
PROTAC Technology as a New Tool for Modern Pharmacotherapy




Abstract
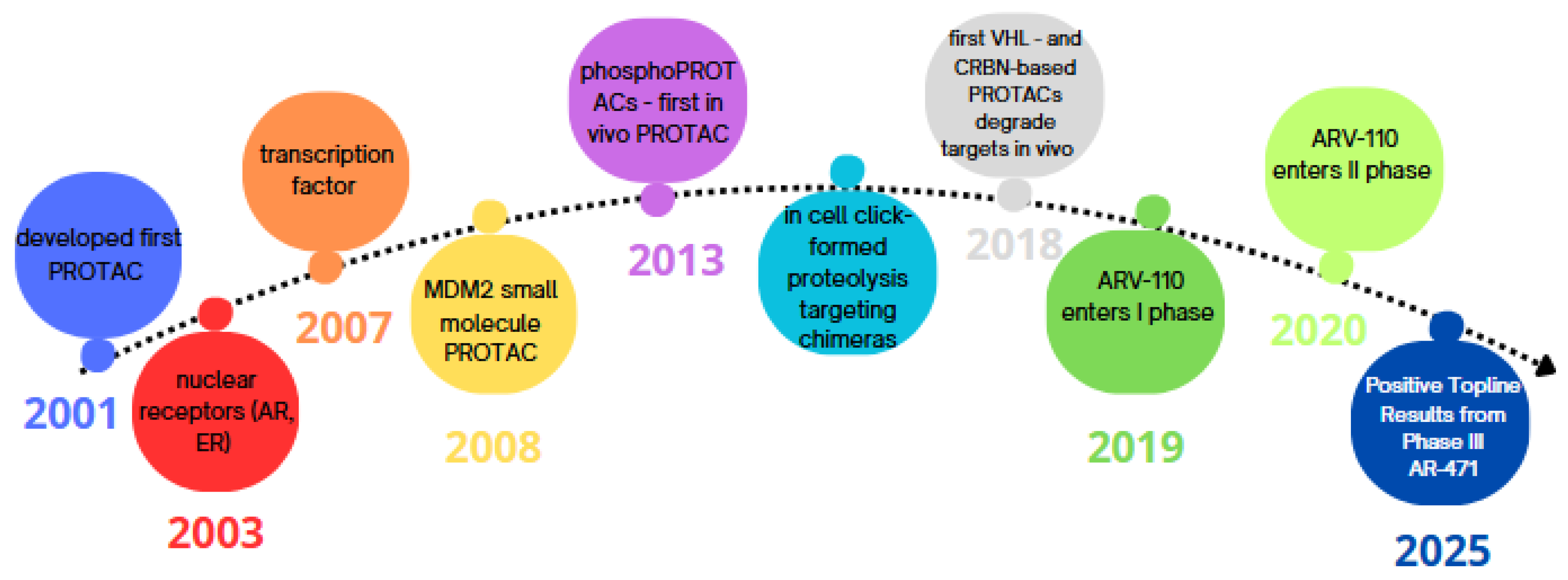
1. Introduction
2. PROTAC Design
2.1. Protein of Interest (POI)
- Pathogenic alterations: the target should exhibit pathogenic gain-of-function alterations, such as overexpression, mutations, or changes in localisation [14].
- Ligand-binding pocket: it should possess a site where the PROTAC molecule can attach [15].
- Ubiquitination site: there should be a location on the target’s surface that allows for the binding of the E3 ubiquitin ligase [16].
- Structure: the target should have a flexible structure that can be processed by the proteasome [17].
2.2. Linker
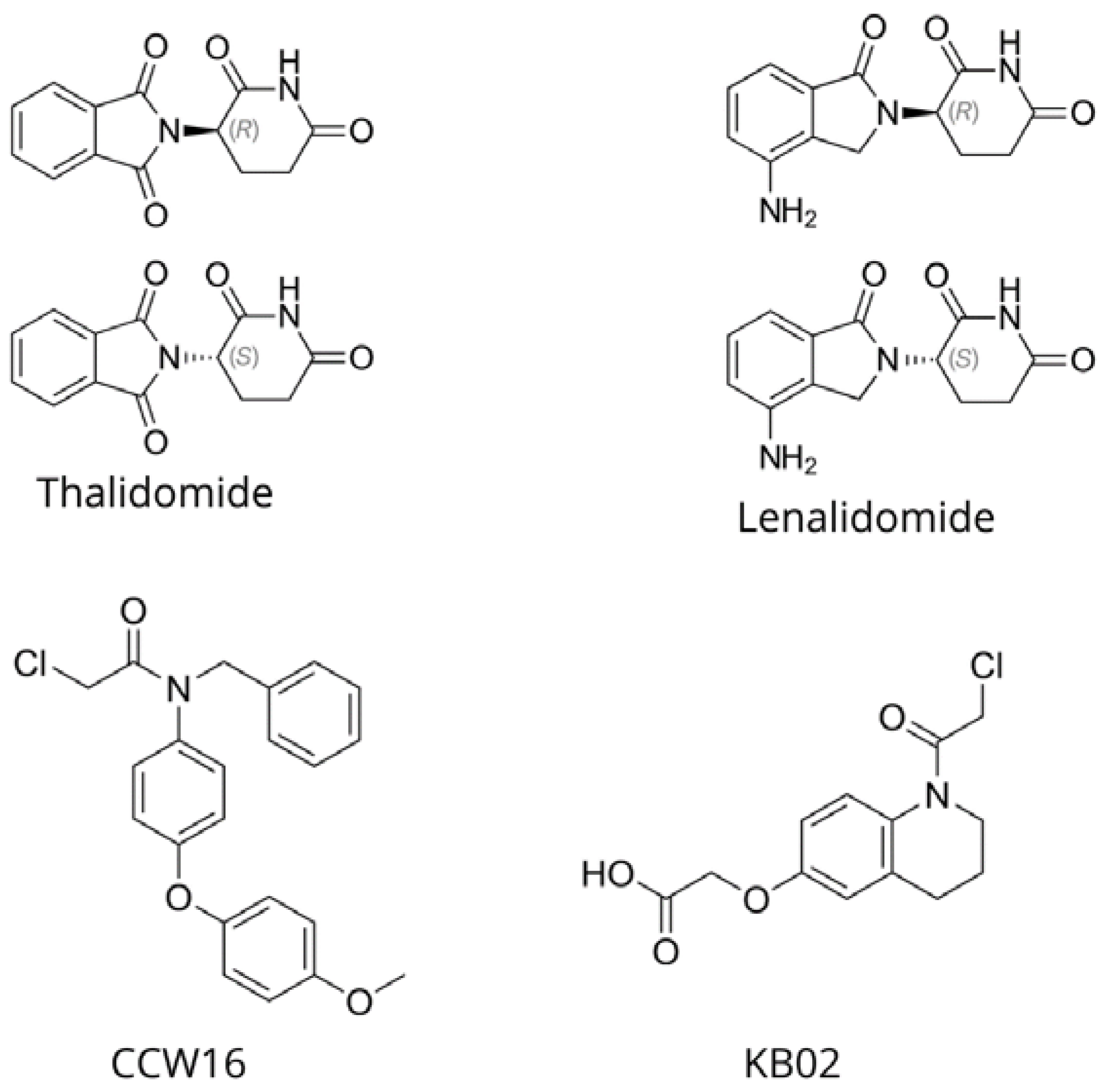
2.3. E3 Ligase
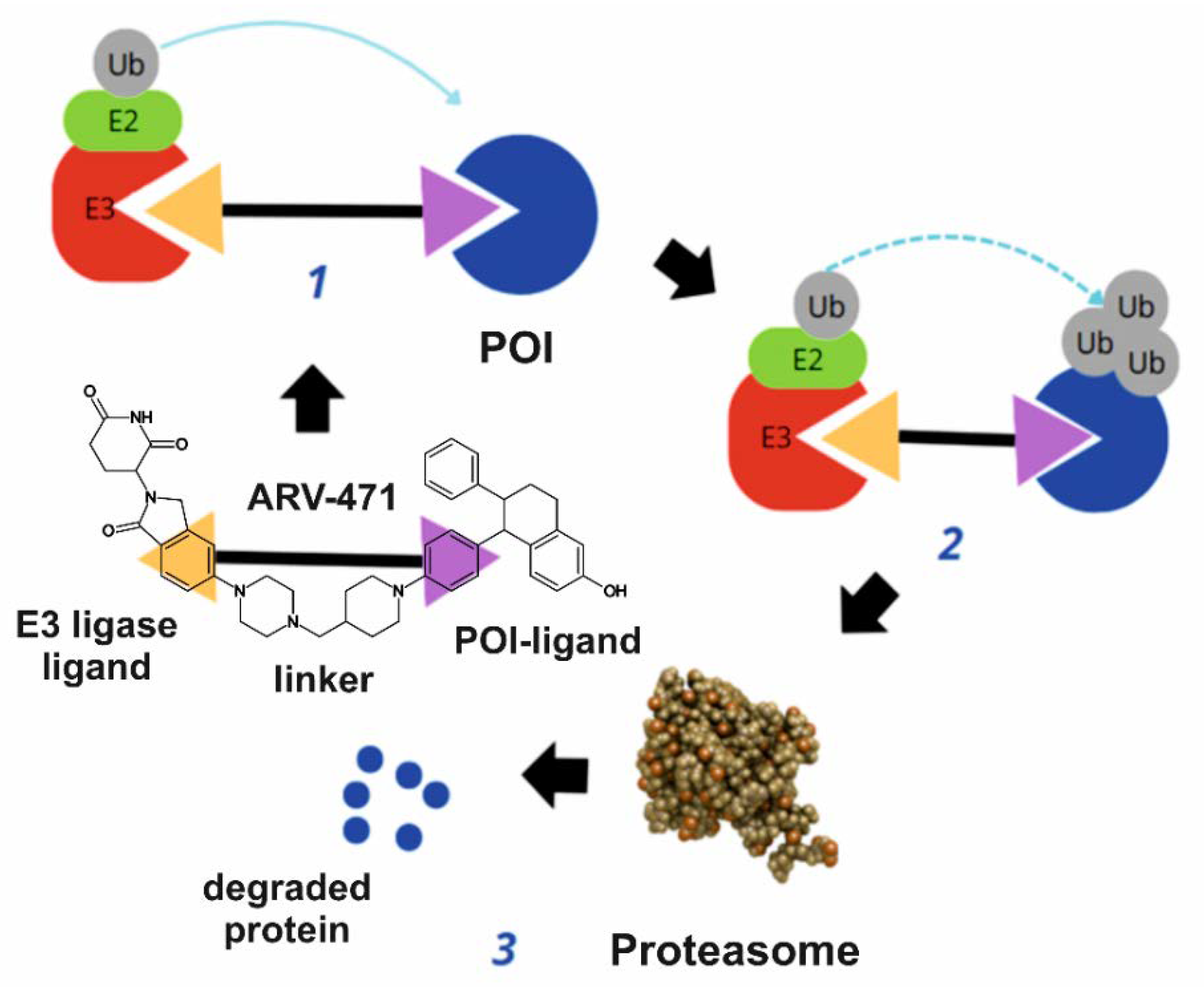
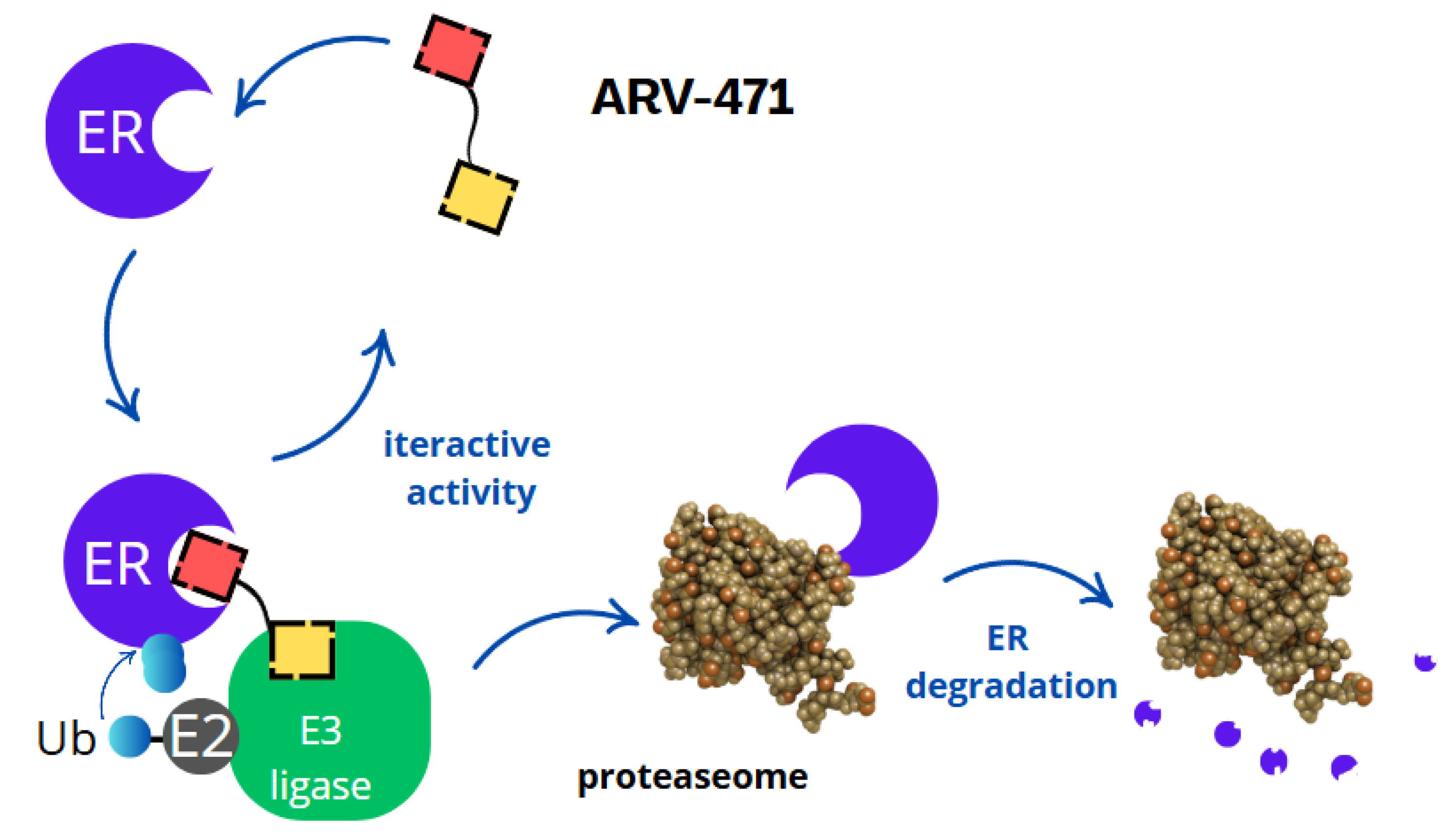
3. Mechanism of Action
- Binding to the oestrogen receptor—ARV-471 contains a ligand that specifically binds to the oestrogen receptor, marking it for degradation.
- Recruitment of E3 ligase—ARV-471 acts as a bridge, connecting the oestrogen receptor with the E3 ligase, an enzyme responsible for tagging proteins with ubiquitin.
- Ubiquitination and proteasomal degradation—the E3 ligase attaches ubiquitin to the oestrogen receptor, signalling its degradation by proteasomes, which break it down into amino acids.
- Inhibition of cancer cell proliferation and induction of apoptosis—the degradation of the oestrogen receptor inhibits tumour growth by blocking gene transcription linked to cancer cell proliferation and inducing programmed cell death (apoptosis).
- Synergistic action and therapeutic advantages—ARV-471 enhances the effects of CDK4/6 inhibitors (e.g., palbociclib) and PI3K/mTOR inhibitors (e.g., everolimus), while also offering efficacy in resistant cases, lower toxicity, and potential use in advanced breast cancer.
