
Electrospray–Mass Spectrometry-Guided Targeted Isolation of Indole Alkaloids from Leaves of Catharanthus roseus by Using High-Performance Countercurrent Chromatography


Abstract

1. Introduction
2. Results and Discussion
2.1. Characterization of C. roseus Crude Extract by LC-ESI-MS Analysis
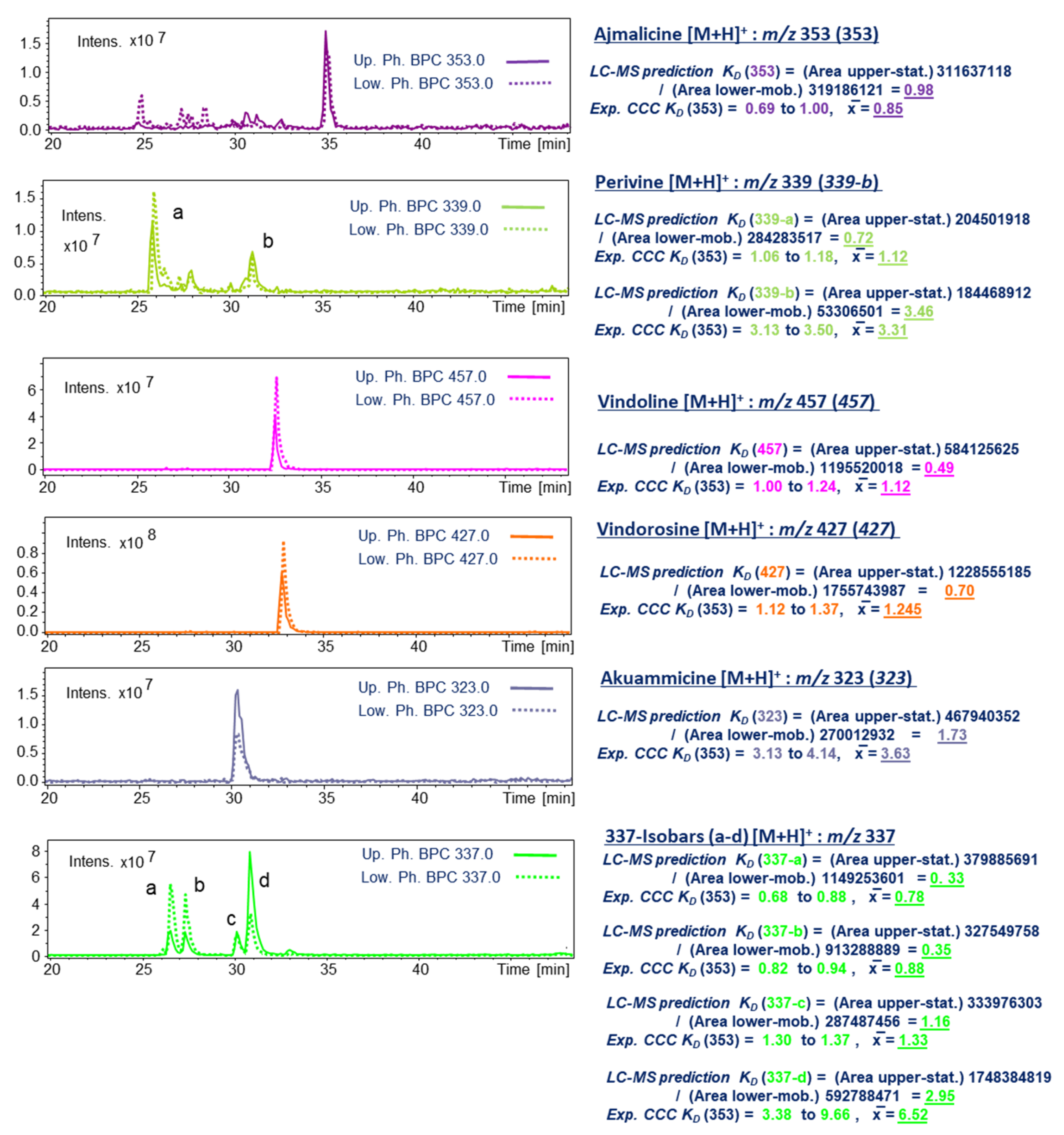
2.2. Solvent System Selection and KD Prediction of Target IAs on HPCCC Using LC-ESI-MS
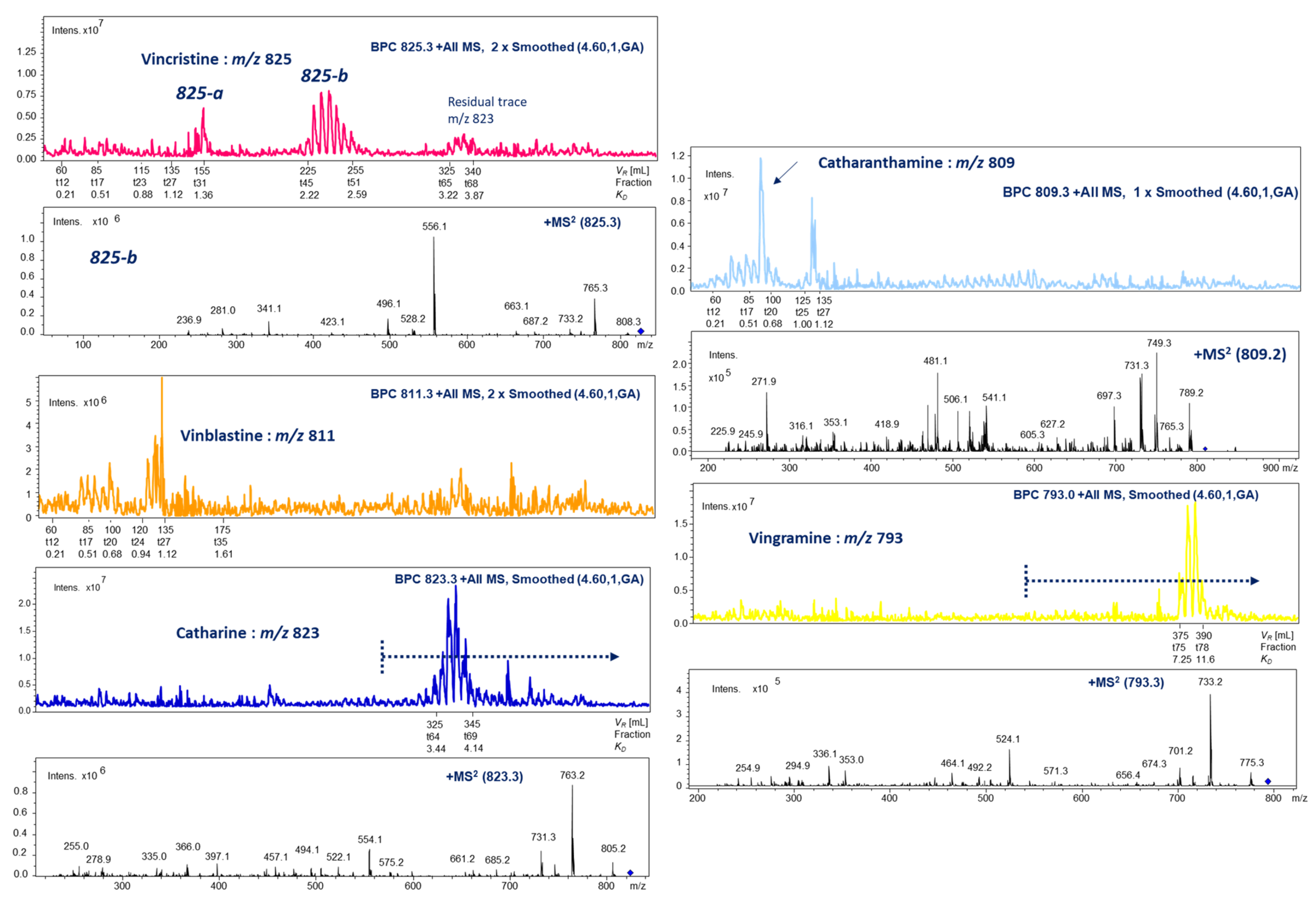
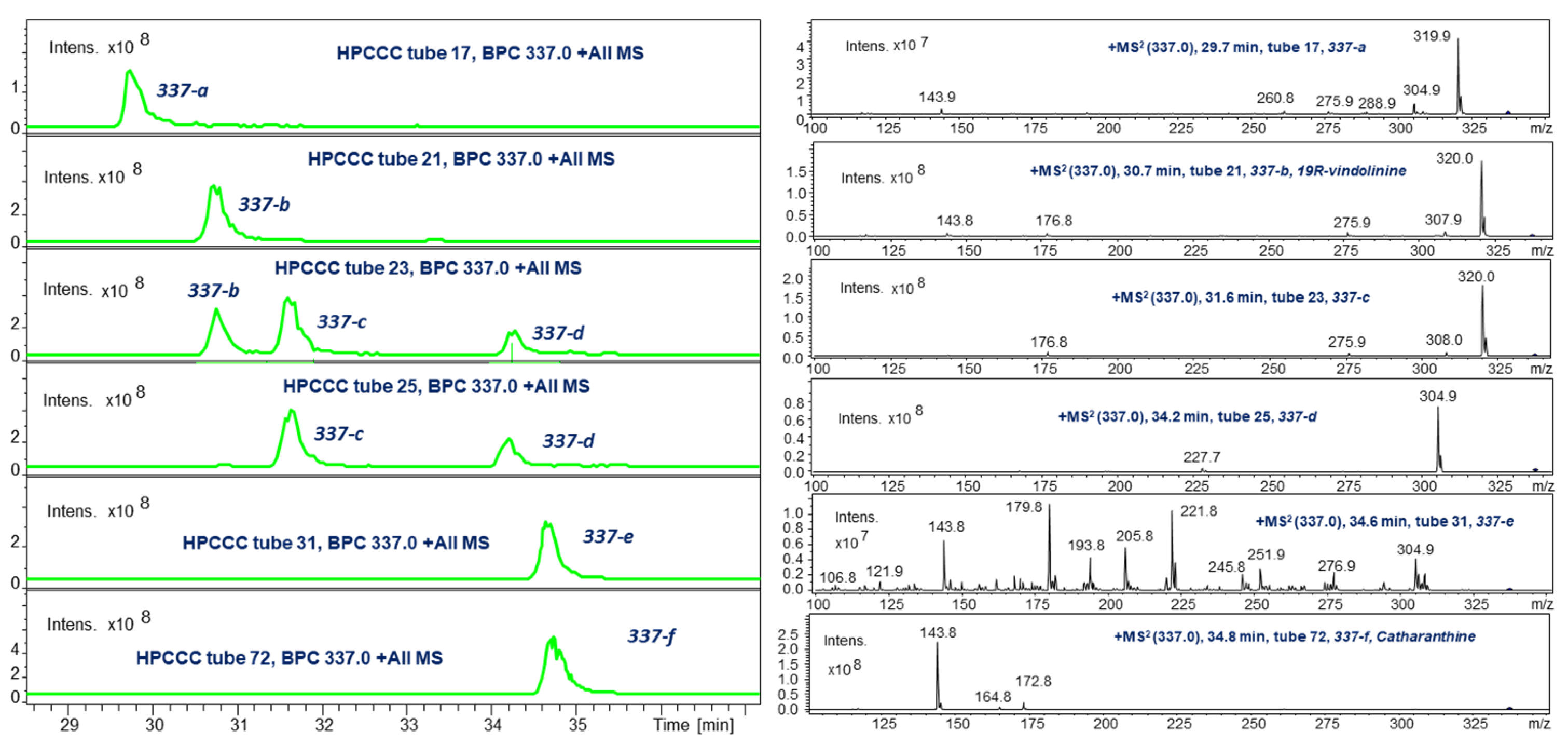
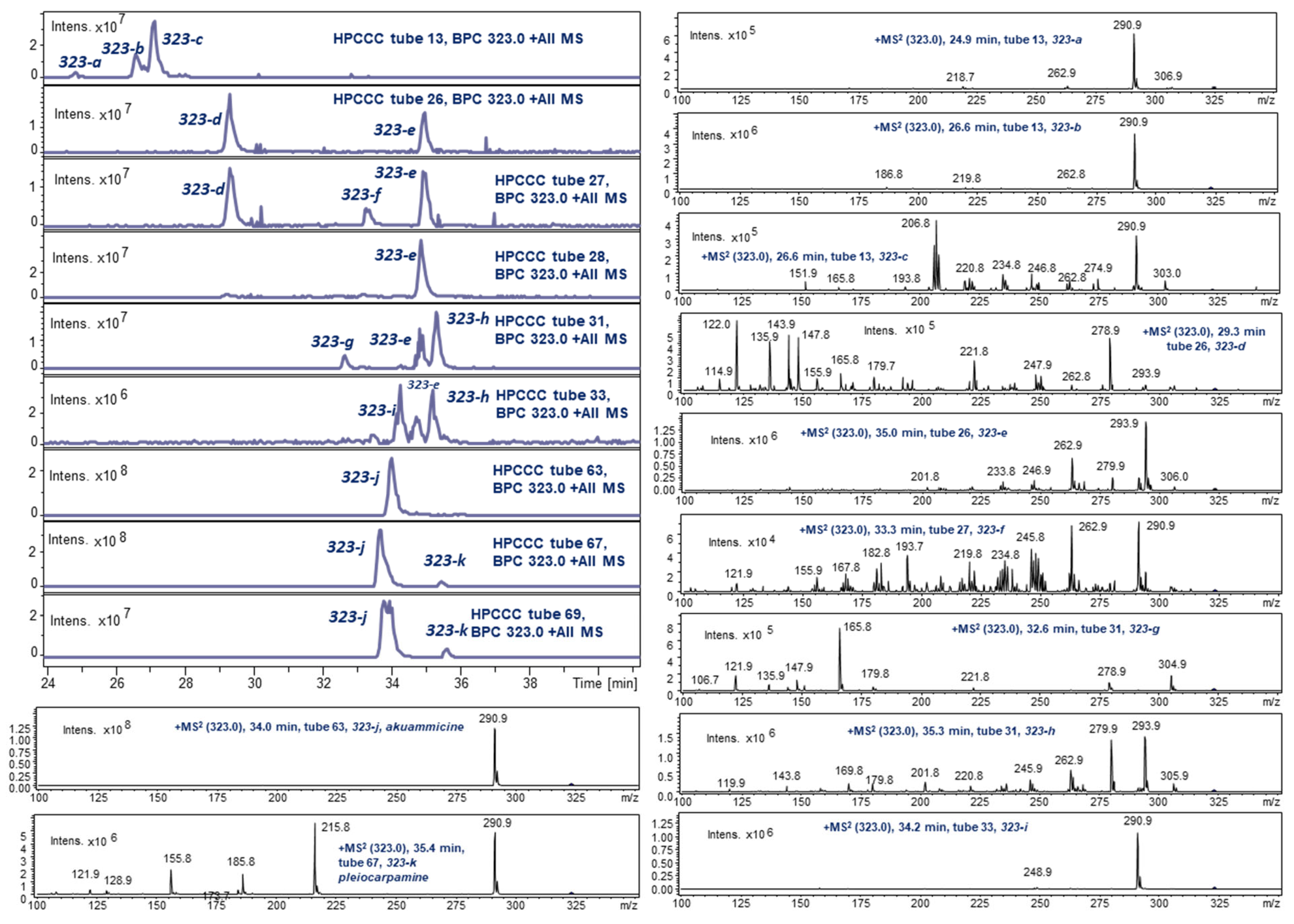
2.3. HPCCC Off-Line ESI-MS/MS Injection Profiles
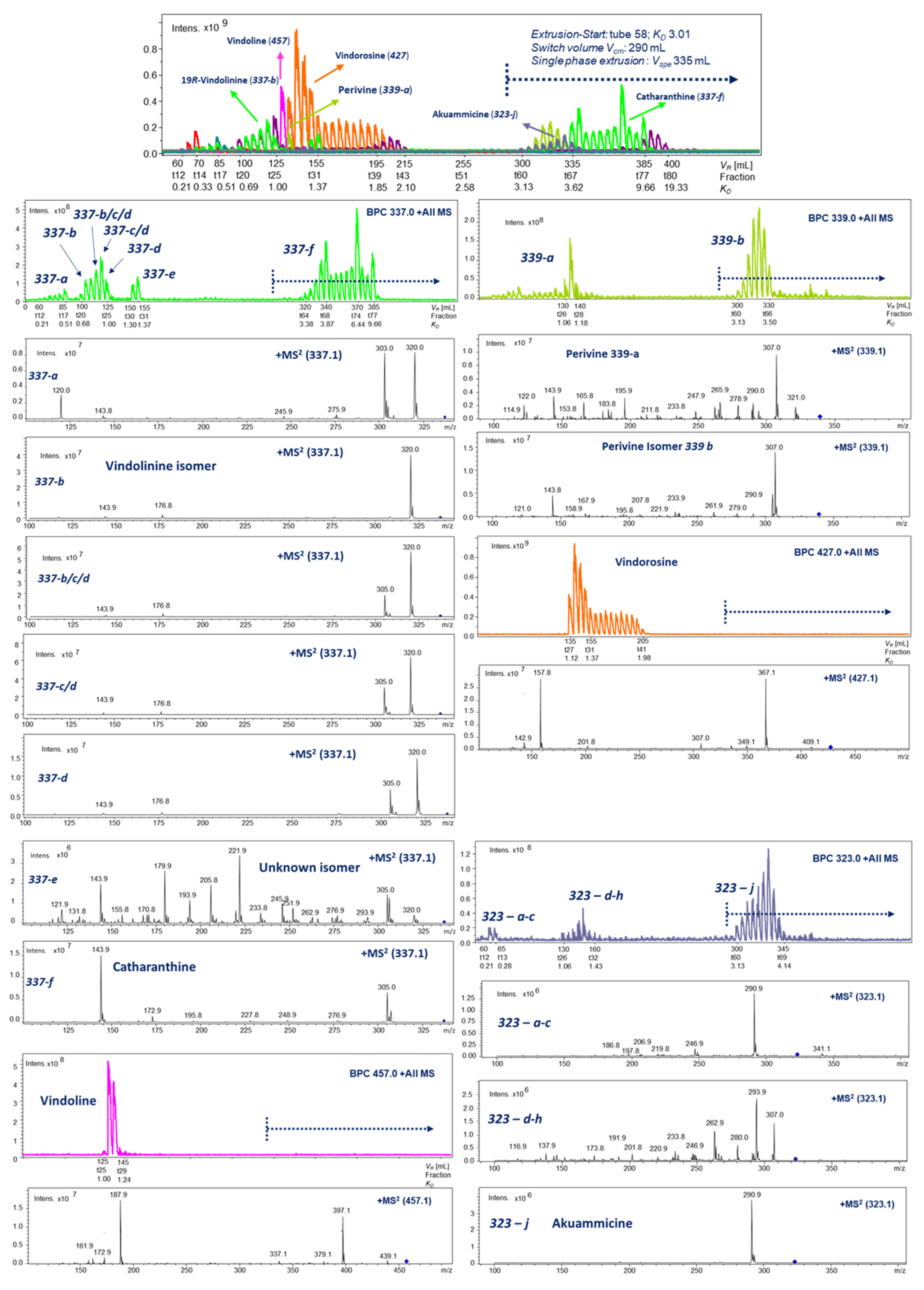
2.4. LC-ESI-MS of Selected HPCCC Fractions for Molecular Weight Isobar Monitoring
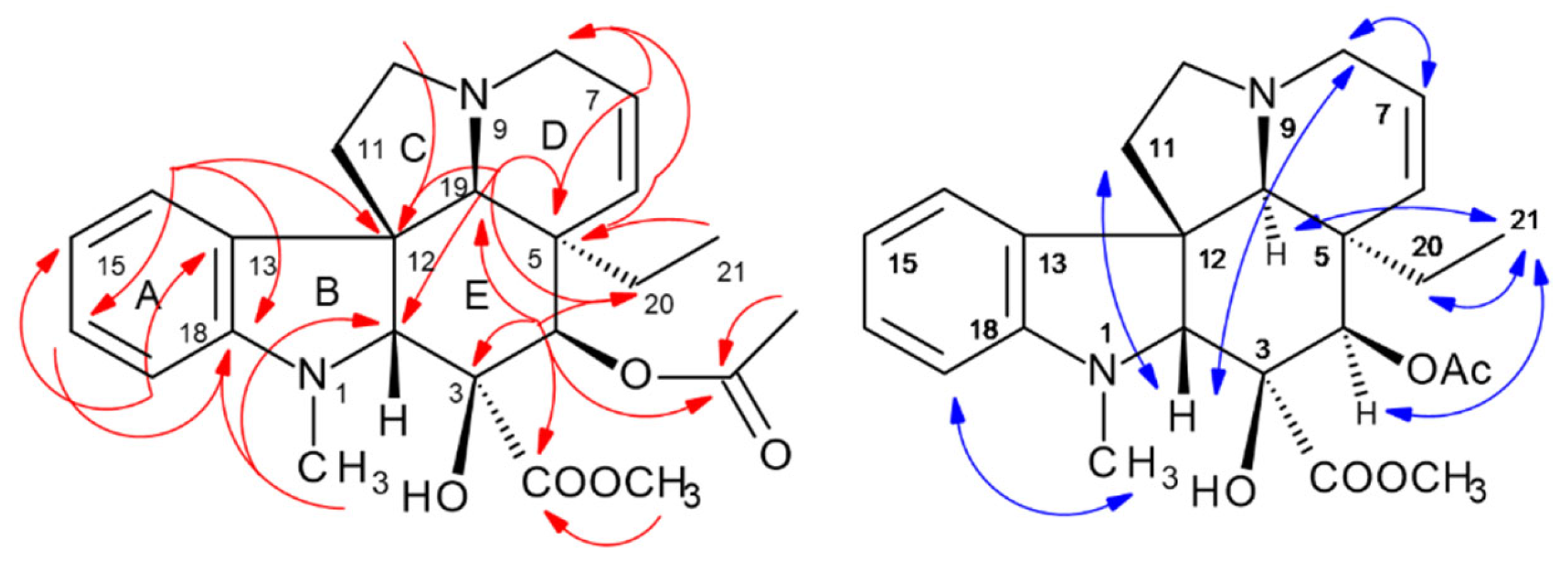
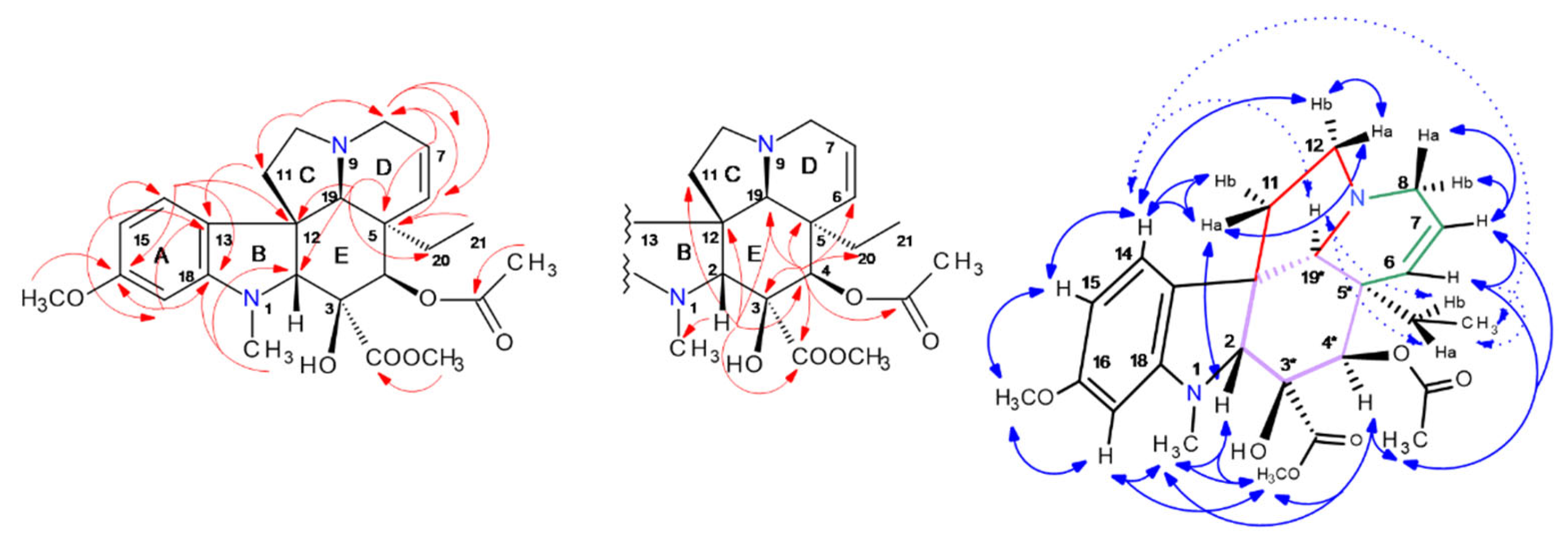
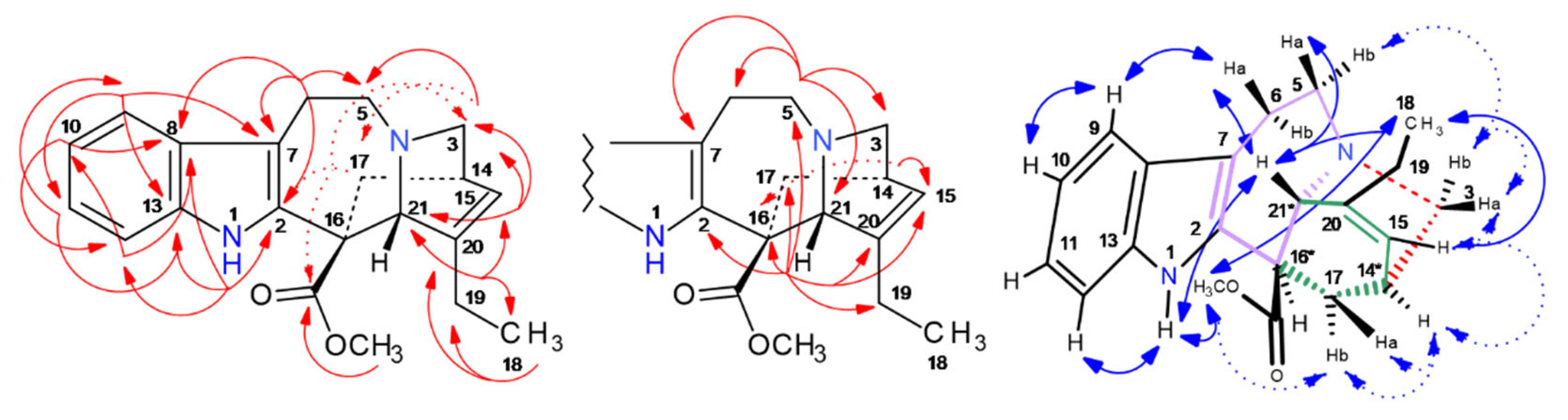
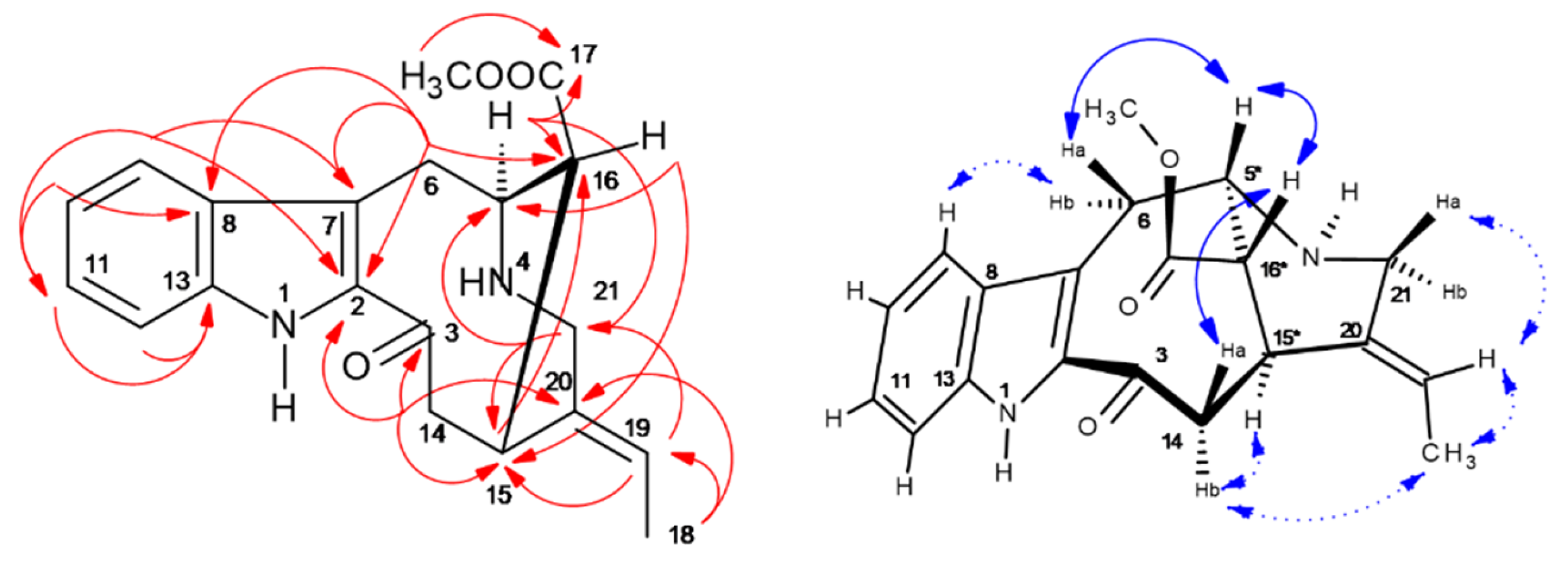
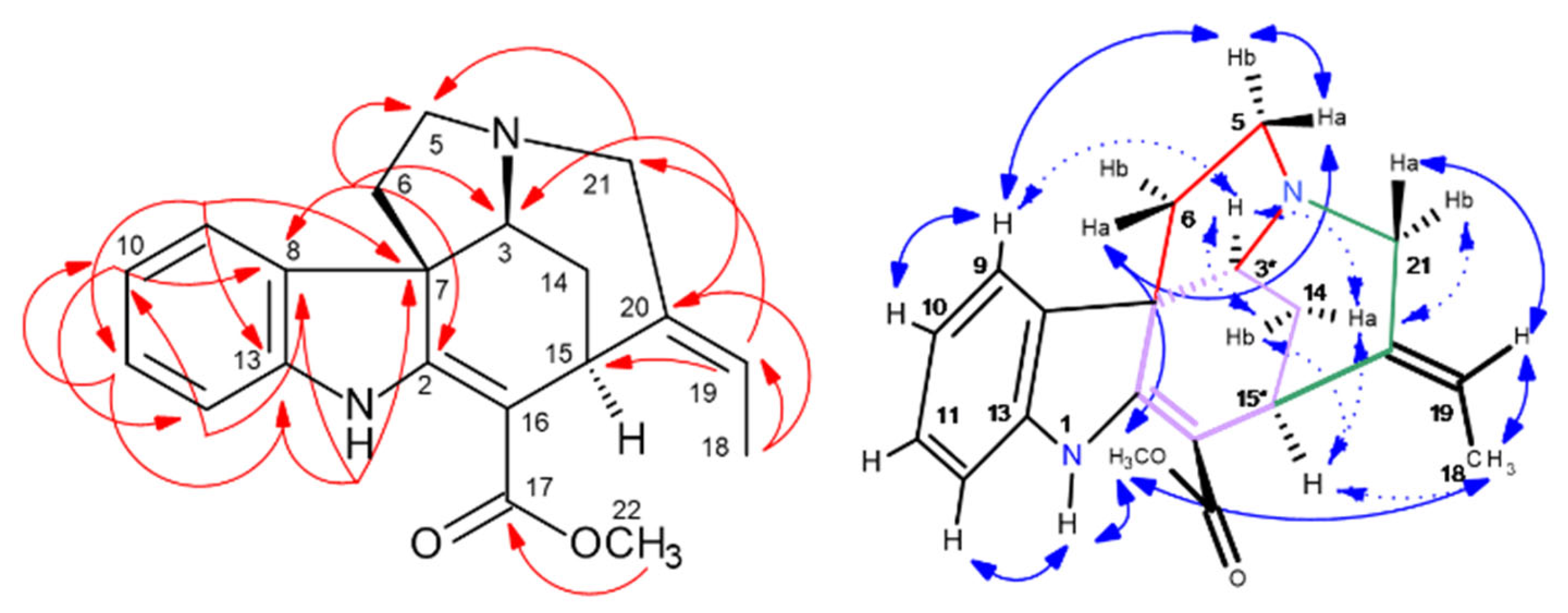
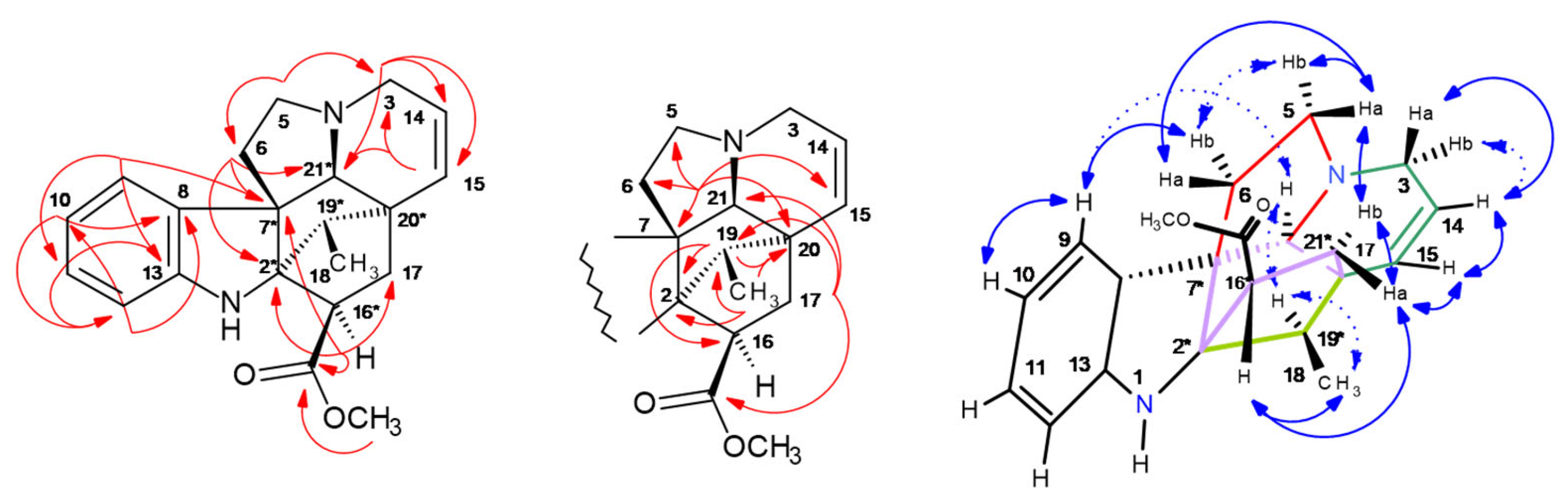
2.5. HPCCC Separation and 1D/2D NMR Structure Elucidation of Indole Alkaloids (IAs)
2.5.1. Vindorosine (427): (Syn. Demethyoxy-Vindoline, Vindolidine); [M + H]+: m/z 427
2.5.2. Vindoline (457): (Syn. Methoxy-Vindorosine); [M + H]+: m/z 457
2.5.3. Catharanthine (337-f): [M + H]+ m/z 337
2.5.4. Perivine (339-a): [M + H]+ m/z 339
2.5.5. Akuammicine (323-j): ([M + H]+: m/z 323
2.5.6. 19R-Vindolinine (337-b): [M + H]+, m/z 337
3. Materials and Methods
3.1. Plant Materials
3.2. Reagents
3.3. Preparation of Alkaloid-Enriched Extracts from the Aerial Plant of C. roseus
3.4. Solvent System Selection by Prediction of Partition Ratio KD Values in the Biphasic HPCCC Solvent System Phase Layers by LC-ESI-MS
