NOTA and NODAGA Radionuclide Complexing Agents: Versatile Approaches for Advancements in Radiochemistry

 , ,
, , 
Abstract
1. NOTA/NODAGA Chemistry
2. Theranostic Isotopes for Usage with NOTA/NODAGA Complexing Agents
3. NOTA/NODAGA Radiometal Complexes of Peptides and Small Molecule Cell-Targeting Agents
3.1. Gastrin-Releasing Peptide Receptor (GRPR)-Targeting Radioligands
3.2. Somatostatin Receptor Subtype 2 (SSTR2)-Targeting Radioligands
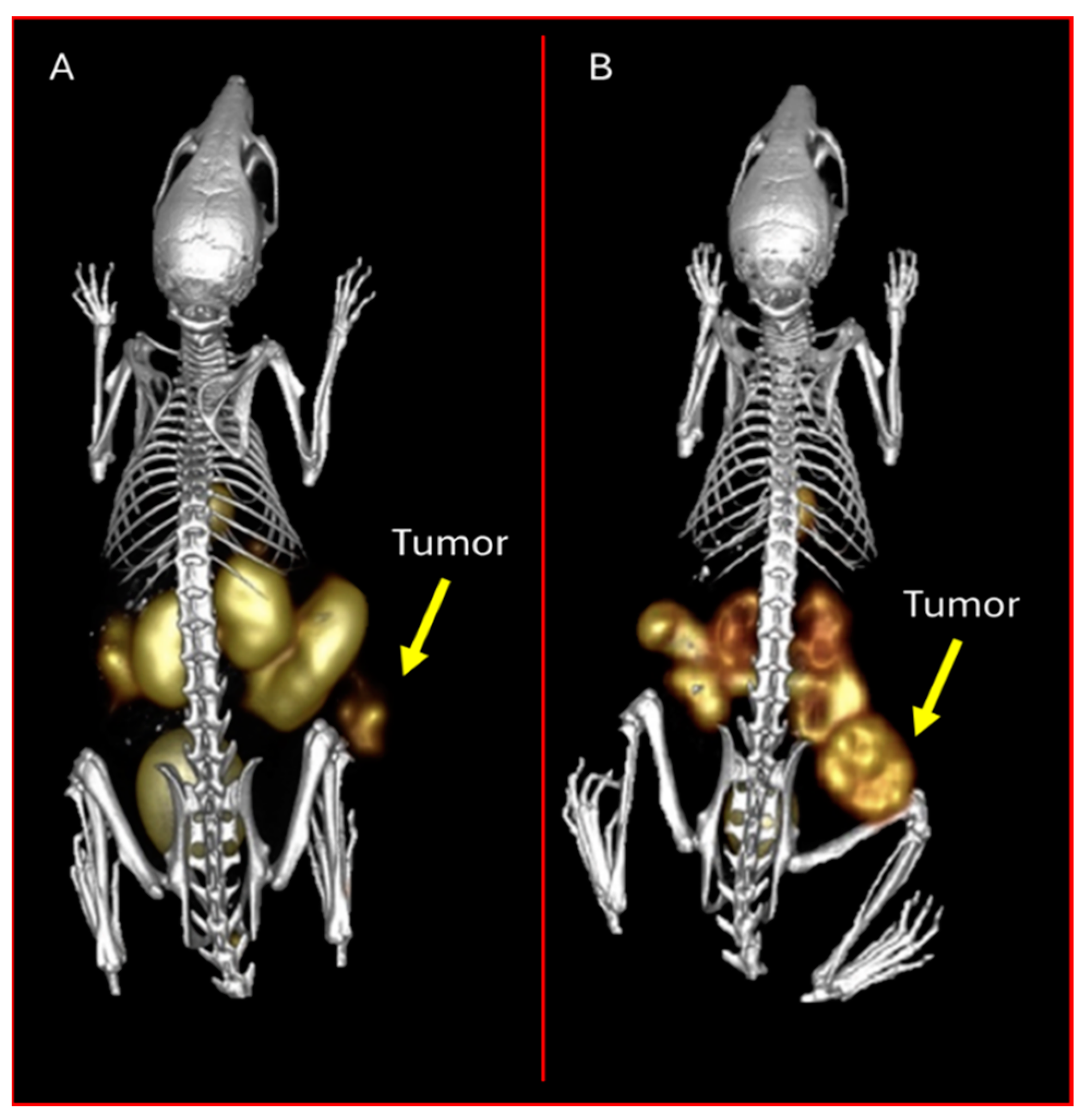
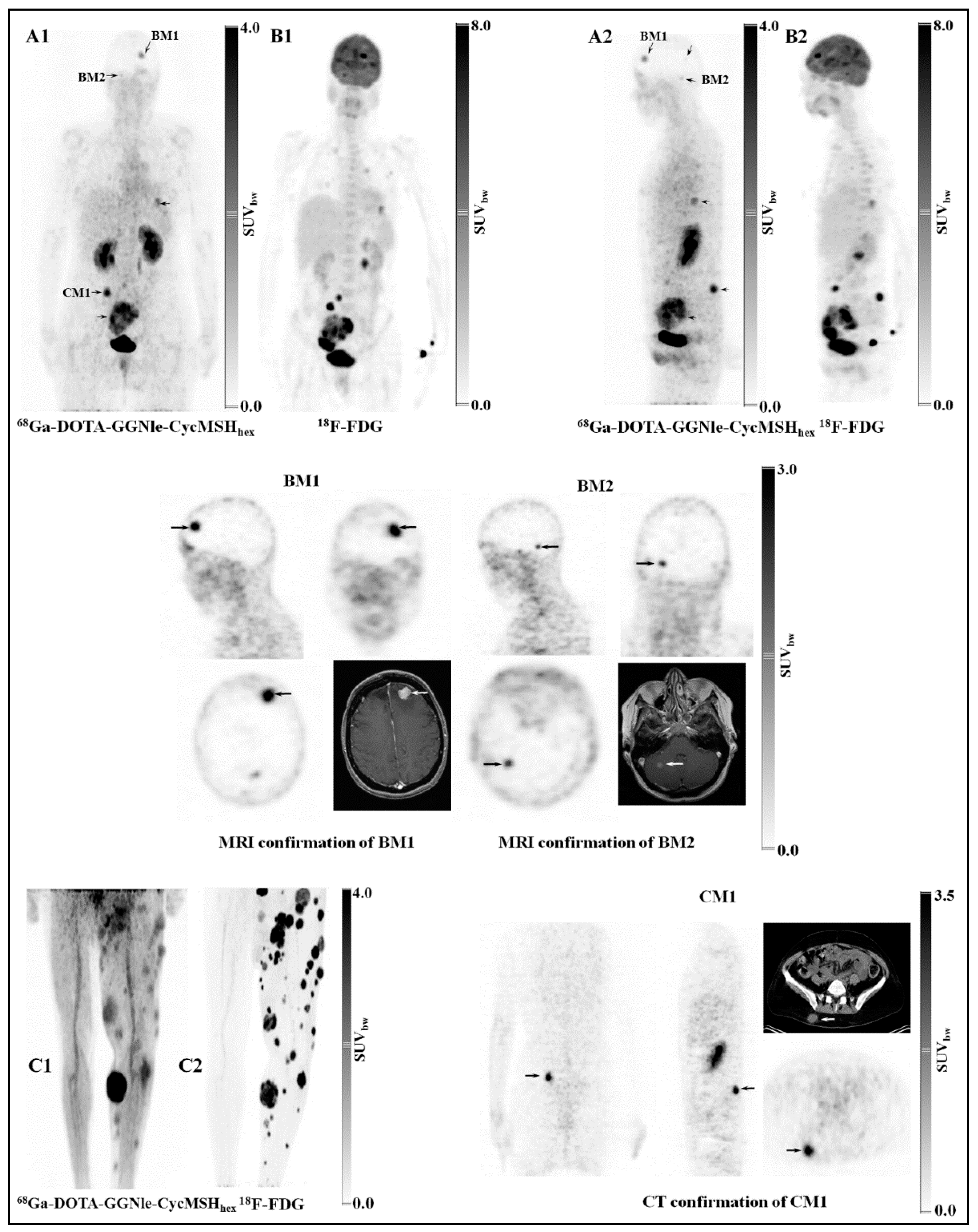
3.3. Melanocortin-1 Receptor (MC1R)-Targeting Radioligands
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kukis, D.L.; Diril, H.; Greiner, D.P.; DeNardo, S.J.; DeNardo, G.L.; Salako, Q.A.; Meares, C.F. A comparative study of copper-67 radiolabeling and kinetic stabilities of antibody-macrocycle chelate conjugates. Cancer 1994, 73, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Kukis, D.L.; Li, M.; Meares, C.F. Selectivity of antibody-chelate conjugates for binding copper in the presence of competing metals. Inorg. Chem. 1993, 32, 3981–3982. [Google Scholar] [CrossRef]
- Pfeifer, A.; Knigge, U.; Binderup, T.; Mortensen, J.; Oturai, P.; Loft, A.; Berthelsen, A.K.; Langer, S.W.; Rasmussen, P.; Elema, D.; et al. 64Cu-DOTATATE PET for Neuroendocrine Tumors: A Prospective Head-to-Head Comparison with 111In-DTPA-Octreotide in 112 Patients. J. Nucl. Med. 2015, 56, 847–854. [Google Scholar] [CrossRef]
- Loft, M.; Carlsen, E.A.; Johnbeck, C.B.; Johannesen, H.H.; Binderup, T.; Pfeifer, A.; Mortensen, J.; Oturai, P.; Loft, A.; Berthelsen, A.K.; et al. Cu-DOTATATE PET in Patients with Neuroendocrine Neoplasms: Prospective, Head-to-Head Comparison of Imaging at 1 Hour and 3 Hours After Injection. J. Nucl. Med. 2021, 62, 73. [Google Scholar] [CrossRef]
- Yang, X.; Mease, R.C.; Pullambhatla, M.; Lisok, A.; Chen, Y.; Foss, C.A.; Wang, Y.; Shallal, H.; Edelman, H.; Hoye, A.T.; et al. [18F]Fluorobenzoyllysinepentanedioic Acid Carbamates: New Scaffolds for Positron Emission Tomography (PET) Imaging of Prostate-Specific Membrane Antigen (PSMA). J. Med. Chem. 2016, 59, 206–218. [Google Scholar] [CrossRef]
- Banerjee, S.R.; Pullambhatla, M.; Byun, Y.; Nimmagadda, S.; Green, G.; Fox, J.J.; Horti, A.; Mease, R.C.; Pomper, M.G. 68Ga-labeled inhibitors of prostate-specific membrane antigen (PSMA) for imaging prostate cancer. J. Med. Chem. 2010, 53, 5333–5341. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenboeck, S.M.; Rauscher, I.; Bluemel, C.; Fendler, W.P.; Rowe, S.P.; Pomper, M.G.; Afshar-Oromieh, A.; Herrmann, K.; Eiber, M. PSMA Ligands for PET Imaging of Prostate Cancer. J. Nucl. Med. 2017, 58, 1545–1552. [Google Scholar] [CrossRef]
- Duan, X.; Liu, F.; Kwon, H.; Byun, Y.; Minn, I.; Cai, X.; Zhang, J.; Pomper, M.G.; Yang, Z.; Xi, Z.; et al. (S)-3-(Carboxyformamido)-2-(3-(carboxymethyl)ureido)propanoic Acid as a Novel PSMA Targeting Scaffold for Prostate Cancer Imaging. J. Med. Chem. 2020, 63, 3563–3576. [Google Scholar] [CrossRef]