4. Clinical Trials
5. New Treatment Possibilities Using PROTACs
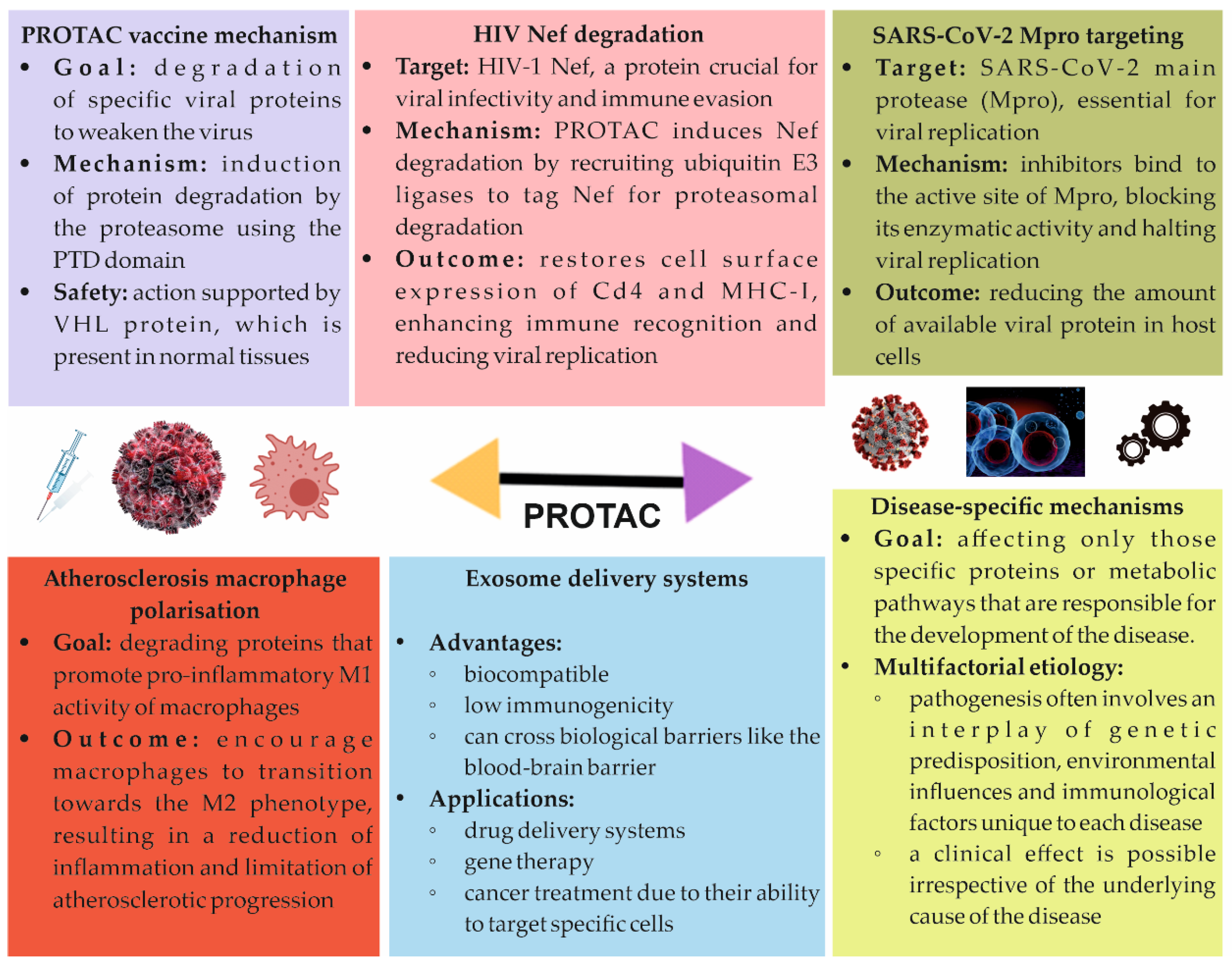
5.1. PROTAC Vaccine
5.2. PROTACs and HIV
5.3. COVID-19 Treatment
5.4. Atherosclerosis Treatment
5.5. The Use of PROTAC in the Treatment of RNA Viral Infections
5.6. Treatment of Alzheimer’s Disease
5.7. PROTAC Technology to Combat Stress Hormone Receptor Activation
5.8. Atopic Dermatitis
5.9. Non-Alcoholic Fatty Liver Disease
5.10. Huntington’s Disease
5.11. Treatment of Immune Disorders
6. New Solutions
6.1. Antibody–PROTAC
6.2. Aptamer-PROTAC Conjugates
6.3. Dual-Target PROTAC
6.4. Folate-Caged PROTAC
6.5. TF–PROTAC
6.6. PhosphoTAC
6.7. PhosTAC
6.8. Photocaged PROTAC
6.9. CLIPTAC
6.10. PROTAC Applications in the Prevention of Virus Threats or Pandemics
7. Study Limitations and Challenges
8. Materials and Methods
9. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics. CA A Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Weng, Z.; Zou, Y. Progress in the controllability technology of PROTAC. Eur. J. Med. Chem. 2024, 265, 116096. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Crews, C.M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [Google Scholar] [CrossRef]
- Li, D.; Yu, D.; Li, Y.; Yang, R. A bibliometric analysis of PROTAC from 2001 to 2021. Eur. J. Med. Chem. 2022, 244, 114838. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Zhang, G.; Luo, W.; Xu, M.; Peng, R.; Du, Z.; Liu, Y.; Bai, Z.; Xiao, X.; Qin, S. PROTAC technology: From drug development to probe technology for target deconvolution. Eur. J. Med. Chem. 2024, 276, 116725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lai, Y.; Pan, J.; Saeed, M.; Li, S.; Zhou, H.; Jiang, X.; Gao, J.; Zhu, Y.; Yu, H.; et al. PROTAC Prodrug-Integrated Nanosensitizer for Potentiating Radiation Therapy of Cancer. Adv. Mater. 2024, 36, 2314132. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, M.; Wang, C.F.; Nagase, T.; Sohma, Y.; Kanai, M.; Hori, Y.; Tomita, T. Proteolytic therapeutic modalities for amyloidoses: Insights into immunotherapy, PROTAC, and photo-oxygenation. Neurotherapeutics 2025, 22, e00548. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, D.; Inuzuka, H.; Wei, W. PROTAC technology for prostate cancer treatment. Acta Mater. Medica 2025, 4, 99–121. [Google Scholar] [CrossRef]
- Monsen, P.J.; Bommi, P.V.; Grigorescu, A.A.; Lauing, K.L.; Mao, Y.; Berardi, P.; Zhai, L.; Ojo, O.; Penco-Campillo, M.; Koch, T.; et al. Rational Design and Optimization of a Potent IDO1 Proteolysis Targeting Chimera (PROTAC). J. Med. Chem. 2025, 68, 4961–4987. [Google Scholar] [CrossRef]
- Simpson, L.M.; Glennie, L.; Brewer, A.; Zhao, J.-F.; Crooks, J.; Shpiro, N.; Sapkota, G.P. Target protein localization and its impact on PROTAC-mediated degradation. Cell Chem. Biol. 2022, 29, 1482–1504.e7. [Google Scholar] [CrossRef]
- Takano, R.; Ohoka, N.; Kurohara, T.; Arakawa, N.; Ohgane, K.; Inoue, T.; Yokoo, H.; Demizu, Y. Clozapine as an E3 Ligand for PROTAC Technology. ACS Med. Chem. Lett. 2025, 16, 258–262. [Google Scholar] [CrossRef]
- Sheng, X.-Y.; Wu, S.-H.; Li, B.-L.; Li, X.-N.; Wu, H.-S.; Cao, J. Advances in the optimization of the linker in proteolysis-targeting chimeras (PROTAC). Acta Pharm. Sinica 2021, 12, 445–455. [Google Scholar]
- Ma, N.; Yang, T.; Mukhaleva, E.; Wei, W.; Vaidehi, N. BPS2025-Innovative strategies in PROTAC design: Evaluating linker efficiency through protein frustration analysis. Biophys. J. 2025, 124, 390a–391a. [Google Scholar] [CrossRef]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current strategies for the design of PROTAC linkers: A critical review. Explor. Target. Anti-tumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, V.; Peintner, S.; Abeje, Y.; Kölling, F.; Meibom, D.; Erdelyi, M.; Kihlberg, J. Linker-Determined Folding and Hydrophobic Interactions Explain a Major Difference in PROTAC Cell Permeability. ACS Med. Chem. Lett. 2025, 16, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Abeje, Y.E.; Wieske, L.H.E.; Poongavanam, V.; Maassen, S.; Atilaw, Y.; Cromm, P.; Lehmann, L.; Erdelyi, M.; Meibom, D.; Kihlberg, J. Impact of Linker Composition on VHL PROTAC Cell Permeability. J. Med. Chem. 2024, 68, 638–657. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Sobhia, M.E. Interplay of PROTAC Complex Dynamics for Undruggable Targets: Insights into Ternary Complex Behavior and Linker Design. ACS Med. Chem. Lett. 2024, 15, 1306–1318. [Google Scholar] [CrossRef]
- Li, L.; Mi, D.; Pei, H.; Duan, Q.; Wang, X.; Zhou, W.; Jin, J.; Li, D.; Liu, M.; Chen, Y. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct. Target. Ther. 2020, 5, 129. [Google Scholar] [CrossRef]
- Ward, C.C.; Kleinman, J.I.; Brittain, S.M.; Lee, P.S.; Chung, C.Y.S.; Kim, K.; Petri, Y.; Thomas, J.R.; Tallarico, J.A.; McKenna, J.M.; et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14, 2430–2440. [Google Scholar] [CrossRef]
- Belcher, B.P.; Ward, C.C.; Nomura, D.K. Ligandability of E3 Ligases for Targeted Protein Degradation Applications. Biochemistry 2021, 62, 588–600. [Google Scholar] [CrossRef]
- Medvar, B.; Raghuram, V.; Pisitkun, T.; Sarkar, A.; Knepper, M.A. Comprehensive database of human E3 ubiquitin ligases: Application to aquaporin-2 regulation. Physiol. Genom. 2016, 48, 502–512. [Google Scholar] [CrossRef]
- Danishuddin; Jamal, M.S.; Song, K.-S.; Lee, K.-W.; Kim, J.-J.; Park, Y.-M. Revolutionizing drug targeting strategies: Integrating artificial intelligence and structure-based methods in PROTAC development. Pharmaceuticals 2023, 16, 1649. [Google Scholar] [CrossRef]
- Palomba, T.; Baroni, M.; Cross, S.; Cruciani, G.; Siragusa, L. ELIOT: A platform to navigate the E3 pocketome and aid the design of new PROTACs. Chem. Biol. Drug Des. 2023, 101, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Zagidullin, A.; Milyukov, V.; Rizvanov, A.; Bulatov, E. Novel approaches for the rational design of PROTAC linkers. Explor. Target. Anti-Tumor Ther. 2020, 1, 381–390. [Google Scholar] [CrossRef]
- Wang, S.; He, F.; Tian, C.; Sun, A. From PROTAC to TPD: Advances and Opportunities in Targeted Protein Degradation. Pharmaceuticals 2024, 17, 100. [Google Scholar] [CrossRef]
