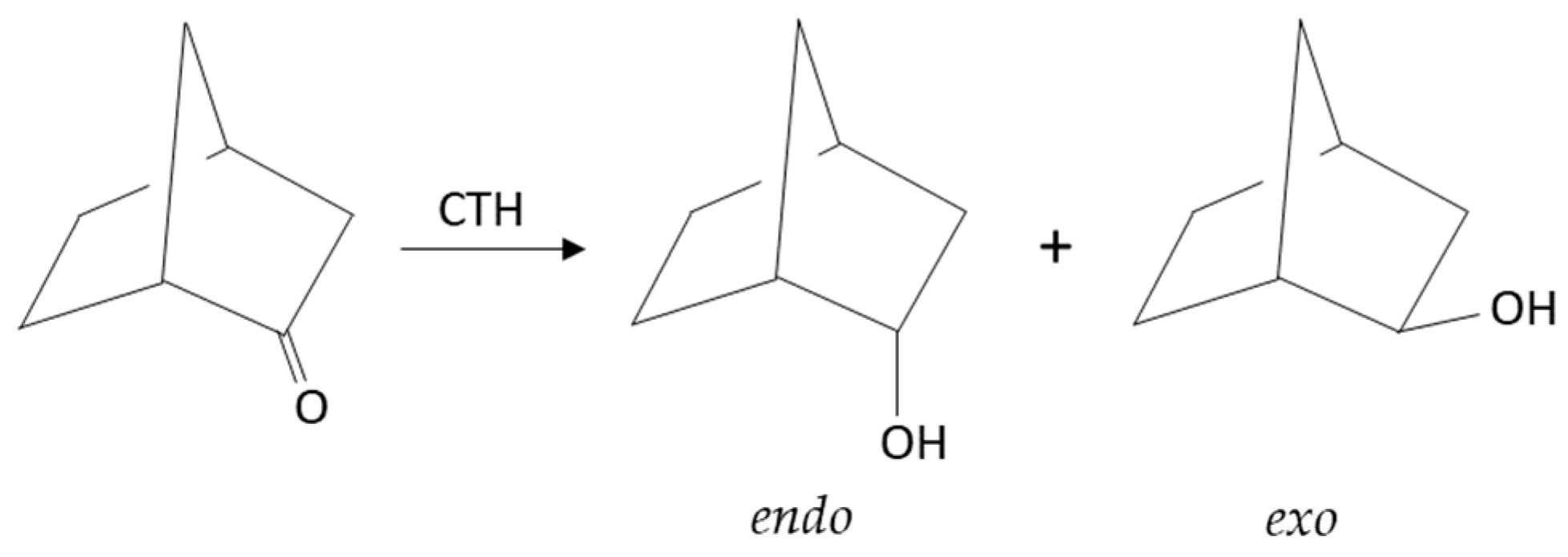
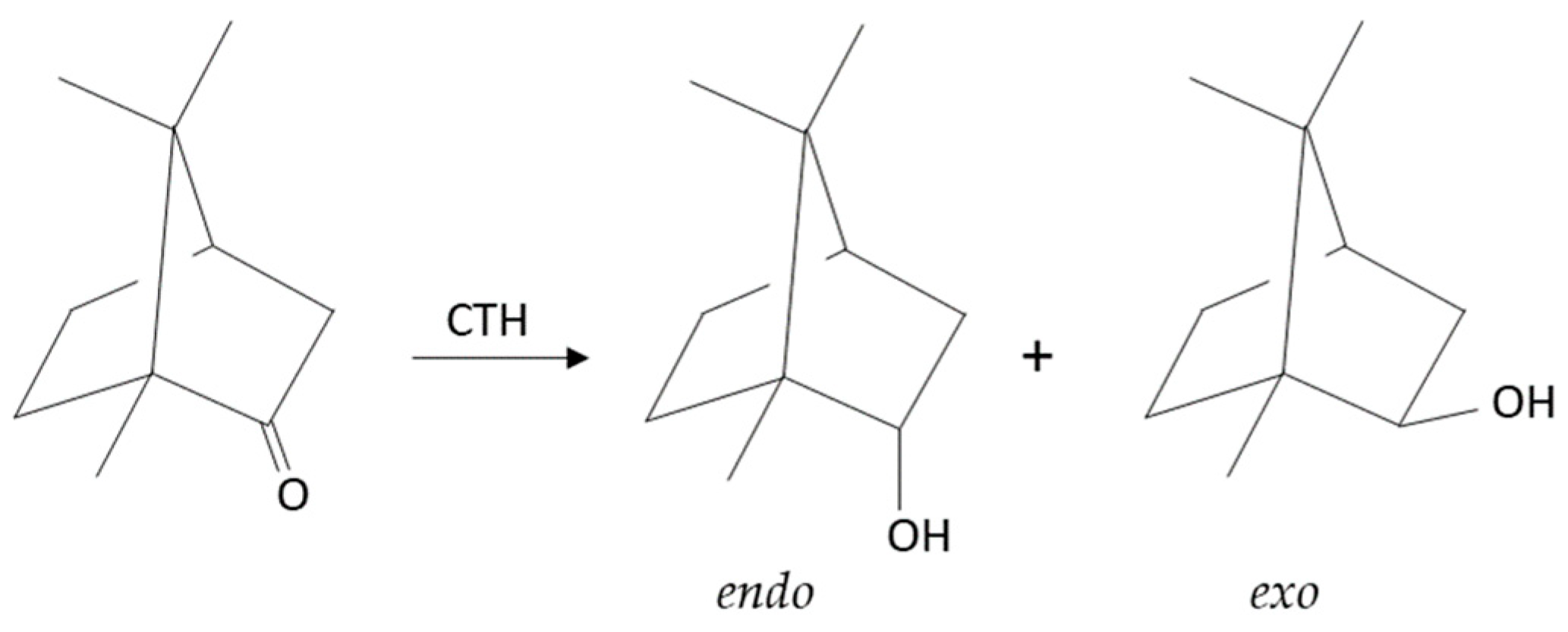
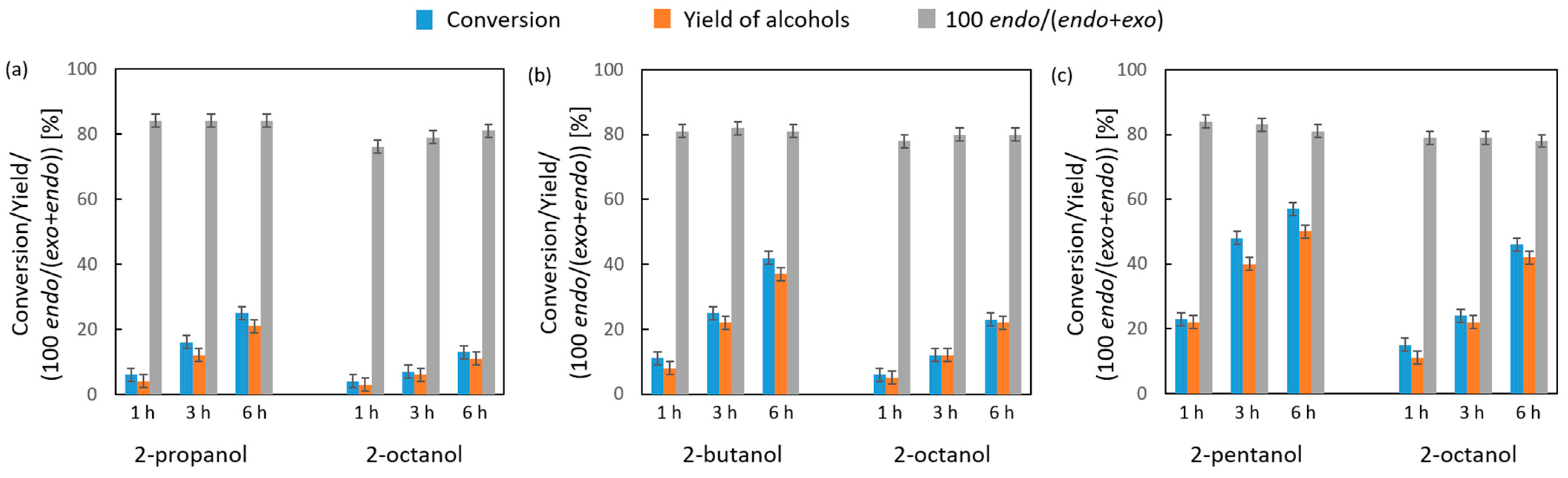
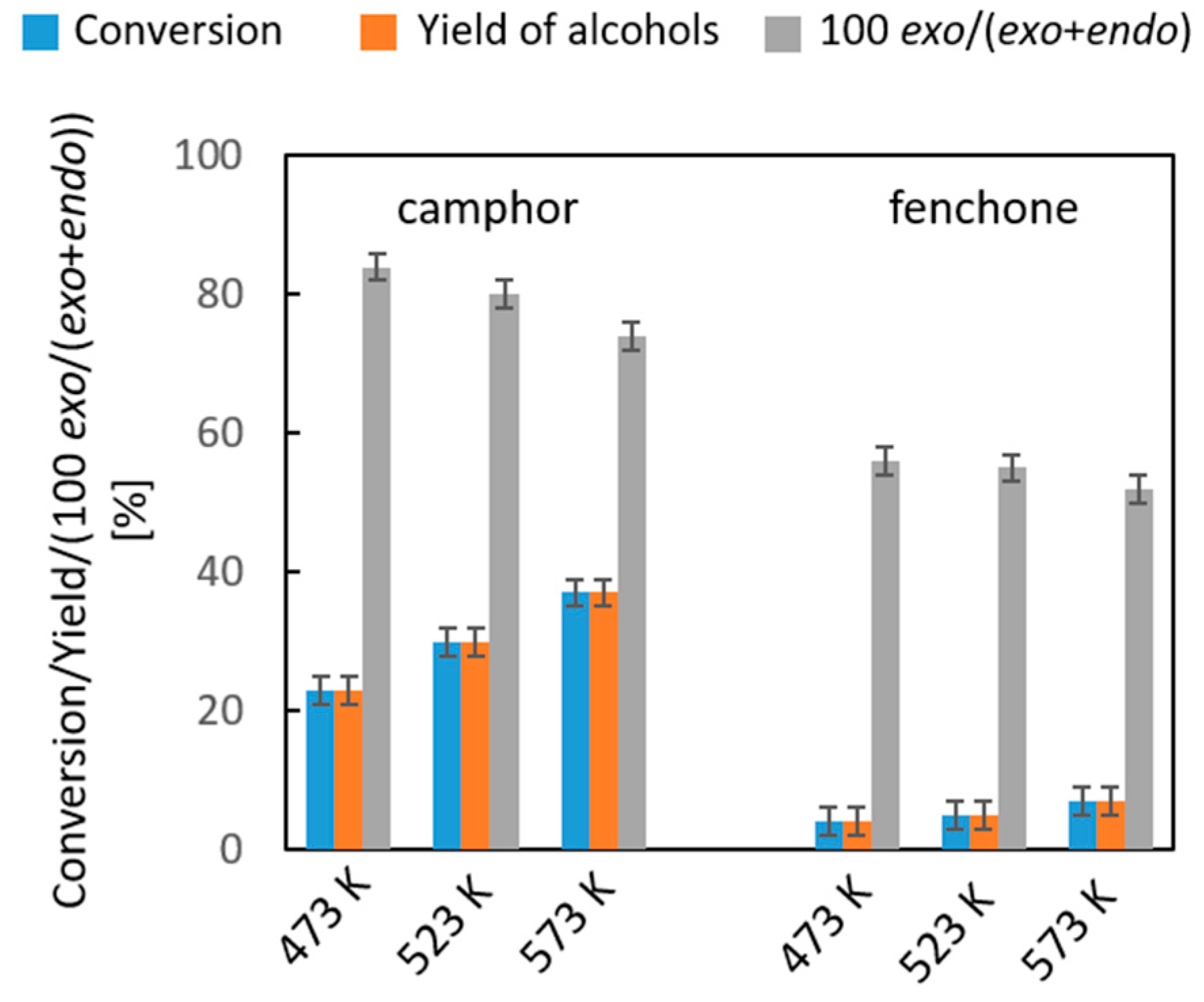
2.2.3. Liquid- and Vapor-Phase Transfer Hydrogenation of Bicyclic Ketones: 2-Norbornanone, Camphor, and Fenchone
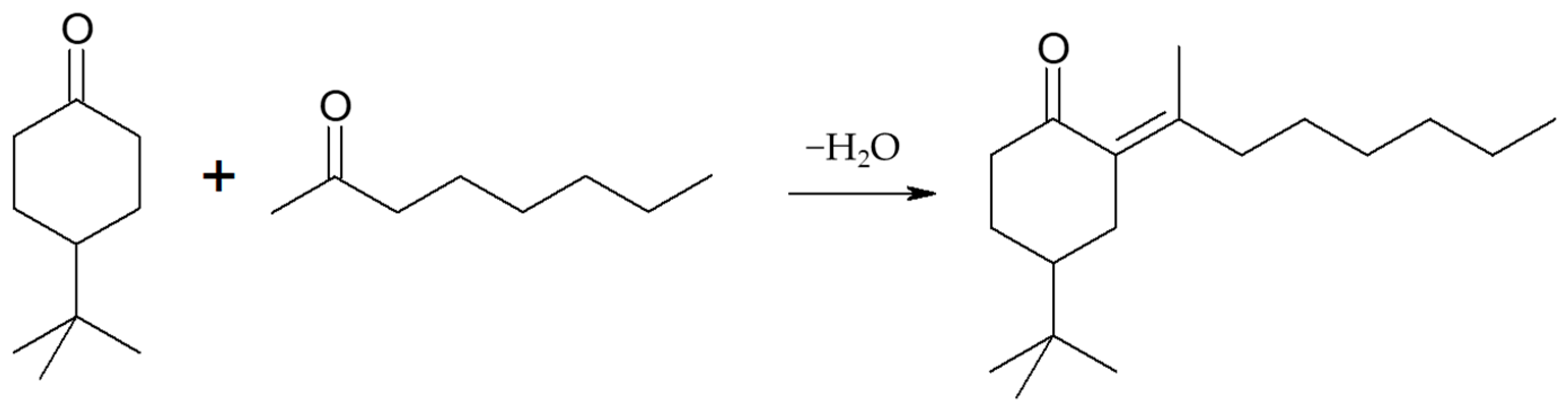
2.2.4. Liquid-Phase Transfer Hydrogenation of 2-Adamantanone
3. Materials and Methods
3.1. Catalysts
3.2. Hydrogen Acceptors
3.3. Hydrogen Donors
3.4. Liquid-Phase Catalytic Activity Measurements
3.5. Vapor-Phase Catalytic Activity Measurements
3.6. Determination of the Composition of Post-Reaction Mixtures
3.7. XRD Analysis
3.8. Scanning Electron Microscopy Coupled with Energy-Dispersive X-Ray Spectroscopy (SEM-EDX)
3.9. Nitrogen Physisorption
3.10. DTA-TGA Measurements
3.11. Strength and Concentration of Acidic/Basic Sites of Studied Metal Oxides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taleb, B.; Jahjah, R.; Cornu, D.; Bechelany, M.; Al Ajami, M.; Kataya, G.; Hijazi, A.; El-Dakdouki, M.H. Exploring Hydrogen Sources in Catalytic Transfer Hydrogenation: A Review of Unsaturated Compound Reduction. Molecules 2023, 28, 7541. [Google Scholar] [CrossRef]
- Chowdhury, R.L.; Bäckvall, J.-E. Efficient ruthenium-catalysed transfer hydrogenation of ketones by propan-2-ol. J. Chem. Soc. Chem. Commun. 1991, 1063–1064. [Google Scholar] [CrossRef]
- Zweifel, T.; Naubron, J.V.; Büttner, T.; Ott, T.; Grützmacher, H. Ethanol as Hydrogen Donor: Highly Efficient Transfer Hydrogenations with Rhodium(I) Amides. Angew. Chem. Int. Ed. 2008, 47, 3245–3249. [Google Scholar] [CrossRef]
- Sklyaruk, J.; Zubar, V.; Borghs, J.C.; Rueping, M. Methanol as the Hydrogen Source in the Selective Transfer Hydrogenation of Alkynes Enabled by a Manganese Pincer Complex. Org. Lett. 2020, 22, 6067–6071. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Wozniak, B.; Savini, A.; Hinze, S.; Tin, S.; de Vries, J.G. Base-Free Iron Catalyzed Transfer Hydrogenation of Esters Using EtOH as Hydrogen Source. Angew. Chem. Int. Ed. 2018, 58, 1129–1133. [Google Scholar] [CrossRef]