3.5. Semi-Preparative Elution–Extrusion HPCCC and Peripheral Devices
3.6. Off-Line ESI-MS/MS Injection Analysis for Semi-Preparative HPCCC Indole-Alkaloid Profiling
3.6.1. General Procedures
3.6.2. ESI-MS/MS Parameter Settings
Off-Line ESI-MS/MS
LC-ESI-MS/MS
3.7. Drying of HPCCC Fractions for Preparative Clean-Up
3.8. Calculation of KD Scale of HPCCC Experiment
3.9. Preparative RP18-HPLC
3.10. Open-Column (CC-SiO2) Separation and TLC Analysis
3.11. NMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HPCCC | High-performance countercurrent chromatography |
| CPC | Centrifugal partition chromatography |
| TLC | Thin-layer chromatography |
| LC-ESI-MS | Liquid chromatography–electrospray–mass-spectrometry |
| IAs | Indole alkaloids |
| NMR | Nuclear magnetic resonance spectroscopy |
| DEPT135 | Distortionless enhancement by polarization transfer |
| HSQC | Hetero single-quantum transfer |
| HMBC | Hetero multiple-bond correlation |
| COSY | Correlation spectroscopy |
| NOESY | Nuclear Overhauser-effect spectroscopy |
References
- Boiteau, P.; Allorge-Boiteau, L. Plantes Médicinales de Madagascar: Cinquante-Huit Plantes Médicinales Utilisées Sur Le Marché de Tananarive (Zoma) à Madagascar; KARTHALA Editions: Paris, France, 1993; pp. 1–135. [Google Scholar]
- Tariq Afta, M.N.; Khan, M.M.A. Catharanthus roseus—Current Research and Future Prospects; Springer: Cham, Switzerland, 2017; pp. 1–424. [Google Scholar]
- Van der Heijden, R.; Jacobs, D.I.; Snoeijer, W.; Hallard, D.; Verpoorte, R. The Catharanthus alkaloids: Pharmacognosy and biotechnology. Curr. Med. Chem. 2004, 11, 607–628. [Google Scholar] [CrossRef] [PubMed]