- Weerakoon, D.; Carbajo, R.J.; De Maria, L.; Tyrchan, C.; Zhao, H. Impact of PROTAC Linker Plasticity on the Solution Conformations and Dissociation of the Ternary Complex. J. Chem. Inf. Model. 2022, 62, 340–349. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Backes, A.; Zech, B.; Felber, B.; Klebl, B.; Müller, G. Small-molecule inhibitors binding to protein kinases. Part I: Exceptions from the traditional pharmacophore approach of type I inhibition. Expert Opin. Drug Discov. 2008, 3, 1409–1425. [Google Scholar] [CrossRef]
- Xiao, M.; Zhao, J.; Wang, Q.; Liu, J.; Ma, L. Recent Advances of Degradation Technologies Based on PROTAC Mechanism. Biomolecules 2022, 12, 1257. [Google Scholar] [CrossRef] [PubMed]
- Graham, H. The mechanism of action and clinical value of PROTACs: A graphical review. Cell. Signal. 2022, 99, 110446. [Google Scholar] [CrossRef]
- Schott, A.F.; Hurvitz, S.; Ma, C.; Hamilton, E.; Nanda, R.; Zahrah, G.; Hunter, N.; Tan, A.R.; Telli, M.; Mesias, J.A.; et al. Abstract GS3-03: GS3-03 ARV-471, a PROTAC® estrogen receptor (ER) degrader in advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer: Phase 2 expansion (VERITAC) of a phase 1/2 study. Cancer Res. 2023, 83 (Suppl. S5), GS3-03. [Google Scholar] [CrossRef]
- Snyder, L.B.; Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Chandler, J.; et al. Abstract 44: The discovery of ARV-471, an orally bioavailable estrogen receptor degrading PROTAC for the treatment of patients with breast cancer. Cancer Res. 2021, 81, 44. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2018, 62, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sun, Y. Strategies for the discovery of oral PROTAC degraders aimed at cancer therapy. Cell Rep. Phys. Sci. 2022, 3, 101062. [Google Scholar] [CrossRef]
- Gough, S.M.; Flanagan, J.J.; Teh, J.; Andreoli, M.; Rousseau, E.; Pannone, M.; Bookbinder, M.; Willard, R.; Davenport, K.; Bortolon, E.; et al. Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models. Clin. Cancer Res. 2024, 30, 3549–3563. [Google Scholar] [CrossRef]
- Nguyen, T.-T.; Kim, J.W.; Choi, H.-I.; Maeng, H.-J.; Koo, T.-S. Development of an LC-MS/MS Method for ARV-110, a PROTAC Molecule, and Applications to Pharmacokinetic Studies. Molecules 2022, 27, 1977. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, A.B.; Scott-Stevens, P.T.; Harling, J.D. Challenges and Opportunities for In Vivo PROTAC Delivery. Futur. Med. Chem. 2022, 14, 119–121. [Google Scholar] [CrossRef]
- Ma, L.; Han, X. Bavdegalutamide (ARV-110): A potent PROTAC androgen receptor degrader for the treatment of metastatic-castration resistant prostate cancer. In Drug Discovery Stories; Elsevier: Amsterdam, The Netherlands, 2025; pp. 357–378. [Google Scholar]
- Vetma, V.; O’Connor, S.; Ciulli, A. Development of PROTAC Degrader Drugs for Cancer. Annu. Rev. Cancer Biol. 2024, 9, 119–140. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.A.; Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef]
- Grigoreva, T.A.; Novikova, D.S.; Melino, G.; Barlev, N.A.; Tribulovich, V.G. Ubiquitin recruiting chimera: More than just a PROTAC. Biol. Direct 2024, 19, 55. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79 (Suppl. S4), P5-04-18. [Google Scholar] [CrossRef]
- Lloyd, M.R.; Wander, S.A.; Hamilton, E.; Razavi, P.; Bardia, A. Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: Current and emerging role. Ther. Adv. Med Oncol. 2022, 14, 17588359221113694. [Google Scholar] [CrossRef]
- Rathkopf, D.; Patel, M.; Choudhury, A.; Rasco, D.; Lakhani, N.; Hawley, J.; Srinivas, S.; Aparicio, A.; Narayan, V.; Runcie, K.; et al. Safety and clinical activity of BMS-986365 (CC-94676), a dual androgen receptor ligand-directed degrader and antagonist, in heavily pretreated patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2025, 36, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Gurney, H.; Underhill, C.; Horvath, L.; Voskoboynik, M.; Li, X.; King, I.; Shao, L.; Dai, Y.; Perabo, F. Preliminary data from a dose-escalation phase 1 study with HP518, an AR PROTAC degrader: Safety, tolerability, pharmacokinetics (PK), and first assessment of anti-tumor activity in patients (Pts) with metastatic castration-resistant prostate cancer (mCRPC). Am. Soc. Clin. Oncol. 2024, 42, 124. [Google Scholar] [CrossRef]
- Li, X.; Mu, P. Restoring our ubiquitination machinery to overcome resistance in cancer therapy. Oncoscience 2024, 11, 43–44. [Google Scholar] [CrossRef]
- Scott, J.S.; Michaelides, I.N.; Schade, M. Property-based optimisation of PROTACs. RSC Med. Chem. 2024, 16, 449–456. [Google Scholar] [CrossRef]
- Peng, X.; Hu, Z.; Zeng, L.; Zhang, M.; Xu, C.; Lu, B.; Tao, C.; Chen, W.; Hou, W.; Cheng, K.; et al. Overview of epigenetic degraders based on PROTAC, molecular glue, and hydrophobic tagging technologies. Acta Pharm. Sin. B 2024, 14, 533–578. [Google Scholar] [CrossRef]
- Kong, N.R.; Jones, L.H. Clinical Translation of Targeted Protein Degraders. Clin. Pharmacol. Ther. 2023, 114, 558–568. [Google Scholar] [CrossRef]
- Park, E.S.; Ahn, J.Y.; Baddour, J.; Chaturvedi, P.; Chiu, M.I.; Cole, K.S.; Crystal, A.S.; Duplessis, M.; Fisher, S.L.; Good, A.C.; et al. Preclinical Evaluation of CFT8919 as a Mutant Selective Degrader of EGFR with L858R Activating Mutations for the Treatment of Non-Small Cell Lung Cancer; Keystone Esymposium Targeting Protein Degradation: From Small Molecules to Complex Organelles; C4 Therapeutics Inc.: Watertown, MA, USA, 2021. [Google Scholar]
- Li, J.; Xu, W.; Yan, P.; Cao, Y.; Hu, M.; Daley, W. Abstract CT128: Phase 1 study of HSK29116, a Bruton tyrosine kinase (BTK) proteolysis-targeting chimera (PROTAC) agent, in patients with relapsed or refractory B-cell malignancies. Cancer Res. 2023, 83 (Suppl. S8), CT128. [Google Scholar] [CrossRef]
- Robbins, D.W.; Noviski, M.; Rountree, R.; Tan, M.; Brathaban, N.; Ingallinera, T.E.; Karr, D.; Kelly, A.; Konst, Z.; Ma, J.; et al. Nx-5948, a Selective Degrader of BTK with Activity in Preclinical Models of Hematologic and Brain Malignancies. Blood 2021, 138, 2251. [Google Scholar] [CrossRef]