- Patil, R.D.; Pratihar, S. Recent developments on selective homogeneous catalytic transfer hydrogenation of C=C using methanol and ethanol. Tetrahedron Lett. 2025, 156, 155439. [Google Scholar] [CrossRef]
- Baidilov, D.; Hayrapetyan, D.; Khalimon, A.Y. Recent advances in homogeneous base-metal-catalyzed transfer hydrogenation reactions. Tetrahedron 2021, 98, 132435. [Google Scholar] [CrossRef]
- Heiden, Z.M.; Rauchfuss, T.B. Homogeneous Catalytic Reduction of Dioxygen Using Transfer Hydrogenation Catalysts. J. Am. Chem. Soc. 2007, 129, 14303–14310. [Google Scholar] [CrossRef]
- Vermaak, V.; Vosloo, H.C.M.; Swarts, A.J. The development and application of homogeneous nickel catalysts for transfer hydrogenation and related reactions. Coord. Chem. Rev. 2024, 507, 215716. [Google Scholar] [CrossRef]
- Anshika, M.F.A.; Sortais, J.-B.; Elangovan, S. Transition-Metal-Catalysed Transfer Hydrogenation Reactions with Glycerol and Carbohydrates as Hydrogen Donors. Eur. J. Org. Chem. 2024, 27, e202301278. [Google Scholar]
- Johnstone, R.A.; Wilby, A.H.; Entwistle, I.D. Heterogeneous catalytic transfer hydrogenation and its relation to other method for reduction of organic compounds. Chem. Rev. 1985, 85, 129–170. [Google Scholar] [CrossRef]
- Chuah, G.K.; Jaenicke, S.; Zhu, Y.Z.; Liu, S.H. Meerwein-Ponndorf-Verley Reduction of Carbonyl Compounds. Curr. Org. Chem. 2006, 10, 1639–1654. [Google Scholar] [CrossRef]
- Ruiz, J.R.; Sanchidrian, C.J. Heterogeneous Catalysis in the Meerwein-Ponndorf-Verley Reduction of Carbonyl Compounds. Curr. Org. Chem. 2007, 11, 1113–1125. [Google Scholar] [CrossRef]
- Gliński, M. Structure-reactivity relationship in transfer hydrogenation of aliphatic ketones over magnesium oxide. React. Kinet. Catal. Lett. 2009, 97, 275–279. [Google Scholar] [CrossRef]
- Gliński, M. Catalytic hydrogen transfer over magnesia. Vapour and liquid phase reduction of various aralkyl ketones. Appl. Catal. A Gen. 2008, 349, 133–139. [Google Scholar] [CrossRef]