- Almagro, L.; Fernández-Pérez, F.; Pedreño, M.A. Indole alkaloids from Catharanthus roseus: Bioproduction and their effect on human health. Molecules 2015, 20, 2973–3000. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, J.; Baggaley, K.H.; Roberts, A.D.; Szabo, L.F. Dictionary of Alkaloids, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 1–2374. [Google Scholar]
- Cannell, R.J.P. James McAlpine, Separation by high-speed countercurrent chromatography. In Methods in Biotechnology 4: Natural Products Isolation; Humana Press: Totowa, NJ, USA, 1998; pp. 247–259. [Google Scholar]
- Berthod, A. Separation and purification of natural products (medicinal herbs) by high-speed countercurrent chromatography. In Countercurrent Chromatography—The Support-Free Liquid Stationary Phase; Barcelo, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2002; Chapter 8; Volume 38, pp. 201–260. [Google Scholar]
- Marston, A.; Hostettmann, K. Developments in the application of counter-current chromatography to plant analysis. J. Chromatogr. A 2006, 1112, 181–194. [Google Scholar] [CrossRef]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Pauli, G.F.; Pro, S.M.; Friesen, J.B. Countercurrent separation of natural products. J. Nat. Prod. 2008, 71, 1489–1508. [Google Scholar] [CrossRef]
- Ito, Y.; Conway, W.D. High-speed countercurrent chromatography. CRC Crit. Rev. Anal. Chem. 1986, 17, 65–143. [Google Scholar] [CrossRef]
- Conway, W.D.; Petroski, R.J. Modern Countercurrent Chromatography. Conway, W.D., Petroski, R.J., Eds.; American Chemical Society: Washington, DC, USA, 1995; pp. 1–238. [Google Scholar]