- Smith, K.; Acuff, S.; Osborne, D. Clinical Workflow Differences in Ga-68 Dotatate vs Cu-64 Dotatate. J. Nucl. Med. 2022, 63, 4094. [Google Scholar]
- Das, S.; Al-Toubah, T.; El-Haddad, G.; Strosberg, J. 177Lu-DOTATATE for the treatment of gastroenteropancreatic neuroendocrine tumors. Expert. Rev. Gastroenterol. Hepatol. 2019, 13, 1023–1031. [Google Scholar] [CrossRef]
- Ray Banerjee, S.; Chen, Z.; Pullambhatla, M.; Lisok, A.; Chen, J.; Mease, R.C.; Pomper, M.G. Preclinical Comparative Study of 68Ga-Labeled DOTA, NOTA, and HBED-CC Chelated Radiotracers for Targeting PSMA. Bioconjugate Chem. 2016, 27, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Ferdani, R.; Shokeen, M.; Anderson, C.J. Comparison of two cross-bridged macrocyclic chelators for the evaluation of 64Cu-labeled-LLP2A, a peptidomimetic ligand targeting VLA-4-positive tumors. Nucl. Med. Biol. 2013, 40, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Copper chelation chemistry and its role in copper radiopharmaceuticals. Curr. Pharm. Des. 2007, 13, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Wadas, T.J.; Wong, E.H.; Weisman, G.R. Cross-bridged macrocyclic chelators for stable complexation of copper radionuclides for PET imaging. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 185–192. [Google Scholar]
- Boswell, C.A.; Sun, X.; Niu, W.; Weisman, G.R.; Wong, E.H.; Rheingold, A.L.; Anderson, C.J. Comparative in Vivo Stability of Copper-64-Labeled Cross-Bridged and Conventional Tetraazamacrocyclic Complexes. J. Med. Chem. 2004, 47, 1465–1474. [Google Scholar] [CrossRef]
- Ferdani, R.; Stigers, D.J.; Fiamengo, A.L.; Wei, L.; Li, B.T.Y.; Golen, J.A.; Rheingold, A.L.; Weisman, G.R.; Wong, E.H.; Anderson, C.J. Synthesis, Cu(ii) complexation, 64Cu-labeling and biological evaluation of cross-bridged cyclam chelators with phosphonate pendant arms. Dalton Trans. 2012, 41, 1938–1950. [Google Scholar] [CrossRef]
- Prasanphanich, A.F.; Nanda, P.K.; Rold, T.L.; Ma, L.; Lewis, M.R.; Garrison, J.C.; Hoffman, T.J.; Sieckman, G.L.; Figueroa, S.D.; Smith, C.J. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc. Natl. Acad. Sci. USA 2007, 104, 12462–12467. [Google Scholar] [CrossRef] [PubMed]
- Prasanphanich, A.F.; Retzloff, L.; Lane, S.R.; Nanda, P.K.; Sieckman, G.L.; Rold, T.L.; Ma, L.; Figueroa, S.D.; Sublett, S.V.; Hoffman, T.J.; et al. In vitro and in vivo analysis of [64Cu-NO2A-8-Aoc-BBN(7-14)NH2]: A site-directed radiopharmaceutical for positron-emission tomography imaging of T-47D human breast cancer tumors. Nucl. Med. Biol. 2009, 36, 171–181. [Google Scholar] [CrossRef]
- Garrison, J.C.; Rold, T.L.; Sieckman, G.L.; Figueroa, S.D.; Volkert, W.A.; Jurisson, S.S.; Hoffman, T.J. In vivo evaluation and small-animal PET/CT of a prostate cancer mouse model using 64Cu bombesin analogs: Side-by-side comparison of the CB-TE2A and DOTA chelation systems. J. Nucl. Med. 2007, 48, 1327–1337. [Google Scholar] [CrossRef]
- Non-HEU Production Technologies for Molybdenum-99 and Technetium-99m; International Atomic Energy Agency: Vienna, Austria, 2013.