- Gliński, M.; Markowska, A.; Wrońska, L.; Jerzak, A.; Tarkowska, M. Highly selective vapor and liquid phase transfer hydrogenation of diaryl and polycyclic ketones into alcohols in the presence of magnesium oxide as catalyst. Catalysts 2021, 11, 574. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Vapour phase transfer hydrogenation of α,β-unsaturated carbonyl compounds. Thermodynamic and experimental studies. Appl. Catal. A Gen. 2016, 511, 131–140. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Description of the structure-chemoselectivity relationship in the transfer hydrogenation of α,β-unsaturated aldehydes and ketones with alcohols in the presence of magnesium oxide. Appl. Catal. A Gen. 2018, 554, 117–124. [Google Scholar] [CrossRef]
- Gliński, M. Highly diastereoselective transfer hydrogenation of 4-tert-butylcyclohexanone in the presence of magnesium oxide. Reac. Kinet. Mechan. Catal. 2010, 99, 93–98. [Google Scholar]
- Gliński, M.; Dubinin, O.; Rostek, K.; Waniek, P. Chemoselective transfer hydrogenation over MgO as the catalyst. Acetylnaphthalenes, diacylbenzenes, acetophenone, benzaldehyde and various aliphatic ketones as hydrogen acceptors. Reactions 2025, 6, 4. [Google Scholar] [CrossRef]
- Dauben, W.G.; Fonken, G.J.; Noyce, D.S. The Stereochemistry of Hydride Reductions. J. Amer. Chem. Soc. 1956, 78, 2579–2582. [Google Scholar] [CrossRef]
- Richer, J.-C. On the Stereochemistry of the Reduction of Cyclic Ketones with Lithium Tri- t-butoxyaluminum Hydride. J. Org. Chem. 1965, 30, 324–325. [Google Scholar] [CrossRef]
- Brown, H.C.; Muzzio, J. Rates of Reaction of Sodium Borohydride with Bicyclic Ketones. Steric Approach Control and Steric Departure Control in the Reactions of Rigid Bicyclic Systems. J. Am. Chem. Soc. 1966, 88, 2811–2822. [Google Scholar] [CrossRef]
- Hach, V.; Fryberg, E.C.; McDonald, E. Epimerization in the NaBH4 reduction of asymmetric ketones. Tetrahedron Lett. 1971, 28, 2629–2632. [Google Scholar] [CrossRef]