- Mato, A.R.; Wierda, W.G.; Ai, W.Z.; Flinn, I.W.; Tees, M.; Patel, M.R.; Patel, K.; O’Brien, S.; Bond, D.A.; Roeker, L.E.; et al. NX-2127-001, a first-in-human trial of NX-2127, a Bruton’s tyrosine kinase-targeted protein degrader, in patients with relapsed or refractory chronic lymphocytic leukemia and B-cell malignancies. Blood 2022, 140, 2329–2332. [Google Scholar] [CrossRef]
- Livingston, J.A.; Blay, J.-Y.; Trent, J.; Valverde, C.; Agulnik, M.; Gounder, M.; Le Cesne, A.; McKean, M.; Wagner, M.J.; Stacchiotti, S. A Phase I Study of FHD-609, a Heterobifunctional degrader of Bromodomain-containing Protein 9, in patients with advanced synovial sarcoma or SMARCB1-deficient tumors. Clin. Cancer Res. 2025, 4, 628–638. [Google Scholar] [CrossRef]
- Jackson, K.L.; Agafonov, R.V.; Carlson, M.W.; Chaturvedi, P.; Cocozziello, D.; Cole, K.; Deibler, R.; Eron, S.J.; Good, A.; Hart, A.A.; et al. Abstract ND09: The discovery and characterization of CFT8634: A potent and selective degrader of BRD9 for the treatment of SMARCB1-perturbed cancers. Cancer Res. 2022, 82 (Suppl. S12), ND09. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Park, W.; Wang, J.S.; Spira, A.I.; Janne, P.A.; Lee, H.-J.; Gill, S.; LoRusso, P.; Herzberg, B.; Goldman, J.W.; et al. Trial in progress: A phase 1, first-in-human, open-label, multicenter, dose-escalation and dose-expansion study of ASP3082 in patients with previously treated advanced solid tumors and KRAS G12D mutations. Am. Soc. Clin. Oncol. 2023, 41, TPS764. [Google Scholar] [CrossRef]
- Wu, Y.; Meibohm, B.; Zhang, T.; Hou, X.; Wang, H.; Sun, X.; Jiang, M.; Zhang, B.; Zhang, W.; Liu, Y.; et al. Translational modelling to predict human pharmacokinetics and pharmacodynamics of a Bruton’s tyrosine kinase-targeted protein degrader BGB-16673. Br. J. Pharmacol. 2024, 181, 4973–4987. [Google Scholar] [CrossRef]
- Patel, M.; Tees, M.; Khan, N.; Awan, F.; Bond, D.; Xu, Q.; Brown, G.; Zhang, H.; Woyach, J. AC676, an Orally Bioavailable BTK Chimeric Degrader in Patients With B-cell Malignancies. Clin. Lymphoma Myeloma Leuk. 2024, 24, S494. [Google Scholar] [CrossRef]
- He, Y.; Koch, R.; Budamagunta, V.; Zhang, P.; Zhang, X.; Khan, S.; Thummuri, D.; Ortiz, Y.T.; Zhang, X.; Lv, D.; et al. DT2216—A Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol. 2020, 13, 1–13. [Google Scholar] [CrossRef]
- Si, L.; Shen, Q.; Li, J.; Chen, L.; Shen, J.; Xiao, X.; Bai, H.; Feng, T.; Ye, A.Y.; Li, L.; et al. Generation of a live attenuated influenza A vaccine by proteolysis targeting. Nat. Biotechnol. 2022, 40, 1370–1377. [Google Scholar] [CrossRef]
- Zhang, C.; Hou, J.; Li, Z.; Shen, Q.; Bai, H.; Chen, L.; Shen, J.; Wang, P.; Su, Y.; Li, J.; et al. PROTAR Vaccine 2.0 generates influenza vaccines by degrading multiple viral proteins. Nat. Chem. Biol. 2025, 1–11. [Google Scholar] [CrossRef]
- Li, Z.; Bai, H.; Xi, X.; Tian, W.; Zhang, J.Z.; Zhou, D.; Si, L. PROTAC vaccine: A new way to live attenuated vaccines. Clin. Transl. Med. 2022, 12, e1081. [Google Scholar] [CrossRef]
- Ahmad, H.; Zia, B.; Husain, H.; Husain, A. Recent advances in PROTAC-based antiviral strategies. Vaccines 2023, 11, 270. [Google Scholar] [CrossRef]
- Luo, D.; Luo, R.; Wang, W.; Deng, R.; Wang, S.; Ma, X.; Pu, C.; Liu, Y.; Zhang, H.; Yu, S.; et al. Discovery of L15 as a novel Vif PROTAC degrader with antiviral activity against HIV-1. Bioorganic Med. Chem. Lett. 2024, 111, 129880. [Google Scholar] [CrossRef]
- Emert-Sedlak, L.A.; Tice, C.M.; Shi, H.; Alvarado, J.J.; Shu, S.T.; Reitz, A.B.; Smithgall, T.E. PROTAC-mediated degradation of HIV-1 Nef efficiently restores cell-surface CD4 and MHC-I expression and blocks HIV-1 replication. Cell Chem. Biol. 2024, 31, 658–668.e14. [Google Scholar] [CrossRef] [PubMed]
- Alugubelli, Y.R.; Xiao, J.; Khatua, K.; Kumar, S.; Sun, L.; Ma, Y.; Ma, X.R.; Vulupala, V.R.; Atla, S.; Blankenship, L.R.; et al. Discovery of First-in-Class PROTAC Degraders of SARS-CoV-2 Main Protease. J. Med. Chem. 2024, 67, 6495–6507. [Google Scholar] [CrossRef]
- Björkegren, J.L.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.; Badimon, L.; Hansson, G.; Deanfield, J.; Bittencourt, M.; Tokgozoglu, L.; Lewis, E. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-E.; Chang, H.-J.; Sung, J.M.; Park, H.-B.; Heo, R.; Rizvi, A.; Lin, F.Y.; Kumar, A.; Hadamitzky, M.; Kim, Y.J.; et al. Effects of statins on coronary atherosclerotic plaques: The PARADIGM study. JACC Cardiovasc. Imaging 2018, 11, 1475–1484. [Google Scholar] [CrossRef]
- Huang, J.-H.; Huang, C.-J.; Yu, L.-N.; Guan, X.-L.; Liang, S.-W.; Li, J.-H.; Liang, L.; Wei, M.-Y.; Zhang, L.-M. Bioinspired PROTAC-induced macrophage fate determination alleviates atherosclerosis. Acta Pharmacol. Sin. 2023, 44, 1962–1976. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, X.; Ning, K.; Guo, H. M1/M2 macrophage-targeted nanotechnology and PROTAC for the treatment of atherosclerosis. Life Sci. 2024, 122811. [Google Scholar] [CrossRef]
- Mukerjee, N.; Maitra, S.; Ghosh, A.; Alexiou, A.; Thorat, N.D. Exosome-mediated PROTAC delivery for treatment of RNA viral infections and zoonosis. Drug Discov. Today 2024, 29, 104044. [Google Scholar] [CrossRef]
- Mukerjee, N.; Mukherjee, D. PROTAC-based therapeutics for targeting HPV oncoproteins in head and neck cancers. Nano TransMed 2025, 4, 100071. [Google Scholar] [CrossRef]
- Wei, J.; Wang, J.; Zhang, J.; Yang, J.; Wang, G.; Wang, Y. Development of inhibitors targeting glycogen synthase kinase-3β for human diseases: Strategies to improve selectivity. Eur. J. Med. Chem. 2022, 236, 114301. [Google Scholar] [CrossRef]
- Guardigni, M.; Pruccoli, L.; Santini, A.; De Simone, A.; Bersani, M.; Spyrakis, F.; Frabetti, F.; Uliassi, E.; Andrisano, V.; Pagliarani, B.; et al. PROTAC-Induced Glycogen Synthase Kinase 3β Degradation as a Potential Therapeutic Strategy for Alzheimer’s Disease. ACS Chem. Neurosci. 2023, 14, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, P.H. Phosphorylated tau targeted small-molecule PROTACs for the treatment of Alzheimer’s disease and tauopathies. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166162. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Tonali, N.; Nencetti, S.; Orlandini, E. Application of PROTAC strategy to TTR-Aβ protein-protein interaction for the development of Alzheimer’s disease drugs. Neural Regen. Res. 2021, 16, 1554–1555. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Meng, L.; Lin, P.; Wu, G. Advancements in PROTAC-based therapies for neurodegenerative diseases. Future Med. Chem. 2025, 17, 591–605. [Google Scholar] [CrossRef]