- Sutherland, I.; Ignatova, S.; Hewitson, P.; Janaway, L.; Wood, P.; Edwards, N.; Harris, G.; Guzlek, H.; Keay, D.; Freebairn, K.; et al. Scalable technology for the extraction of pharmaceutics (STEP): The transition from academic knowhow to industrial reality. J. Chromatogr. A 2011, 1218, 6114–6121. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, B.; Chen, L.; Luo, H.; Fisher, D.; Sutherland, I.A.; Wei, Y. How to realize the linear scale-up process for rapid purification using high-performance counter-current chromatography. J. Chromatogr. A 2008, 1194, 192–198. [Google Scholar] [CrossRef]
- das Neves Costa, F.; Neves Vieira, M.; Garrard, I.; Hewitson, P.; Jerz, G.; Guimarães Leitão, G.; Ignatova, S. Schinus terebinthifolius countercurrent chromatography (Part II): Intra-apparatus scale-up and inter-apparatus method transfer. J. Chromatogr. A 2016, 1466, 76–83. [Google Scholar] [CrossRef]
- Renault, J.-H.; Nuzillard, J.-M.; Le Crouérour, G.; Thépenier, P.; Zèches-Hanrot, M.; Le Men-Olivier, L. Isolation of indole alkaloids from Catharanthus roseus by centrifugal partition chromatography in the pH-zone refining mode. J. Chromatogr. A 1999, 849, 421–431. [Google Scholar] [CrossRef]
- Kotland, A.; Chollet, S.; Diard, C.; Autret, J.-M.; Meucci, J.; Renault, J.-H.; Marchal, L. Industrial case study on alkaloids purification by pH-zone refining centrifugal partition chromatography, J. Chromatogr. A 2016, 1474, 59–70. [Google Scholar] [CrossRef]