- Ashby, E.C.; Sevenair, J.P.; Dobbs, F.R. Concerning the Stereoselectivity of Lithium Tri-tert-butoxyaluminum Hydride. J. Org. Chem. 1971, 36, 197–199. [Google Scholar] [CrossRef]
- Yoon, N.M.; Kim, K.E.; Kang, J. Potassium Triphenylborohydride. A New Reducing Agent for the Reduction of Carbonyl Compounds with an Exceptional Stereo- and Chemoselectivity. J. Org. Chem. 1986, 51, 226–229. [Google Scholar] [CrossRef]
- Okano, T.; Matsuoka, M.; Konishi, H.; Kiji, J. Meerwein-Ponndorf-Verley Reduction of Ketones and Aldehydes Catalyzed by Lanthanide Tri-2-propoxides. Chem. Lett. 1987, 16, 181–184. [Google Scholar] [CrossRef]
- Huffman, J.W.; Charles, J.T. The Metal-Ammonia Reduction of Ketones. J. Am. Chem. Soc. 1968, 90, 6486–6492. [Google Scholar] [CrossRef]
- Ramana, D.V.; Pillai, C.N. Hydrogen Transfer Reactions: Part II—Stereochemistry of the Reduction of (-)-Menthone by Alcohols Catalysed by Alumina. Indian J. Chem. 1970, 8, 1106–1108. [Google Scholar]
- Shibagaki, M.; Takahashi, K.; Kuno, H.; Kawakami, H.; Matsushita, H. Vapor-phase Reduction of Aldehydes and Ketones with 2-Propanol over Hydrous Zirconium Oxide. Chem. Lett. 1988, 17, 1633–1636. [Google Scholar] [CrossRef]
- Creyghton, E.J.; Ganeshie, S.D.; Downing, R.S.; van Bekkum, H. Stereoselective Meerwein-Ponndorf-Verley and Oppenauer catalysed by zeolite BEA. J. Mol. Catal. A Chem. 1997, 115, 457–472. [Google Scholar] [CrossRef]
- Corma, A.; Domine, M.E.; Valencia, S. Water-resistant Lewis acid catalysts: Meerwein-Ponndorf-Verley and Oppenauer reactions catalysed by tin-beta zeolite. J. Catal. 2003, 215, 294–304. [Google Scholar] [CrossRef]
- Corma, A.; Domine, M.E.; Nemeth, L.; Valencia, S. Al-free Sn-Beta Zeolite as a Catalyst for the Selective Reduction of Carbonyl Compounds (Meerwein-Ponndorf-Verley Reaction). J. Amer. Chem. Soc. 2002, 124, 3194–3195. [Google Scholar] [CrossRef] [PubMed]