- Alves, S.; Correia, J.D.G.; Gano, L.; Rold, T.L.; Prasanphanich, A.; Haubner, R.; Rupprich, M.; Alberto, R.; Decristoforo, C.; Santos, I.; et al. In vitro and in vivo evaluation of a novel 99mTc(CO)3-pyrazolyl conjugate of cyclo-(Arg-Gly-Asp-D-Tyr-Lys). Bioconjugate Chem. 2007, 18, 530–537. [Google Scholar] [CrossRef]
- Giblin, M.F.; Wang, N.; Hoffman, T.J.; Jurisson, S.S.; Quinn, T.P. Design and characterization of α-melanotropin peptide analogs cyclized through rhenium and technetium metal coordination. Proc. Natl. Acad. Sci. USA 1998, 95, 12814–12818. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Giblin, M.F.; Wang, N.; Jurisson, S.S.; Quinn, T.P. In vivo evaluation of 99mTc/188Re-labeled linear alpha-melanocyte stimulating hormone analogs for specific melanoma targeting. Nucl. Med. Biol. 1999, 26, 687–693. [Google Scholar] [CrossRef]
- Chen, J.Q.; Cheng, Z.; Hoffman, T.J.; Jurisson, S.S.; Quinn, T.P. Melanoma-targeting properties of 99mtechnetium-labeled cyclic α-melanocyte-stimulating hormone peptide analogues. Cancer Res. 2000, 60, 5649–5658. [Google Scholar]
- Jaouen, G.; Vessieres, A.; Top, S.; Ismail, A.A.; Butler, I.S. Metal carbonyl fragments as a new class of markers in molecular biology. J. Am. Chem. Soc. 1985, 107, 4778–4780. [Google Scholar] [CrossRef]
- Alberto, R. The “Carbonyl Story” and Beyond; Experiences, Lessons and Implications. Chembiochem 2020, 21, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Badar, A.; Williams, J.; de Rosales, R.T.; Tavaré, R.; Kampmeier, F.; Blower, P.J.; Mullen, G.E. Optimising the radiolabelling properties of technetium tricarbonyl and His-tagged proteins. EJNMMI Res. 2014, 4, 14. [Google Scholar] [CrossRef]
- Katti, K.V.; Gali, H.; Smith, C.J.; Berning, D.E. Design and development of functionalized water-soluble phosphines: Catalytic and biomedical implications. Acc. Chem. Res. 1999, 32, 9–17. [Google Scholar] [CrossRef]
- Karacay, H.; McBride, W.J.; Sharkey, R.M.; Cardillo, T.; Smith, C.J.; Goldenberg, D.M. 18F Labeling of a Peptide for PET Imaging of Receptor-expressing Tumors. J. Nucl. Med. 2009, 50 (Suppl. S2), 1567. [Google Scholar]
- Decristoforo, C.; Santos, I.; Pietzsch, H.J.; Kuenstler, J.U.; Duatti, A.; Smith, C.J.; Rey, A.; Alberto, R.; Von Guggenberg, E.; Haubner, R. Comparison of in vitro and in vivo properties of [99mTc]cRGD peptides labeled using different novel Tc-cores. Q. J. Nucl. Med. Mol. Imaging 2007, 51, 33–41. [Google Scholar]
- Smith, C.J.; Volkert, W.A.; Hoffman, T.J. Gastrin releasing peptide (GRP) receptor targeted radiopharmaceuticals: A concise update. Nucl. Med. Biol. 2003, 30, 861–868. [Google Scholar] [CrossRef]
- Alves, S.; Correia, J.D.G.; Santos, I.; Veerendra, B.; Sieckman, G.L.; Hoffman, T.J.; Rold, T.L.; Figueroa, S.D.; Retzloff, L.; McCrate, J.; et al. Pyrazolyl conjugates of bombesin: A new tridentate ligand framework for the stabilization of fac-[M(CO)3]+ moiety. Nucl. Med. Biol. 2006, 33, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Paulo, A.; Correia, J.D.G.; Gano, L.; Smith, C.J.; Hoffman, T.J.; Santos, I. Pyrazolyl derivatives as bifunctional chelators for labeling tumor-seeking peptides with the fac-[M(CO)3]+ moiety (M = 99mTc, Re): Synthesis, characterization, and biological behavior. Bioconjugate Chem. 2005, 16, 438–449. [Google Scholar] [CrossRef]
- McBride, W.J.; D’Souza, C.A.; Sharkey, R.M.; Karacay, H.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Improved 18F labeling of peptides with a fluoride-aluminum- chelate complex. Bioconjugate Chem. 2010, 21, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Overbey, D.; Watkinson, L.D.; Smith, C.J.; Daibes-Figueroa, S.; Hoffman, T.J.; Forte, L.R.; Volkert, W.A.; Giblin, M.F. Comparative evaluation of three 64Cu-labeled E. coli heat-stable enterotoxin analogues for PET imaging of colorectal cancer. Bioconjugate Chem. 2010, 21, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Mastren, T.; Silvers, W.; Hassan, G.; Öz, O.K.; Sun, X. Copper-67 radioimmunotheranostics for simultaneous immunotherapy and immuno-SPECT. Sci. Rep. 2021, 11, 3622. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Haberkorn, U.; Eder, M.; Eisenhut, M.; Zechmann, C.M. [68Ga]Gallium-labelled PSMA ligand as superior PET tracer for the diagnosis of prostate cancer: Comparison with 18F-FECH. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1085–1086. [Google Scholar] [CrossRef]
- Wei, L.; Miao, Y.; Gallazzi, F.; Quinn, T.P.; Welch, M.J.; Vavere, A.L.; Lewis, J.S. Gallium-68-labeled DOTA-rhenium-cyclized α-melanocyte-stimulating hormone analog for imaging of malignant melanoma. Nucl. Med. Biol. 2007, 34, 945–953. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, X.; Gallazzi, F.; Miao, Y.; Jin, X.; Brechbiel, M.W.; Xu, H.; Clifford, T.; Welch, M.J.; Lewis, J.S.; et al. Melanoma imaging using 111In-, 86Y- and 68Ga-labeled CHX-A″-Re(Arg11)CCMSH. Nucl. Med. Biol. 2009, 36, 345–354. [Google Scholar] [CrossRef]
- Hoffman, T.J.; Gali, H.; Smith, C.J.; Sieckman, G.L.; Hayes, D.L.; Owen, N.K.; Volkert, W.A. Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J. Nucl. Med. 2003, 44, 823–831. [Google Scholar]