- Ito, Y. pH-zone-refining counter-current chromatography: Origin, mechanism, procedure and applications. J. Chromatogr. A 2013, 1271, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Friesen, B.J.; Pauli, G.F. GUESS—A Generally Useful Estimate of Solvent Systems for CCC. J. Liq. Chromatogr. Relat. Technol. 2005, 28, 2777–2806. [Google Scholar] [CrossRef]
- Berthod, A.; Ruiz-Angel, M.J.; Carda-Broch, S. Elution-extrusion countercurrent chromatography. Use of the liquid nature of the stationary phase to extend the hydrophobicity window. Anal. Chem. 2003, 75, 5886–5894. [Google Scholar] [CrossRef] [PubMed]
- Jerz, G.; Elnakady, Y.A.; Braun, A.; Jäckel, K.; Sasse, F.; Al Ghamdi, A.A.; Omar, M.O.M.; Winterhalter, P. Preparative mass-spectrometry profiling of bioactive metabolites in Saudi-Arabian propolis fractionated by high-speed countercurrent chromatography and off-line atmospheric pressure chemical ionization mass-spectrometry injection. J. Chromatogr. A 2014, 1347, 17–29. [Google Scholar] [CrossRef]
- Grecco, S.S.; Letsyo, E.; Tempone, A.G.; Ghilardi Lago, J.H.; Jerz, G. Electrospray mass-spectrometry guided target isolation of neolignans from Nectandra leucantha (Lauraceae) by high performance- and spiral-coil countercurrent chromatography. J. Chromatogr. A 2019, 1608, 460422. [Google Scholar] [CrossRef]
- Thi Tran, T.M.; Nguyen Thanh, B.; Moussa-Ayoub, T.E.; Rohn, S.; Jerz, G. Profiling of polar metabolites in fruits of Opuntia stricta var. dillenii by ion-pair high-performance countercurrent chromatography and off-line electrospray mass-spectrometry injection. J. Chromatogr. A 2019, 1601, 274–287. [Google Scholar]