- Kargbo, R.B. Treatment of Cancer and Alzheimer’s Disease by PROTAC Degradation of EGFR. ACS Med. Chem. Lett. 2019, 10, 1098–1099. [Google Scholar] [CrossRef]
- Gazorpak, M.; Hugentobler, K.M.; Paul, D.; Germain, P.-L.; Kretschmer, M.; Ivanova, I.; Frei, S.; Mathis, K.; Rudolf, R.; Barrenechea, S.M.; et al. Harnessing PROTAC technology to combat stress hormone receptor activation. Nat. Commun. 2023, 14, 8177. [Google Scholar] [CrossRef]
- Geiger, T.M.; Walz, M.; Meyners, C.; Kuehn, A.; Dreizler, J.K.; Sugiarto, W.O.; Maciel, E.V.S.; Zheng, M.; Lermyte, F.; Hausch, F. Entdeckung eines potenten PROTAC ermöglicht die gezielte Ausschaltung der Gerüstfunktionen von FKBP51. Angew. Chem. 2024, 136, e202309706. [Google Scholar] [CrossRef]
- Wu, J.; Li, L.; Zhu, Q.; Zhang, T.; Miao, F.; Cui, Z.; Dong, G.; Tai, Z.; Chen, Z. JAK1/JAK2 degraders based on PROTAC for topical treatment of atopic dermatitis. Biomed. Pharmacother. 2024, 171, 116167. [Google Scholar] [CrossRef]
- Colleoni, A.; Fassi, E.; Miluzio, A.; Albani, M.; Lecchi, D.; Tempra, G.; De Amici, M.; Grazioso, G.; Biffo, S.; Matera, C. Targeted degraders of eIF6: A novel strategy to remodulate liver pathological lipidic metabolism. In Proceedings of the Merck Young Chemists’ Symposium (MYCS), Rimini, Italy, 13–15 November 2024. [Google Scholar]
- Colleoni, A.; Fassi, E.; Miluzio, A.; Tempra, G.; Albani, M.; De Amici, M.; Grazioso, G.; Biffo, S.; Matera, C. Exploiting the Potential of Computational Approaches in Medicinal Chemistry: CADD of Novel eIF6 Binders for the Development of anti-HCC PROTACs. In Proceedings of the WIDEnzymes, Lecco, Italy, 27–31 January 2025. [Google Scholar]
- Qi, M.; Zhong, H.; Cheng, Z.; Chen, S.; Xiao, H.; Shang, J.; Chen, L.; Sun, J. Discovery of NAFLD-Improving Agents by Promoting the Degradation of Keap1. J. Med. Chem. 2023, 66, 9184–9200. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, J.; Long, Y.; Wu, W.; Zhu, Y. Direct degradation and stabilization of proteins: New horizons in treatment of nonalcoholic steatohepatitis. Biochem. Pharmacol. 2024, 220, 115989. [Google Scholar] [CrossRef]
- Winzker, M.; Friese, A.; Koch, U.; Janning, P.; Ziegler, S.; Waldmann, H. Development of a pdeδ-targeting PROTACs that impair lipid metabolism. Angew. Chem. 2020, 132, 5644–5650. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Xing, D.; Zhang, R. PROTACs technology for targeting non-oncoproteins: Advances and perspectives. Bioorganic Chem. 2021, 114, 105109. [Google Scholar] [CrossRef] [PubMed]
- Tashima, T. Proteolysis-Targeting Chimera (PROTAC) Delivery into the Brain across the Blood-Brain Barrier. Antibodies 2023, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Farrell, K.; Jarome, T.J. Is PROTAC technology really a game changer for central nervous system drug discovery? Expert Opin. Drug Discov. 2021, 16, 833–840. [Google Scholar] [CrossRef]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Jude, J.A.; Panettieri, R.A. IRAK4: Potential therapeutic target for airway disease exacerbations. Trends Pharmacol. Sci. 2025, 46, 201–203. [Google Scholar] [CrossRef]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target. Ther. 2022, 7, 181. [Google Scholar] [CrossRef]
- Dragovich, P.S. Degrader-antibody conjugates. Chem. Soc. Rev. 2022, 51, 3886–3897. [Google Scholar] [CrossRef]
- Chan, K.; Sathyamurthi, P.S.; Queisser, M.A.; Mullin, M.; Shrives, H.; Coe, D.M.; Burley, G.A. Antibody-proteolysis targeting chimera conjugate enables selective degradation of receptor-interacting serine/threonine-protein kinase 2 in HER2+ cell lines. Bioconjugate Chem. 2023, 34, 2049–2054. [Google Scholar] [CrossRef]
- Wang, L.; Ke, Y.; He, Q.; Paerhati, P.; Zhuang, W.; Yue, Y.; Liu, J.; Zhang, J.; Huang, L.; Yin, Q.; et al. A novel ROR1-targeting antibody-PROTAC conjugate promotes BRD4 degradation for solid tumor treatment. Theranostics 2025, 15, 1238–1254. [Google Scholar] [CrossRef]
- He, S.; Gao, F.; Ma, J.; Ma, H.; Dong, G.; Sheng, C. Aptamer-protac conjugates (apcs) for tumor-specific targeting in breast cancer. Angew. Chem. 2021, 133, 23487–23493. [Google Scholar] [CrossRef]
- Liu, Y.; Qian, X.; Ran, C.; Li, L.; Fu, T.; Su, D.; Xie, S.; Tan, W. Aptamer-Based Targeted Protein Degradation. ACS Nano 2023, 17, 6150–6164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yan, S.; Liu, Y.; Du, Z.; Min, Q.; Qin, S. PROTACs coupled with oligonucleotides to tackle the undruggable. Bioanalysis 2025, 17, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Dong, G.; Wang, W.; Sheng, C. Precise Modulation of Protein Degradation by Smart PROTACs. ChemBioChem 2025, 26, e202400682. [Google Scholar] [CrossRef]
- Chen, H.; Liu, J.; Kaniskan, H.U.M.; Wei, W.; Jin, J. Folate-guided protein degradation by immunomodulatory imide drug-based molecular glues and proteolysis targeting chimeras. J. Med. Chem. 2021, 64, 12273–12285. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Kaniskan, H.U.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs enable targeted degradation of transcription factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef]
- Fan, L.; Tong, W.; Wei, A.; Mu, X. Progress of proteolysis-targeting chimeras (PROTACs) delivery system in tumor treatment. Int. J. Biol. Macromol. 2024, 275, 133680. [Google Scholar] [CrossRef]
- Zografou-Barredo, N.A.; Hallatt, A.J.; Goujon-Ricci, J.; Cano, C. A beginner’s guide to current synthetic linker strategies towards VHL-recruiting PROTACs. Bioorganic Med. Chem. 2023, 88–89, 117334. [Google Scholar] [CrossRef]
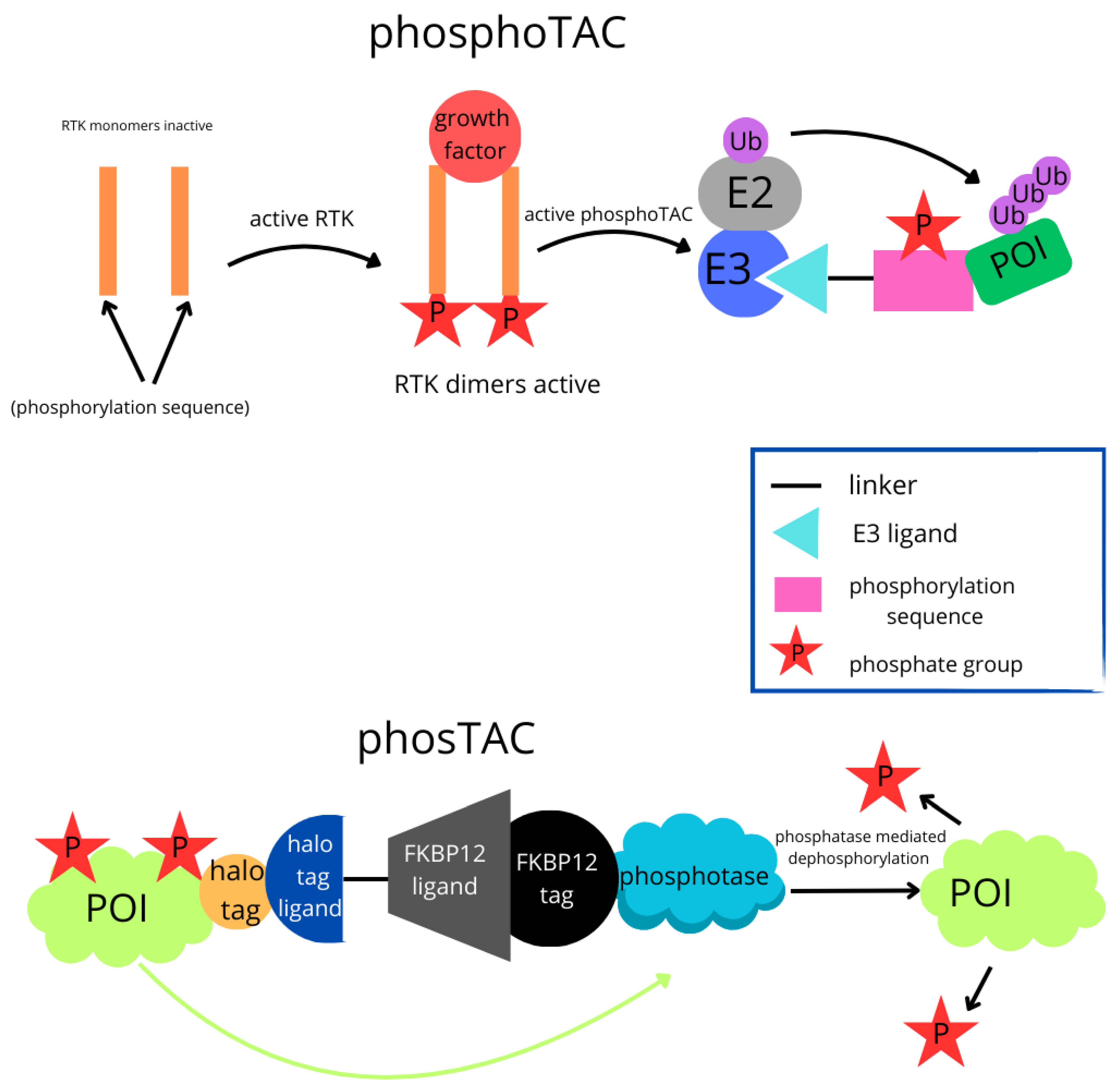
- Hines, J.; Gough, J.D.; Corson, T.W.; Crews, C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA 2013, 110, 8942–8947. [Google Scholar] [CrossRef]
- Uliassi, E.; Bolognesi, M.L.; Milelli, A. Targeting Tau Protein with Proximity Inducing Modulators: A New Frontier to Combat Tauopathies. ACS Pharmacol. Transl. Sci. 2025, 8, 654–672. [Google Scholar] [CrossRef]