- Smith, C.J.; Sieckman, G.L.; Owen, N.K.; Hayes, D.L.; Mazuru, D.G.; Kannan, R.; Volkert, W.A.; Hoffman, T.J. Radiochemical investigations of gastrin-releasing peptide receptor-specific [99mTc(X)(CO)3-Dpr-Ser-Ser-Ser-Gln-Trp-Ala-Val-Gly-His-Leu-Met-(NH2)] in PC-3, tumor-bearing, rodent models: Syntheses, radiolabeling, and in vitro/in vivo studies where Dpr = 2,3-diaminopropionic acid and X = H2O or P(CH2OH)3. Cancer Res. 2003, 63, 4082–4088. [Google Scholar]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International Union of Pharmacology. LXVIII. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Fleischmann, A.; Mäcke, H.R.; Reubi, J.C. Targeting GRPR in urological cancers—From basic research to clinical application. Nat. Rev. Urol. 2013, 10, 235–244. [Google Scholar] [CrossRef]
- Nagasaki, S.; Nakamura, Y.; Maekawa, T.; Akahira, J.; Miki, Y.; Suzuki, T.; Ishidoya, S.; Arai, Y.; Sasano, H. Immunohistochemical analysis of gastrin-releasing peptide receptor (GRPR) and possible regulation by estrogen receptor βcx in human prostate carcinoma. Neoplasma 2012, 59, 224–232. [Google Scholar] [CrossRef]
- Sun, B.; Halmos, G.; Schally, A.V.; Wang, X.; Martinez, M. Presence of receptors for bombesin/gastrin-releasing peptide and mRNA for three receptor subtypes in human prostate cancers. Prostate 2000, 42, 295–303. [Google Scholar] [CrossRef]
- Markwalder, R.; Reubi, J.C. Gastrin-releasing peptide receptors in the human prostate: Relation to neoplastic transformation. Cancer Res. 1999, 59, 1152–1159. [Google Scholar] [PubMed]
- Fleischmann, A.; Waser, B.; Reubi, J.C. High expression of gastrin-releasing peptide receptors in the vascular bed of urinary tract cancers: Promising candidates for vascular targeting applications. Endocr. Relat. Cancer 2009, 16, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Beer, M.; Montani, M.; Gerhardt, J.; Wild, P.J.; Hany, T.F.; Hermanns, T.; Müntener, M.; Kristiansen, G. Profiling gastrin-releasing peptide receptor in prostate tissues: Clinical implications and molecular correlates. Prostate 2012, 72, 318–325. [Google Scholar] [CrossRef]
- Baratto, L.; Jadvar, H.; Iagaru, A. Prostate Cancer Theranostics Targeting Gastrin-Releasing Peptide Receptors. Mol. Imaging Biol. 2018, 20, 501–509. [Google Scholar] [CrossRef]
- Mansi, R.; Nock, B.A.; Dalm, S.U.; Busstra, M.B.; van Weerden, W.M.; Maina, T. Radiolabeled bombesin analogs. Cancers 2021, 13, 5766. [Google Scholar] [CrossRef]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Mäcke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef]
- de Castiglione, R.; Gozzini, L. Bombesin receptor antagonists. Crit. Rev. Oncol./Hematol. 1996, 24, 117–151. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Wang, X.; Forrer, F.; Kneifel, S.; Tamma, M.L.; Waser, B.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Evaluation of a 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-conjugated bombesin-based radioantagonist for the labeling with single-photon emission computed tomography, positron emission tomography, and therapeutic radionuclides. Clin. Cancer Res. 2009, 15, 5240–5249. [Google Scholar] [CrossRef]
- Wieser, G.; Popp, I.; Christian Rischke, H.; Drendel, V.; Grosu, A.-L.; Bartholomä, M.; Weber, W.A.; Mansi, R.; Wetterauer, U.; Schultze-Seemann, W. Diagnosis of recurrent prostate cancer with PET/CT imaging using the gastrin-releasing peptide receptor antagonist 68 Ga-RM2: Preliminary results in patients with negative or inconclusive [18 F] Fluoroethylcholine-PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1463–1472. [Google Scholar] [CrossRef]
- Minamimoto, R.; Sonni, I.; Hancock, S.; Vasanawala, S.; Loening, A.; Gambhir, S.S.; Iagaru, A. Prospective Evaluation of (68)Ga-RM2 PET/MRI in Patients with Biochemical Recurrence of Prostate Cancer and Negative Findings on Conventional Imaging. J. Nucl. Med. 2018, 59, 803–808. [Google Scholar] [CrossRef]
- Kurth, J.; Krause, B.J.; Schwarzenböck, S.M.; Bergner, C.; Hakenberg, O.W.; Heuschkel, M. First-in-human dosimetry of gastrin-releasing peptide receptor antagonist [177Lu]Lu-RM2: A radiopharmaceutical for the treatment of metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Wieghardt, K.; Bossek, U.; Chaudhuri, P.; Herrmann, W.; Menke, B.C.; Weiss, J. 1,4,7-Triazacyclononane-N,N′,N″-Triacetate (TCTA), a hexadentate ligand for divalent and trivalent metal ions. Crystal structures of [CrIII(TCTA)], [FeIII(TCTA)], and Na[CuII(TCTA)]·2NaBr·8H2O. Inorg. Chem. 1982, 21, 4308–4314. [Google Scholar] [CrossRef]
- Delgado, R.; Sun, Y.; Motekaitis, R.J.; Martell, A.E. Stabilities of divalent and trivalent metal ion complexes of macrocyclic triazatriacetic acids. Inorg. Chem. 1993, 32, 3320–3326. [Google Scholar] [CrossRef]
- Lane, S.R.; Nanda, P.; Rold, T.L.; Sieckman, G.L.; Figueroa, S.D.; Hoffman, T.J.; Jurisson, S.S.; Smith, C.J. Optimization, biological evaluation and microPET imaging of copper-64-labeled bombesin agonists, [64Cu-NO2A-(X)-BBN(7-14)NH2], in a prostate tumor xenografted mouse model. Nucl. Med. Biol. 2010, 37, 751–761. [Google Scholar] [CrossRef]
- Makris, G.; Shegani, A.; Kankanamalage, P.H.; Kuchuk, M.; Bandari, R.P.; Smith, C.J.; Hennkens, H.M. Preclinical evaluation of novel 64Cu-labeled gastrin-releasing peptide receptor bioconjugates for PET imaging of prostate cancer. Bioconjugate Chem. 2021, 32, 1290–1297. [Google Scholar] [CrossRef]
- Chambers, C.G.; Liles, G.; Watkinson, L.; Carmack, T.; Smith, C.J. University of Missouri Department of Chemistry: Columbia, MO, USA, 2025; manuscript in preparation.