- Surup, F.; Thi Tran, T.M.; Pfütze, S.; Budde, J.; Moussa-Ayoub, T.E.; Rohn, S.; Jerz, G. Opuntisines, 14-membered cyclopeptide alkaloids from fruits of Opuntia stricta var. dillenii isolated by high-performance countercurrent chromatography. Food Chem. 2021, 334, 127552. [Google Scholar]
- Ishikawa, H.; Elliot, G.I.; Velcicky, J.; Choi, Y.; Boger, D.L. Total Synthesis of (−)- and ent-(+)-Vindoline and Related Alkaloids. J. Am. Chem. Soc. 2006, 128, 10596–10612. [Google Scholar] [CrossRef]
- Ruszkowska, J.; Chrobak, R.; Maurin, J.K.; Czarnocki, Z. Vindoline and 16-demethoxyvindoline: Two Catharanthus-derived alkaloids. Acta Cryst. 2004, C60, o377–o380. [Google Scholar] [CrossRef]
- Wenkert, E.; Cochran, D.W.; Hagaman, E.W.; Schell, F.M.; Neuss, N.; Katner, A.S.; Potier, P.; Kan, C.; Plat, M.; Koch, M.; et al. Carbon-13 nuclear magnetic resonance spectroscopy of naturally occurring substances. XIX. Aspidosperma alkaloids. J. Am. Chem. Soc. 1973, 95, 4990–4995. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Tai, Y.; Sun, C.; Pan, Y. Rapid identification of Vinca alkaloids by direct-injection electrospray ionisation tandem mass spectrometry and confirmation by high-performance liquid chromatography–mass spectrometry. Phytochem. Anal. 2005, 16, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Moncrief, J.W.; Lipscomb, W.N.J. Structures of leurocristine (vincristine) and vincaleukoblastine. X-ray analysis of leurocristine methiodide. J. Am. Chem. Soc. 1965, 87, 4963–4964. [Google Scholar] [CrossRef]