- Picquart, M.; López, T.; Gómez, R.; Torres, E.; Moreno, A.; Garcia, J. Dehydration and Crystallization Process in Sol–Gel Zirconia Thermal and Spectroscopic Study. J. Therm. Anal. Calor. 2004, 76, 755–761. [Google Scholar] [CrossRef]
- Winstein, S.; Holness, N.J. Neighboring Carbon and Hydrogen. XIX. t-Butylcyclohexyl Derivatives. Quantitative Conformational Analysis. J. Am. Chem. Soc. 1955, 77, 5562–5578. [Google Scholar] [CrossRef]
- Eliel, E.L.; Ro, H.S. Conformational Analysis. III. Epimerization Equilibria of Alkylcyclohexanols. J. Amer. Chem. Soc. 1957, 79, 5992–5994. [Google Scholar] [CrossRef]
- Szöllösi, G.; Bartók, M. Vapour-phase heterogeneous catalytic transfer hydrogenation of alkyl methyl ketones on MgO: Prevention of the deactivation of MgO in the presence of carbon tetrachloride. Appl. Catal. A Gen. 1998, 169, 263–269. [Google Scholar] [CrossRef]
- Wilcox Jr, C.F.; Sexton, M.; Wilcox, M.F. Equilibration of Bicyclic Alcohols. J. Org. Chem. 1963, 28, 1079–1082. [Google Scholar] [CrossRef]
- Coulombeau, A.; Rassat, A. Réduction de la fenchone, épimérisation des fenchols. Bull. Soc. Chim. France 1965, 3338–3343. [Google Scholar]
- Gliński, M.; Ulkowska, U. Reactivity of alcohols in chemoselective transfer hydrogenation of acrolein over magnesium oxide as the catalyst. Catal. Lett. 2011, 141, 293–299. [Google Scholar] [CrossRef]
- Motsui, S.; Saito, H.; Yamashita, Y.; Kaminaga, M.; Senda, Y. Stereochemistry and mechanism of catalytic hydrogenation of substituted cyclohexanones. Tetrahedron 1973, 29, 1531–1539. [Google Scholar] [CrossRef]
- Kleinfelder, C.D.; von Schleyer, P. 2-Norbornanone. Org. Synth. 1962, 42, 79–81. [Google Scholar]
- Iwanek, E.; Ulkowska, U.; Gliński, M. Surface studies of magnesium oxide-based catalysts modified with X2 or MgX2 (X = Br, I). Surf. Interface Anal. 2015, 47, 1001–1008. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyclic Ketone | Reducing Agent | Catalyst | Mode of Reaction 1 | Ref. |
|---|---|---|---|---|