- Taylor, R.M.; Severns, V.; Brown, D.C.; Bisoffi, M.; Sillerud, L.O. Prostate cancer targeting motifs: Expression of ανβ3, neurotensin receptor 1, prostate specific membrane antigen, and prostate stem cell antigen in human prostate cancer cell lines and xenografts. Prostate 2012, 72, 523–532. [Google Scholar] [CrossRef]
- Shallal, H.M.; Minn, I.; Banerjee, S.R.; Lisok, A.; Mease, R.C.; Pomper, M.G. Heterobivalent Agents Targeting PSMA and Integrin-αvβ3. Bioconjugate Chem. 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Z.B.; Cao, Q.; Liu, S.; Wang, F.; Chen, X. Small-animal PET of tumors with 64Cu-labeled RGD-bombesin heterodimer. J. Nucl. Med. 2009, 50, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Niu, G.; Wang, F.; Chen, X. 68Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. E J. Nucl. Med. Mol. Imag. 2009, 36, 1483–1494. [Google Scholar] [CrossRef]
- Bandari, R.P.; Jiang, Z.; Reynolds, T.S.; Bernskoetter, N.E.; Szczodroski, A.F.; Bassuner, K.J.; Kirkpatrick, D.L.; Rold, T.L.; Sieckman, G.L.; Hoffman, T.J.; et al. Synthesis and biological evaluation of copper-64 radiolabeled [DUPA-6-Ahx-(NODAGA)-5-Ava-BBN(7-14)NH2], a novel bivalent targeting vector having affinity for two distinct biomarkers (GRPr/PSMA) of prostate cancer. Nucl. Med. Biol. 2014, 41, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Abouzayed, A.; Yim, C.B.; Mitran, B.; Rinne, S.S.; Tolmachev, V.; Larhed, M.; Rosenström, U.; Orlova, A. Synthesis and Preclinical Evaluation of Radio-Iodinated GRPR/PSMA Bispecific Heterodimers for the Theranostics Application in Prostate Cancer. Pharmaceutics 2019, 11, 358. [Google Scholar] [CrossRef]
- Mitran, B.; Varasteh, Z.; Abouzayed, A.; Rinne, S.S.; Puuvuori, E.; De Rosa, M.; Larhed, M.; Tolmachev, V.; Orlova, A.; Rosenström, U. Bispecific GRPR-Antagonistic Anti-PSMA/GRPR Heterodimer for PET and SPECT Diagnostic Imaging of Prostate Cancer. Cancers 2019, 11, 1371. [Google Scholar] [CrossRef]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Haberkorn, U.; Eisenhut, M.; Kopka, K. Preclinical evaluation of a bispecific low-molecular heterodimer targeting both PSMA and GRPR for improved PET imaging and therapy of prostate cancer. Prostate 2014, 74, 659–668. [Google Scholar] [CrossRef]
- Makris, G.; Bandari, R.P.; Kuchuk, M.; Jurisson, S.S.; Smith, C.J.; Hennkens, H.M. Development and preclinical evaluation of 99m Tc-and 186 Re-labeled NOTA and NODAGA bioconjugates demonstrating matched pair targeting of GRPR-expressing tumors. Mol. Imaging Biol. 2021, 23, 52–61. [Google Scholar] [CrossRef]
- Dijkgraaf, I.; Franssen, G.M.; McBride, W.J.; D’Souza, C.A.; Laverman, P.; Smith, C.J.; Goldenberg, D.M.; Oyen, W.J.; Boerman, O.C. PET of tumors expressing gastrin-releasing peptide receptor with an 18F-labeled bombesin analog. J. Nucl. Med. 2012, 53, 947–952. [Google Scholar] [CrossRef]