- Kumar, S.; Bajpai, V.; Singh, A.; Kumar, B. Identification, characterization and distribution of terpene indole alkaloids in ethanolic extracts of Catharanthus roseus using high-performance liquid chromatography/electrospray ionization quadrupole time-of-flight tandem mass spectrometry and the study of their geographical variation. Rapid Commun. Mass Spectrom. 2018, 32, 319–332. [Google Scholar]
- Balsevich, J.; Hogge, L.R.; Berry, A.J.; Games, D.E.; Mylchreest, I.C. Analysis of indole alkaloids from leaves of Catharanthus roseus by means of supercritical fluid chromatography/mass spectrometry. J. Nat. Prod. 1988, 51, 1173–1177. [Google Scholar] [CrossRef]
- Yahyazadeh, M.; Jerz, G.; Selmar, D.; Winterhalter, P.; Jones, P. Crystal structure of akuammicine, an indole alkaloid from Catharanthus roseus. Acta Cryst. Sect. E 2017, E73, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Ahond, A.; Janot, M.-M.; Langlois, N.; Lukacs, G.; Potier, P.; Rasoanaivo, P.; Sangare, M.; Neuss, N.; Plat, M.; Men, J.L.; et al. On the structure of vindolinine. J. Am. Chem. Soc. 1974, 96, 633–634. [Google Scholar] [CrossRef]
- He, L.; Yang, L.; Tan, R.; Zhao, S.; Hu, Z. Enhancement of vindoline production in suspension culture of the Catharanthus roseus cell line C20hi by light and methyl jasmonate elicitation. Anal. Sci. 2011, 27, 1243–1248. [Google Scholar] [CrossRef]
- Ur-Rahman, A.; Bashir, M.; Kaleem, S.; Fatima, T. 16-Epi-19S-vindolinine, Catharanthus roseus. Phytochemistry 1983, 22, 1021–1023. [Google Scholar] [CrossRef]
- Keever, J.; Voyksner, R.D.; Tyczkowska, K.L. Quantitative determination of ceftiofur in milk by liquid chromatography–electrospray mass spectrometry. J. Chromatogr. A 1998, 794, 57–62. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 19R-Vindolinine (337-b) | -NH | -COOCH3 | ||||