| x-methylcyclohexanones | LiAlH4, NaBH4, Al(Oi-C3H7)3, NaBH(OCH3)3 | none | LP | [21] |
| LiAlH(O-t-Bu)3 | none | LP | [22] | |
| M0 + NH3 (liq) M0 = Li, Na, K, and Rb | none | LP | [28] | |
| LiAlH(OCH3)3 | none | LP | [25] | |
| KHBPh3 | none | LP | [26] | |
| 2-propanol, 2-butanol | Al-free Sn-Beta zeolite | LP | [33] | |
| menthone | LiAlH4, NaBH4, Al(Oi-C3H7)3, NaBH(OCH3)3 | none | LP | [21] |
| Aliphatic alcohols | Al2O3 + 2 wt% Na | VP | [29] | |
| 2-Propanol | ZrO2·nH2O | VP | [30] | |
| 4-t-butylcyclohexanone | LiAlH(O-t-Bu)3 | none | LP | [22] |
| KHBPh3 | none | LP | [26] | |
| 2-propanol | Gd(Oi-C3H7)3 | LP | [27] | |
| 2-propanol | ZrO2·nH2O | VP | [30] | |
| 2-propanol | Zeolite BEA (Si/Al = 12) | VP | [31] | |
| 2-propanol, 2-butanol | Al-free Sn-Beta zeolite | LP | [32] | |
| Aliphatic alcohols | MgO | LP, VP | [19] | |
| 2-norbornanone | LiAlH(OCH3)3 | none | LP | [25] |
| KHBPh3 | none | LP | [26] | |
| M0 + NH3 (liq) M0 = Li, Na, K, and Rb | none | LP | [28] | |
| camphor | LiAlH(OCH3)3 | none | LP | [25] |
| KHBPh3 | none | LP | [26] | |
| M0 + NH3 (liq) M0 = Li, Na, K, and Rb | none | LP | [28] | |
| 2-Propanol | ZrO2·nH2O | VP | [30] |
| Catalyst | 2θ [°]/(hkl) | Crystallite Size [nm] | SBET [m2·g−1] |
|---|---|---|---|
| Al2O3 | 67.1/(042) | 12 | 103 |
| MgO | 42.9/(200) | 12 | 100 |
| ZrO2·nH2O | -- | -- 1 | 235 |
| ZrO2 | 28.2/(111) | 18 | 32 |
| Cr2O3 | 54.9/(116) | 20 | 21 |
| MnO2 | 37.3/(101) | 22 | 2 |
| SiO2 | -- | -- 1 | 253 |
| TiO2 | 25.3/(101) | 23 2 | 35 |
| 27.4/(110) | 31 3 | ||
| ZnO | 36.2/(101) | 33 | 4 |
| CeO2 | 47.6/(220) | 13 | 7 |
| Catalyst 1 | H0 | H− | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| −5.6 | −3.0 | 0.8 | 4.8 | 7.2 | 9.3 | 15.0 | 18.4 | 22.3 | 26.5 | 33.0 | |
| Al2O3 | − | + | + | + | + | − | − | − | − | − | − |
| MgO | − | − | − | − | + | + | + | + | + | + | − |
| ZrO2·nH2O | − | + | + | + | + | + | + | + | + | − | − |
| ZrO2 | − | − | + | + | + | + | + | − | − | − | − |
| CeO2 | − | − | + | + | + | + | + | − | − | − | − |