- Ambrosini, V.; Kunikowska, J.; Baudin, E.; Bodei, L.; Bouvier, C.; Capdevila, J.; Cremonesi, M.; de Herder, W.W.; Dromain, C.; Falconi, M.; et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur. J. Cancer 2021, 146, 56–73. [Google Scholar] [CrossRef]
- Hicks, R.J.; Kwekkeboom, D.J.; Krenning, E.; Bodei, L.; Grozinsky-Glasberg, S.; Arnold, R.; Borbath, I.; Cwikla, J.; Toumpanakis, C.; Kaltsas, G.; et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Neoplasms: Peptide Receptor Radionuclide Therapy with Radiolabelled Somatostatin Analogues. Neuroendocrinology 2017, 105, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Goldner, W.S.; Benson, A.B.; Bergsland, E.; Blaszkowsky, L.S.; Brock, P.; Chan, J.; Das, S.; Dickson, P.V.; Fanta, P.; et al. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc Netw. 2021, 19, 839–868. [Google Scholar] [CrossRef] [PubMed]
- Krenning, E.; Koolj, P.; Bakker, W.; Breeman, W.; Postema, P.; Kwekkeboom, D.J.; Oei, H.; de Jong, M.; Visser, T.; Reijs, A. Radiotherapy with a radiolabeled somatostatin analogue,[111In-DTPA-D-Phe1]-octreotide. A case history. N. Y. Acad. Sci. Ann. 1994, 733, 496–506. [Google Scholar] [CrossRef]
- Krenning, E.P.; Breeman, W.A.; Kooij, P.P.; Lameris, J.; Bakker, W.H.; Koper, J.; Ausema, L.; Reubi, J.C.; Lamberts, S.W. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet 1989, 333, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Michael, M.; Hicks, R.J. 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 1390–1391. [Google Scholar]
- Strosberg, J.; Wolin, E.; Chasen, B.; Kulke, M.; Bushnell, D.; Caplin, M.; Baum, R.P.; Kunz, P.; Hobday, T.; Hendifar, A.; et al. Health-Related Quality of Life in Patients With Progressive Midgut Neuroendocrine Tumors Treated With 177Lu-Dotatate in the Phase III NETTER-1 Trial. J. Clin. Oncol. 2018, 36, 2578–2584. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Kam, B.L.; van Essen, M.; Teunissen, J.J.; van Eijck, C.H.; Valkema, R.; de Jong, M.; de Herder, W.W.; Krenning, E.P. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2010, 17, R53–R73. [Google Scholar] [CrossRef]
- Rylova, S.N.; Stoykow, C.; Del Pozzo, L.; Abiraj, K.; Tamma, M.L.; Kiefer, Y.; Fani, M.; Maecke, H.R. The somatostatin receptor 2 antagonist 64Cu-NODAGA-JR11 outperforms 64Cu-DOTA-TATE in a mouse xenograft model. PLoS ONE 2018, 13, e0195802. [Google Scholar] [CrossRef]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: The chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef]
- Eisenwiener, K.P.; Prata, M.I.; Buschmann, I.; Zhang, H.W.; Santos, A.C.; Wenger, S.; Reubi, J.C.; Mäcke, H.R. NODAGATOC, a new chelator-coupled somatostatin analogue labeled with [67/68Ga] and [111In] for SPECT, PET, and targeted therapeutic applications of somatostatin receptor (hsst2) expressing tumors. Bioconjugate Chem. 2002, 13, 530–541. [Google Scholar] [CrossRef]