- Zhao, C.; Dekker, F.J. Novel Design Strategies to Enhance the Efficiency of Proteolysis Targeting Chimeras. ACS Pharmacol. Transl. Sci. 2022, 5, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, P.-H.; Li, W.; Douglas, T.; Hines, J.; Liu, Y.; Crews, C.M. Targeted dephosphorylation of tau by phosphorylation targeting chimeras (PhosTACs) as a therapeutic modality. J. Am. Chem. Soc. 2023, 145, 4045–4055. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, P.-H.; Li, W.; Krone, M.; Zheng, S.; Saarbach, J.; Velasco, I.U.; Hines, J.; Liu, Y.; Crews, C.M. EGFR targeting PhosTACs as a dual inhibitory approach reveals differential downstream signaling. Sci. Adv. 2024, 10, eadj7251. [Google Scholar] [CrossRef]
- Negi, A.; Voisin-Chiret, A.S. Strategies to reduce the on-target platelet toxicity of Bcl-xL inhibitors: PROTACs, SNIPERs and prodrug-based approaches. ChemBioChem 2022, 23, e202100689. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.; Kesari, K.K.; Voisin-Chiret, A.S. Light-Activating PROTACs in Cancer: Chemical Design, Challenges, and Applications. Appl. Sci. 2022, 12, 9674. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef]
- Verma, S.; Manna, D. Controlling PROTACs with Light. ChemMedChem 2020, 15, 1258–1261. [Google Scholar] [CrossRef]
- Yang, C.; Tripathi, R.; Wang, B. Click chemistry in the development of PROTACs. RSC Chem. Biol. 2023, 5, 189–197. [Google Scholar] [CrossRef]
- Noblejas-López, M.d.M.; Tébar-García, D.; López-Rosa, R.; Alcaraz-Sanabria, A.; Cristóbal-Cueto, P.; Pinedo-Serrano, A.; Rivas-García, L.; Galán-Moya, E.M. TACkling cancer by targeting selective protein degradation. Pharmaceutics 2023, 15, 2442. [Google Scholar] [CrossRef]
- Liang, J.; Wu, Y.; Lan, K.; Dong, C.; Wu, S.; Li, S.; Zhou, H.-B. Antiviral PROTACs: Opportunity borne with challenge. Cell Insight 2023, 2, 100092. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Pandemics Throughout History. Front. Microbiol. 2021, 11, 631736. [Google Scholar] [CrossRef]
- Brüssow, H. The beginning and ending of a respiratory viral pandemic-lessons from the Spanish flu. Microb. Biotechnol. 2022, 15, 1301–1317. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.N.S.; Pingarilho, M.; Pimentel, V.; Torneri, A.; Seabra, S.G.; Libin, P.J.K.; Abecasis, A.B. A Tale of Three Recent Pandemics: Influenza, HIV and SARS-CoV-2. Front. Microbiol. 2022, 13, 889643. [Google Scholar] [CrossRef] [PubMed]
- Hatchett, R.; Moreira, M. We can stop the next pandemic, but only if we act now. Telegraph. 2024. [Google Scholar]
- Guedeney, N.; Cornu, M.; Schwalen, F.; Kieffer, C.; Voisin-Chiret, A.S. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 2022, 28, 103395. [Google Scholar] [CrossRef]
- Oleinikovas, V.; Gainza, P.; Ryckmans, T.; Fasching, B.; Thomä, N.H. From Thalidomide to Rational Molecular Glue Design for Targeted Protein Degradation. Annu. Rev. Pharmacol. Toxicol. 2024, 64, 291–312. [Google Scholar] [CrossRef]
- Dong, G.; Ding, Y.; He, S.; Sheng, C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64, 10606–10620. [Google Scholar] [CrossRef]
- Li, H.; Dong, J.; Cai, M.; Xu, Z.; Cheng, X.-D.; Qin, J.-J. Protein degradation technology: A strategic paradigm shift in drug discovery. J. Hematol. Oncol. 2021, 14, 1–23. [Google Scholar] [CrossRef]
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef]
- Zeng, S.; Huang, W.; Zheng, X.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Principle | Description |
|---|---|
| Linker Length | PEG increases solubility and compatibility with biological systems. |
| Linker Composition | Hydrophobic linkers can improve membrane permeability, which is crucial for targeting intracellular proteins. Alkyl chains can improve membrane permeability. Hydrophobic linkers may have solubility issues. |
| Attachment Points | Attachment points of the linker to the ligand are crucial for proper formation of the ternary complex Optimisation of length and attachment points can significantly impact protein degradation efficiency. |
| Linker Permeability | Chemical composition and flexibility of the linker are key for forming folded conformations, which correlate with high cellular permeability. |
| Dimension | PROTAC | Traditional Inhibitors |
|---|---|---|
| Mechanism | Targeted protein degradation via the ubiquitin–proteasome system | Inhibition of protein function by binding to active sites |
| Catalytic Efficiency | Catalytic; one PROTAC molecule can degrade multiple target proteins | Non-catalytic; requires continuous binding to inhibit protein function |
| Target Scope | Can target “undruggable” proteins lacking active sites | Limited to proteins with well-defined active sites |
| Clinical Advantages | Lower doses are required, the degradation is sustained, and the off-target effects are reduced | Effective for proteins with accessible active sites and well-established methods |
| Resistance Mechanisms | Can overcome resistance due to mutations in active sites | Resistance often arises from mutations in the protein’s active site |
| Dosing | Lower doses are required, as PROTACs act catalytically; one PROTAC molecule can degrade multiple targets | Requires continuous presence at high concentrations to maintain therapeutic effect |
| Dosing Frequency | The sustained degradation effect has resulted in a reduced dosing frequency | Frequent dosing is necessary to ensure the inhibitor remains bound to the target protein |
| Clinical Trial Number | Phase of Clinical Trial | Study Status | Conditions | Enrolment |
|---|---|---|---|---|
| NCT05930925 | Phase 1 | Completed | Healthy | 12 |
| NCT05732428 | Phase 1 | Completed | Breast cancer | 9 |
| NCT04072952 | Phase 1 Phase 2 | Active, not recruiting | Breast cancer | 217 |
| NCT05501769 | Phase 1 | Active, not recruiting | Breast cancer | 32 |
| NCT05548127 | Phase 1 Phase 2 | Active, not recruiting | Breast cancer | 37 |
| NCT05652660 | Phase 1 | Completed | Healthy | 12 |
| NCT05573555 | Phase 1 Phase 2 | Recruiting | Breast cancer | 47 |
| NCT05673889 | Phase 1 | Completed | Healthy | 24 |