| TiO2 | − | − | + | + | + | − | − | − | − | − | − |
| ZnO | − | − | − | + | + | + | − | − | − | − | − |
| SiO2 | − | − | − | + | + | − | − | − | − | − | − |
| Catalyst | Acidic Sites | Basic Sites | ||
|---|---|---|---|---|
| [µmol·g−1] | [µmol·m−2] | [µmol·g−1] | [µmol·m−2] | |
| Al2O3 | 242.0 | 2.3 | 541.4 | 5.3 |
| MgO | 76.6 | 0.8 | 1174.8 | 11.7 |
| ZrO2·nH2O | 398.7 | 1.7 | 732.2 | 3.1 |
| ZrO2 | 100.9 | 3.2 | 119.4 | 3.7 |
| Cr2O3 | 179.7 | 8.6 | 51.0 | 2.4 |
| MnO2 | 42.8 | 21.4 | 0.4 | 0.2 |
| SiO2 | 321.1 | 1.3 | 3.4 | ~0 |
| TiO2 | 147.8 | 4.2 | 114.5 | 3.3 |
| ZnO | 40.6 | 10.2 | 32.3 | 8.1 |
| CeO2 | 121.4 | 17.3 | 186.6 | 26.7 |
| Catalyst | Time [h] | Conversion [%] | Yield of Alcohols [%] | [100 trans/(trans + cis)] 1 [%] |
|---|---|---|---|---|
| SiO2 | 3 | 1 | 1 | n.d. |
| 6 | 2 2 | 2 | n.d. | |
| CeO2 | 3 | 2 | 2 | n.d. |
| 6 | 4 | 4 | n.d. | |
| ZnO | 3 | 5 | 5 | 58 |
| 6 | 9 | 8 | 58 | |
| MnO2 | 3 | 5 | 5 | 60 |
| 6 | 10 | 9 | 61 | |
| TiO2 | 3 | 6 | 6 | 86 |
| 6 | 12 | 11 | 87 | |
| Cr2O3 | 3 | 12 | 12 | 83 |
| 6 | 23 | 22 | 82 | |
| ZrO2 | 1 | 78 | 77 | 90 |
| 3 | 88 | 87 | 89 | |
| ZrO2·nH2O | 1 | 73 | 71 | 87 |
| 3 | 92 | 89 | 86 | |
| Al2O3 | 1 | 91 | 74 | 87 |
| 3 | 96 | 66 | 88 | |
| MgO | 1 | 95 | 88 | 79 |
| 3 | 98 | 86 | 71 |
| Catalyst | Time [h] | Conversion [%] | Yield of Alcohols [%] | [100 trans/(trans + cis] 1 [%] |
|---|---|---|---|---|
| ZrO2 | 1 | 20 | 19 | 95 |
| 3 | 47 | 45 | 96 | |
| ZrO2·nH2O | 1 | 36 | 36 | 95 |
| 3 | 70 | 69 | 95 | |
| Al2O3 | 1 | 47 | 47 | 91 |
| 3 | 78 | 77 | 90 | |
| MgO | 1 | 80 2 | 79 | 97 |
| 3 | 86 | 82 | 97 |
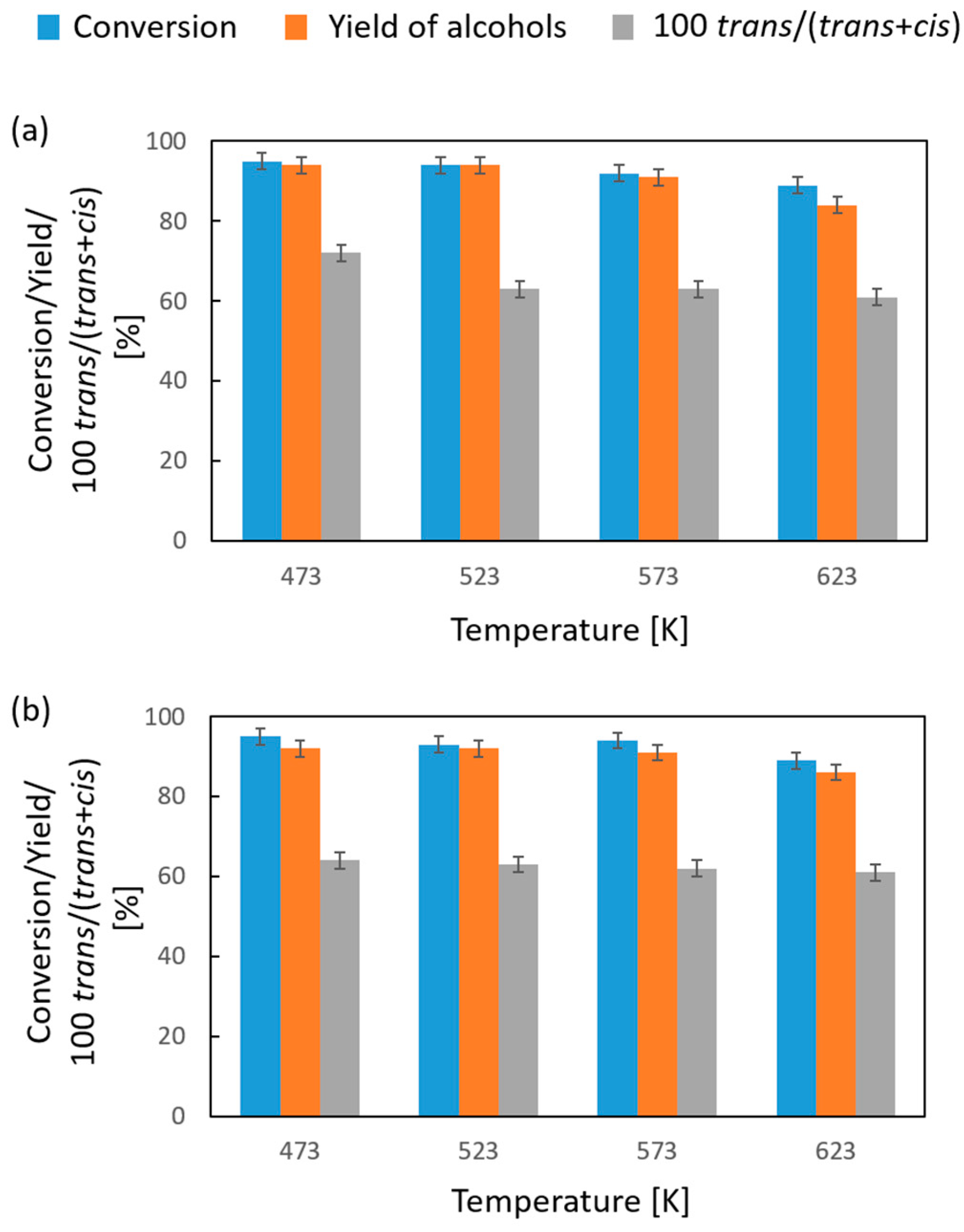
| D/A | T [K] | Conversion [%] | Yield of Alcohols [%] | [100 trans/(trans + cis)] 1 [%] |
|---|---|---|---|---|
| 3 | 473 | 85 | 82 | 67 |
| 523 | 84 | 81 | 65 | |
| 573 | 84 | 81 | 64 | |
| 623 | 84 | 81 | 63 | |
| 6 | 473 | 97 | 92 | 63 |
| 523 | 96 | 91 | 62 | |
| 573 | 96 | 91 | 60 | |
| 623 | 95 | 90 | 59 |