| 2 | Vindoline (457) | -N-CH3 | -COOCH3 | -OH | -OAc | -CH2-CH3 | |
| 3 | Perivine (339-a) | -NH | -COOCH3 | -CH2-CH3 | |||
| 4 | Vindorosine (427) | -N-CH3 | -COOCH3 | -OH | -OAc | -CH2-CH3 | -O-CH3 |
| 5 | Akuammicine (323-j) | -NH | -COOCH3 | ||||
| 6 | Catharanthine (337-f) | -NH | -CO | -OH | -OAc | -CH2-CH3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yahyazadeh, M.; Selmar, D.; Jerz, G. Electrospray–Mass Spectrometry-Guided Targeted Isolation of Indole Alkaloids from Leaves of Catharanthus roseus by Using High-Performance Countercurrent Chromatography. Molecules 2025, 30, 2115. https://doi.org/10.3390/molecules30102115
Yahyazadeh M, Selmar D, Jerz G. Electrospray–Mass Spectrometry-Guided Targeted Isolation of Indole Alkaloids from Leaves of Catharanthus roseus by Using High-Performance Countercurrent Chromatography. Molecules. 2025; 30(10):2115. https://doi.org/10.3390/molecules30102115
Chicago/Turabian StyleYahyazadeh, Mahdi, Dirk Selmar, and Gerold Jerz. 2025. "Electrospray–Mass Spectrometry-Guided Targeted Isolation of Indole Alkaloids from Leaves of Catharanthus roseus by Using High-Performance Countercurrent Chromatography" Molecules 30, no. 10: 2115. https://doi.org/10.3390/molecules30102115
APA StyleYahyazadeh, M., Selmar, D., & Jerz, G. (2025). Electrospray–Mass Spectrometry-Guided Targeted Isolation of Indole Alkaloids from Leaves of Catharanthus roseus by Using High-Performance Countercurrent Chromatography. Molecules, 30(10), 2115. https://doi.org/10.3390/molecules30102115