| NCT06125522 | Phase 1 Phase 2 | Recruiting | Breast cancer | 67 |
| NCT05463952 | Phase 1 | Active, not recruiting | Breast neoplasms | 6 |
| NCT05549505 | Phase 2 | Completed | Breast cancer | 152 |
| NCT05909397 | Phase 3 | Active, not recruiting | Breast cancer | 1180 |
| NCT05654623 | Phase 3 | Active, not recruiting | Advanced breast cancer | 624 |
| NCT06347861 | Phase 1 | Completed | Healthy | 52 |
| NCT06645938 | Phase 1 | Recruiting | Healthy | 12 |
| NCT05538312 | Phase 1 | Completed | Healthy | 12 |
| NCT06206837 | Phase 1 Phase 2 | Recruiting | Breast cancer | 65 |
| NCT06256510 | Phase 1 | Completed | Healthy participants | 15 |
| NCT06005688 | Phase 1 | Completed | Healthy participants | 12 |
| NCT01042379 | Phase 2 | Recruiting | Angiosarcoma Breast cancer Breast neoplasms Breast tumours | 5000 |
| NCT06275841 | Phase 1 | Completed | Healthy | 12 |
| Indications | Degrader | Target | Ref. |
|---|---|---|---|
| Breast cancer | ARV-471 | ER | [50] |
| AC-682 | ER | [51] | |
| Prostate cancer | ARV-110 | AR | [48] |
| CC-94676 | AR | [52] | |
| HP-518 | AR | [53] | |
| AC0176 | AR | [54] | |
| Cancer and solid tumours | CFT-1946 | BRAF-V600 | [55] |
| RNK-05047 | BRD4 | [56] | |
| Multiple myeloma | CFT-7455 | IKZF1/3 | [57] |
| Non-small cell lung cancer | CFT8919 | EGFR | [58] |
| B-cell lymphoma | HSK29116 | BTK | [59] |
| B-cell malignant tumour | NX-5948 | BTK | [60] |
| NX-2127 | BTK | [61] | |
| Synovial sarcoma | FHD-609 | BRD9 | [62] |
| Advanced synovial sarcoma | CFT8634 | BRD9 | [63] |
| Pancreatic cancer and solid tumours | ASP3082 | KRAS G12D | [64] |
| B cell malignancy | BGB-16673 | BTK | [65] |
| Haematologic cancers | AC0676 | BTK | [66] |
| Soft tissue sarcoma | CFT8634 | BRD9 | [63] |
| Solid and blood tumour | DT2216 | BCL-XL | [67] |
| Technology Type | Schematic Representation | Therapeutic Implications |
|---|---|---|
| Antibody-PROTAC | 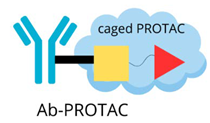 | The precise targeting ability of antibodies with the protein-degradation function of PROTAC: - selective breakdown of disease-causing proteins in specific tissues or cell types, - improving therapeutic accuracy, - minimising unintended side effects. |
| Aptamer-PROTAC Conjugates | 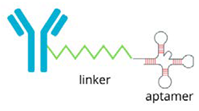 | The precise binding ability of aptamers and the protein-degrading function of PROTAC to selectively degrade disease-associated proteins: - improves therapeutic accuracy, - minimises unintended effects, - holds promise for treating a range of diseases, such as cancer. |
| Dual-Target PROTAC |  | This dual engagement strategy is designed to simultaneously engage two different proteins: - enhancing the degradation of disease-related targets, - increasing therapeutic efficacy, - reducing the likelihood of resistance, - offering potential for treating multifactorial diseases like cancer. |
| Folate-Caged PROTAC |  | This targeted approach utilises folate receptors, which are highly expressed in many cancers, to selectively deliver and degrade disease-causing proteins: - enhancing therapeutic precision, - minimising off-target effects, - holding promise for improving cancer treatment outcomes. |
| TF-PROTAC | 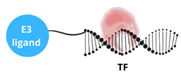 | This approach enables the degradation of transcription factors that lack small molecule binding sites and target transcription factors by combining a DNA oligonucleotide with an E3 ligase ligand: - enhancing therapeutic precision, - reducing off-target effects, - offering potential for treating cancers and other diseases. |
| PhosphoTAC | 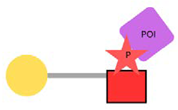 | This approach is designed to degrade specific proteins by leveraging phosphorylation. When activated by receptor tyrosine kinases (RTKs), PhosphoTAC recruits proteins such as FRS2α and PI3K, leading to their ubiquitination and subsequent degradation. |
| PhosTAC |  | Small bifunctional molecules that promote dephosphorylation by bringing a phosphatase close to the target protein. |
| Photocaged PROTAC |  | This approach is designed to degrade target proteins upon exposure to specific wavelengths of light: - precise spatial and temporal control of protein degradation, making it a promising strategy for treating diseases like cancer, - minimising off-target effects, - reducing toxicity. |
| CLIPTAC | 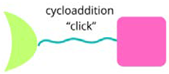 | Utilise click chemistry to assemble bifunctional molecules within cells, enhancing targeted protein degradation. |
| Aspect | PROTAC | Molecular Glues | Hydrophobic Tagging |
|---|---|---|---|
| Mechanism | Link target protein to E3 ubiquitin ligase for ubiquitination and degradation | Stabilize interaction between E3 ubiquitin ligase and target protein for degradation | Attach hydrophobic tag to target protein, causing misfolding and degradation |
| Advantages | High specificity Catalytic nature, requiring lower doses | Simplicity Broad applicability | Simplicity Versatility |
| Specificity | High specificity | Moderate specificity | Lower specificity |
| Complexity | High complexity in synthesis and optimisation | Moderate complexity | Lower complexity |
| Mechanism Type | Relies on ubiquitin–proteasome system | Relies on ubiquitin–proteasome system | Does not rely on ubiquitin–proteasome system |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubryń, N.; Fijałkowski, Ł.; Nowaczyk, J.; Jamil, A.; Nowaczyk, A. PROTAC Technology as a New Tool for Modern Pharmacotherapy. Molecules 2025, 30, 2123. https://doi.org/10.3390/molecules30102123
Kubryń N, Fijałkowski Ł, Nowaczyk J, Jamil A, Nowaczyk A. PROTAC Technology as a New Tool for Modern Pharmacotherapy. Molecules. 2025; 30(10):2123. https://doi.org/10.3390/molecules30102123
Chicago/Turabian StyleKubryń, Natalia, Łukasz Fijałkowski, Jacek Nowaczyk, Amer Jamil, and Alicja Nowaczyk. 2025. "PROTAC Technology as a New Tool for Modern Pharmacotherapy" Molecules 30, no. 10: 2123. https://doi.org/10.3390/molecules30102123
APA StyleKubryń, N., Fijałkowski, Ł., Nowaczyk, J., Jamil, A., & Nowaczyk, A. (2025). PROTAC Technology as a New Tool for Modern Pharmacotherapy. Molecules, 30(10), 2123. https://doi.org/10.3390/molecules30102123