- Froidevaux, S.; Heppeler, A.; Eberle, A.N.; Meier, A.M.; Häusler, M.; Beglinger, C.; Béhé, M.; Powell, P.; Mäcke, H.R. Preclinical comparison in AR4-2J tumor-bearing mice of four radiolabeled 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-somatostatin analogs for tumor diagnosis and internal radiotherapy. Endocrinology 2000, 141, 3304–3312. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; McBride, W.J.; Sharkey, R.M.; Eek, A.; Joosten, L.; Oyen, W.J.; Goldenberg, D.M.; Boerman, O.C. A novel facile method of labeling octreotide with 18F-fluorine. J. Nucl. Med. 2010, 51, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Makris, G.; Kuchuk, M.; Gallazzi, F.; Jurisson, S.S.; Smith, C.J.; Hennkens, H.M. Somatostatin receptor targeting with hydrophilic [99mTc/186Re]Tc/Re-tricarbonyl NODAGA and NOTA complexes. Nucl. Med. Biol. 2019, 71, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Makris, G.; Radford, L.L.; Kuchuk, M.; Gallazzi, F.; Jurisson, S.S.; Smith, C.J.; Hennkens, H.M. NOTA and NODAGA [99mTc]Tc- and [186Re]Re-Tricarbonyl Complexes: Radiochemistry and First Example of a [99mTc]Tc-NODAGA Somatostatin Receptor-Targeting Bioconjugate. Bioconjugate Chem. 2018, 29, 4040–4049. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Z.; Mo, C.; Han, Y.; Liu, Q.; Tang, G. Synthesis and preclinical evaluation of [18F] AlF-NOTA-Asp2-PEG2-JR11 as a novel antagonist radioligand for PET imaging of somatostatin receptor. Eur. J. Nucl. Med. Mol. Imaging 2024, 52, 1189–1199. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef]
- Siegrist, W.; Solca, F.; Stutz, S.; Giuffrè, L.; Carrel, S.; Girard, J.; Eberle, A.N. Characterization of receptors for alpha-melanocyte-stimulating hormone on human melanoma cells. Cancer Res. 1989, 49, 6352–6358. [Google Scholar]
- Tatro, J.B.; Wen, Z.; Entwistle, M.L.; Atkins, M.B.; Smith, T.J.; Reichlin, S.; Murphy, J.R. Interaction of an alpha-melanocyte-stimulating hormone-diphtheria toxin fusion protein with melanotropin receptors in human melanoma metastases. Cancer Res. 1992, 52, 2545–2548. [Google Scholar]
- Yang, J.; Xu, J.; Gonzalez, R.; Lindner, T.; Kratochwil, C.; Miao, Y. 68Ga-DOTA-GGNle-CycMSH(hex) targets the melanocortin-1 receptor for melanoma imaging. Sci. Transl. Med. 2018, 10, eaau4445. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, T.K.; Sanfilippo, P.J.; Hruby, V.J.; Engel, M.H.; Heward, C.B.; Burnett, J.B.; Hadley, M.E. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: A highly potent alpha-melanotropin with ultralong biological activity. Proc. Natl. Acad. Sci. USA 1980, 77, 5754–5758. [Google Scholar] [CrossRef]
- Miao, Y.; Quinn, T.P. Advances in Receptor-Targeted Radiolabeled Peptides for Melanoma Imaging and Therapy. J. Nucl. Med. 2021, 62, 313–318. [Google Scholar] [CrossRef]