| Catalyst | Time [h] | Conversion [%] | Yield of Alcohols [%] | [100 trans/(trans + cis)] 1 [%] |
|---|---|---|---|---|
| Al2O3 | 1 | 15 | 15 | 22 |
| 6 | 56 | 55 | 21 | |
| MgO | 1 | 68 | 64 | 5 |
| 6 | 76 | 69 | 7 | |
| MgO 2 | 1 | 66 | 64 | 19 |
| 6 | 86 | 82 | 18 | |
| MgO 3 | 1 | 80 | 78 | 22 |
| 6 | 96 | 92 | 21 |
| Catalyst | Time [h] | Conversion [%] | Yield of Alcohols [%] | [100 endo/(endo + exo)] 1 [%] |
|---|---|---|---|---|
| MOx 2 | 6 | <0.2 | n.d. | -- |
| Cr2O3 | 1 | 13 | 13 | 78 |
| 6 | 16 | 16 | 77 | |
| Al2O3 | 1 | 18 | 14 | 80 |
| 6 | 64 | 54 | 79 | |
| MgO | 1 | 31 | 30 | 79 |
| 6 | 72 | 67 | 75 | |
| ZrO2 | 1 | 32 | 30 | 79 |
| 6 | 74 | 71 | 73 | |
| ZrO2·nH2O | 1 | 43 | 39 | 74 |
| 6 | 82 | 79 | 70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gliński, M.; Armusiewicz, D.; Łukasik-Kwaśniewska, K.; Materowski, M.; Rułka, A.; Iwanek, E.M.; Kucharska, M. Diastereoselective Transfer Hydrogenation of Cyclic and Bicyclic Ketones over Selected Metal Oxides as Catalysts. Molecules 2025, 30, 2153. https://doi.org/10.3390/molecules30102153
Gliński M, Armusiewicz D, Łukasik-Kwaśniewska K, Materowski M, Rułka A, Iwanek EM, Kucharska M. Diastereoselective Transfer Hydrogenation of Cyclic and Bicyclic Ketones over Selected Metal Oxides as Catalysts. Molecules. 2025; 30(10):2153. https://doi.org/10.3390/molecules30102153
Chicago/Turabian StyleGliński, Marek, Dorota Armusiewicz, Karolina Łukasik-Kwaśniewska, Michał Materowski, Adam Rułka, Ewa M. Iwanek (nee Wilczkowska), and Monika Kucharska. 2025. "Diastereoselective Transfer Hydrogenation of Cyclic and Bicyclic Ketones over Selected Metal Oxides as Catalysts" Molecules 30, no. 10: 2153. https://doi.org/10.3390/molecules30102153
APA StyleGliński, M., Armusiewicz, D., Łukasik-Kwaśniewska, K., Materowski, M., Rułka, A., Iwanek, E. M., & Kucharska, M. (2025). Diastereoselective Transfer Hydrogenation of Cyclic and Bicyclic Ketones over Selected Metal Oxides as Catalysts. Molecules, 30(10), 2153. https://doi.org/10.3390/molecules30102153