- Guo, H.; Miao, Y. Cu-64-labeled lactam bridge-cyclized α-MSH peptides for PET imaging of melanoma. Mol. Pharm. 2012, 9, 2322–2330. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Xu, J.; Gonzalez, R.; Miao, Y. Novel 64Cu-Labeled NOTA-Conjugated Lactam-Cyclized Alpha-Melanocyte-Stimulating Hormone Peptides with Enhanced Tumor to Kidney Uptake Ratios. Mol. Pharm. 2022, 19, 2535–2541. [Google Scholar] [CrossRef]
- Qiao, Z.; Xu, J.; Fisher, D.R.; Gonzalez, R.; Miao, Y. Introduction of a Polyethylene Glycol Linker Improves Uptake of 67Cu-NOTA-Conjugated Lactam-Cyclized Alpha-Melanocyte-Stimulating Hormone Peptide in Melanoma. Cancers 2023, 15, 2755. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Xu, J.; Gonzalez, R.; Miao, Y. Effects of Polyethylene Glycol and 8-Aminooctanoic Acid Linkers on Melanoma Uptake of [99mTc]Tc-Tricarbonyl-NOTA-Conjugated Lactam-Cyclized α-MSH Peptides. Bioconjugate Chem. 2023, 34, 934–940. [Google Scholar] [CrossRef]
- Qiao, Z.; Xu, J.; Gonzalez, R.; Miao, Y. Novel Al18F-NOTA-Conjugated Lactam-Cyclized α-Melanocyte-Stimulating Hormone Peptides with Enhanced Melanoma Uptake. Bioconjugate Chem. 2022, 33, 982–990. [Google Scholar] [CrossRef]
- Guo, H.; Gallazzi, F.; Miao, Y. Gallium-67-Labeled Lactam Bridge-Cyclized Alpha-MSH Peptides with Enhanced Melanoma Uptake and Reduced Renal Uptake. Bioconjugate Chem. 2012, 23, 1341–1348. [Google Scholar] [CrossRef]
- Xu, J.; Qiao, Z.; Gonzalez, R.; Miao, Y. Facile preparation of a novel Ga-67-labeled NODAGA-conjugated lactam-cyclized alpha-MSH peptide at room temperature for melanoma targeting. Bioorg Med. Chem. Lett. 2020, 30, 127627. [Google Scholar] [CrossRef]
- Guo, H.; Gallazzi, F.; Miao, Y. Design and Evaluation of New Tc-99m-Labeled Lactam Bridge-Cyclized Alpha-MSH Peptides for Melanoma Imaging. Mol. Pharm. 2013, 10, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Miao, Y. Introduction of an 8-aminooctanoic acid linker enhances uptake of 99mTc-labeled lactam bridge-cyclized α-MSH peptide in melanoma. J. Nucl. Med. 2014, 55, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Xu, J.; Gonzalez, R.; Miao, Y. Novel [99mTc]-Tricarbonyl-NOTA-Conjugated Lactam-Cyclized Alpha-MSH Peptide with Enhanced Melanoma Uptake and Reduced Renal Uptake. Mol. Pharm. 2020, 17, 3581–3588. [Google Scholar] [CrossRef] [PubMed]
- Palangka, C.; Hanaoka, H.; Yamaguchi, A.; Murakami, T.; Tsushima, Y. Al18F-labeled alpha-melanocyte-stimulating hormone (α-MSH) peptide derivative for the early detection of melanoma. Ann. Nucl. Med. 2019, 33, 733–739. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Lin, K.-S.; Lau, J.; Zeisler, J.; Colpo, N.; Perrin, D.M.; Bénard, F. Melanoma Imaging Using 18F-Labeled α-Melanocyte-Stimulating Hormone Derivatives with Positron Emission Tomography. Mol. Pharm. 2018, 15, 2116–2122. [Google Scholar] [CrossRef]

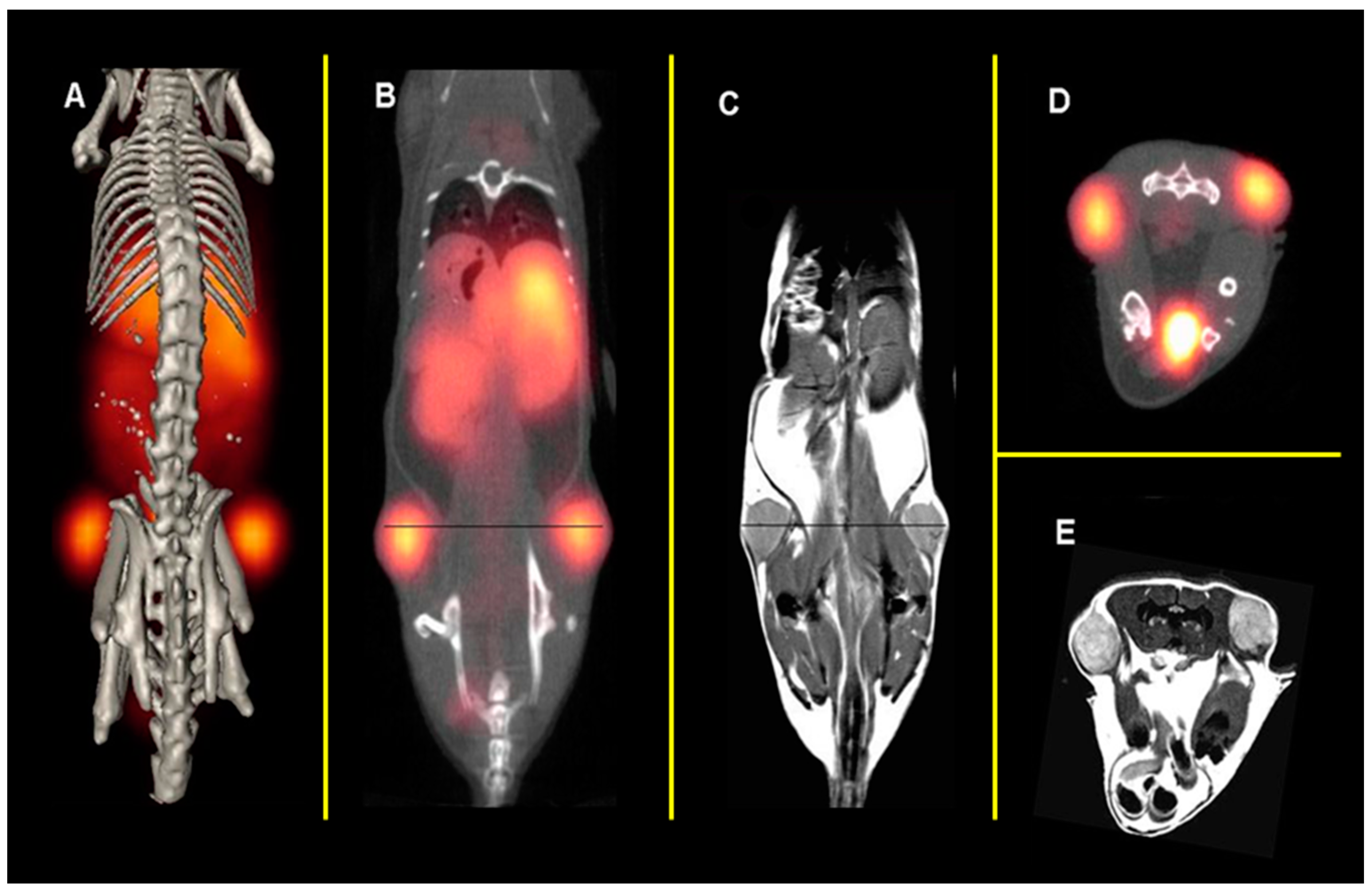
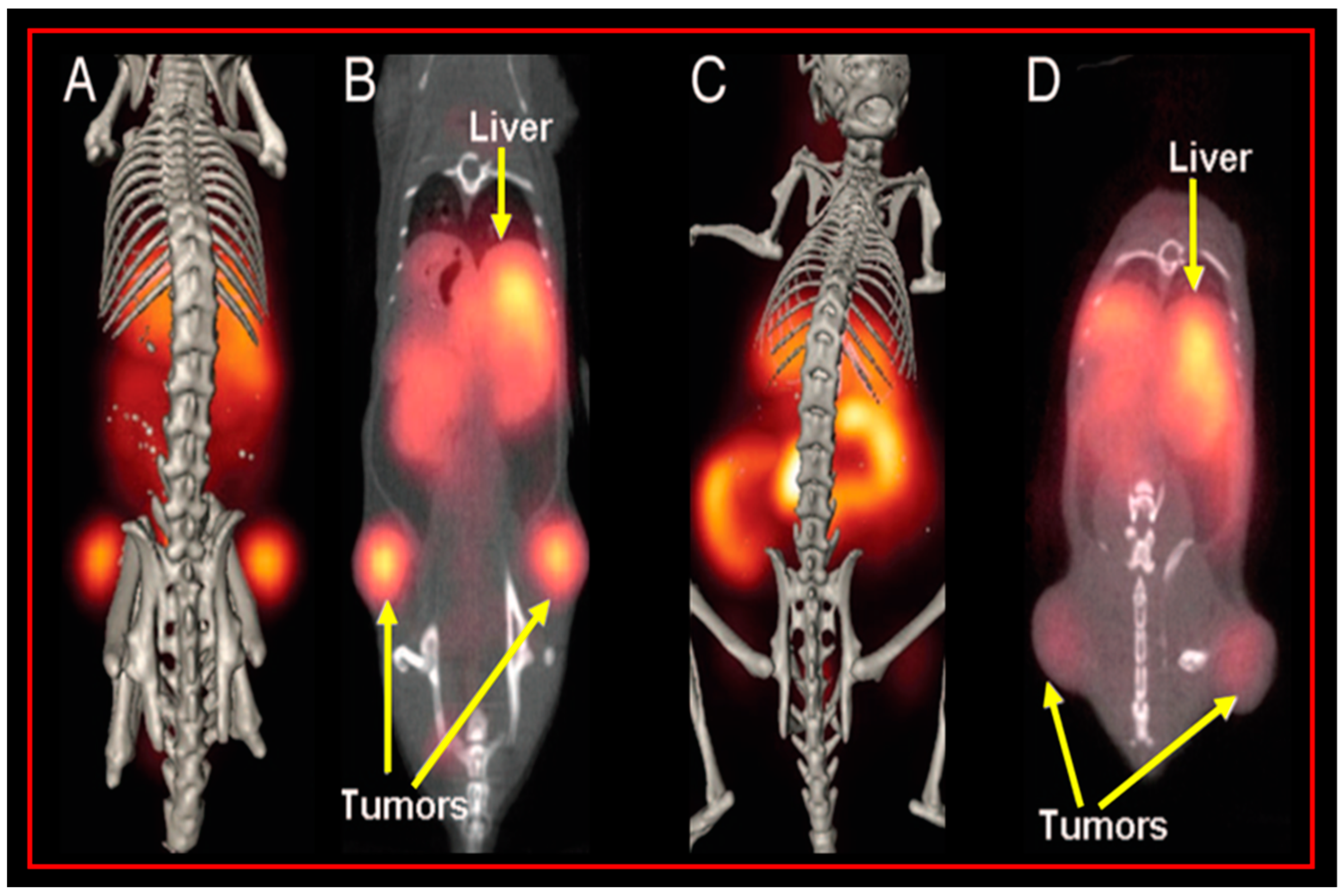
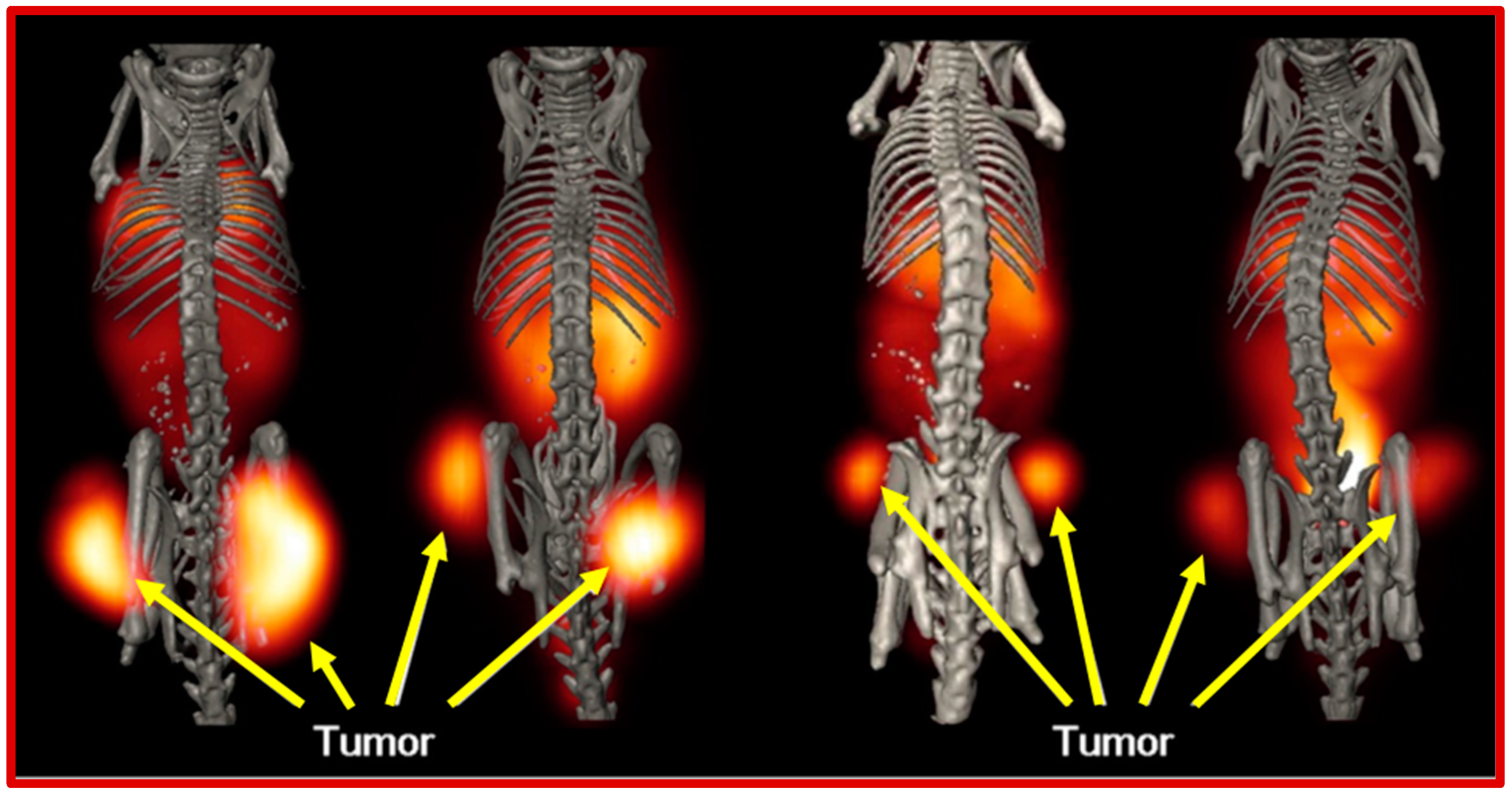
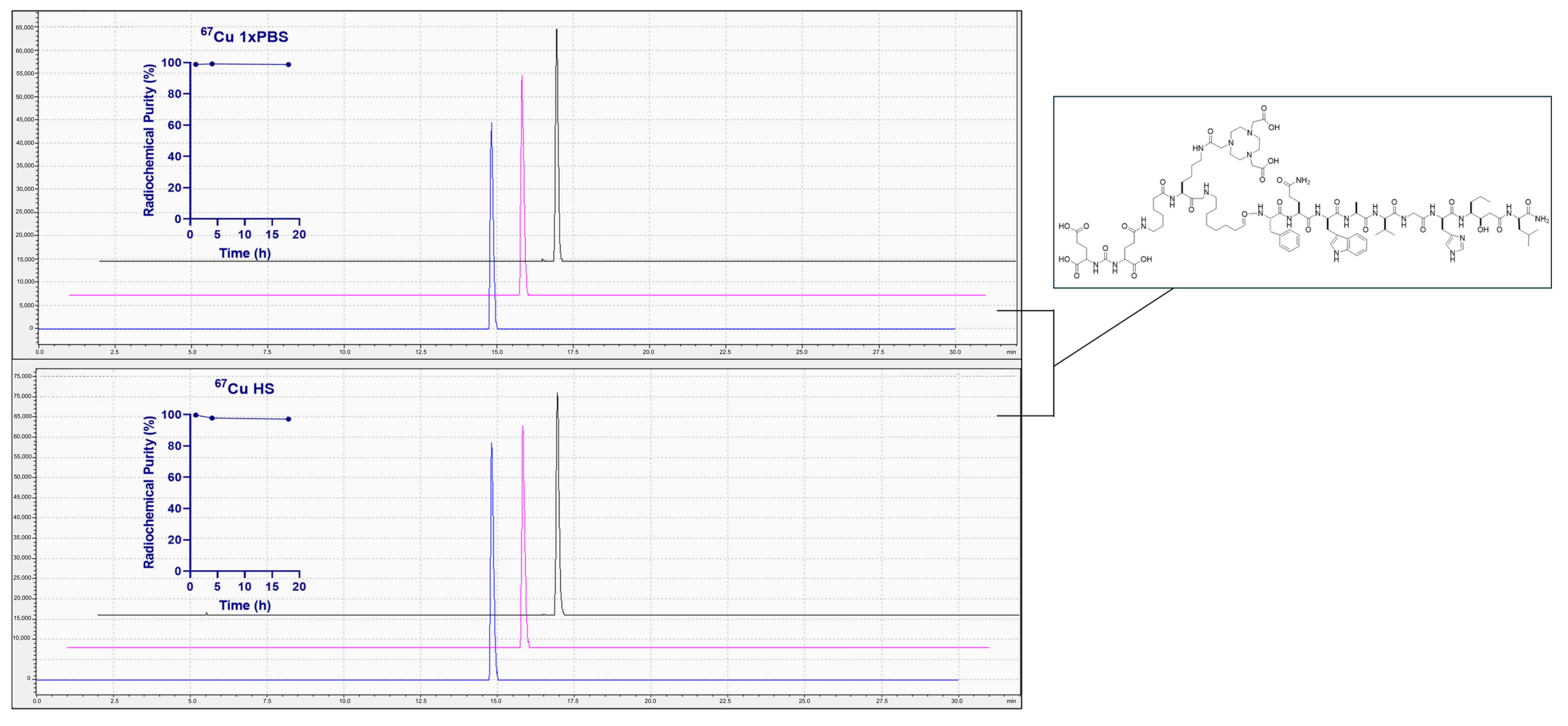
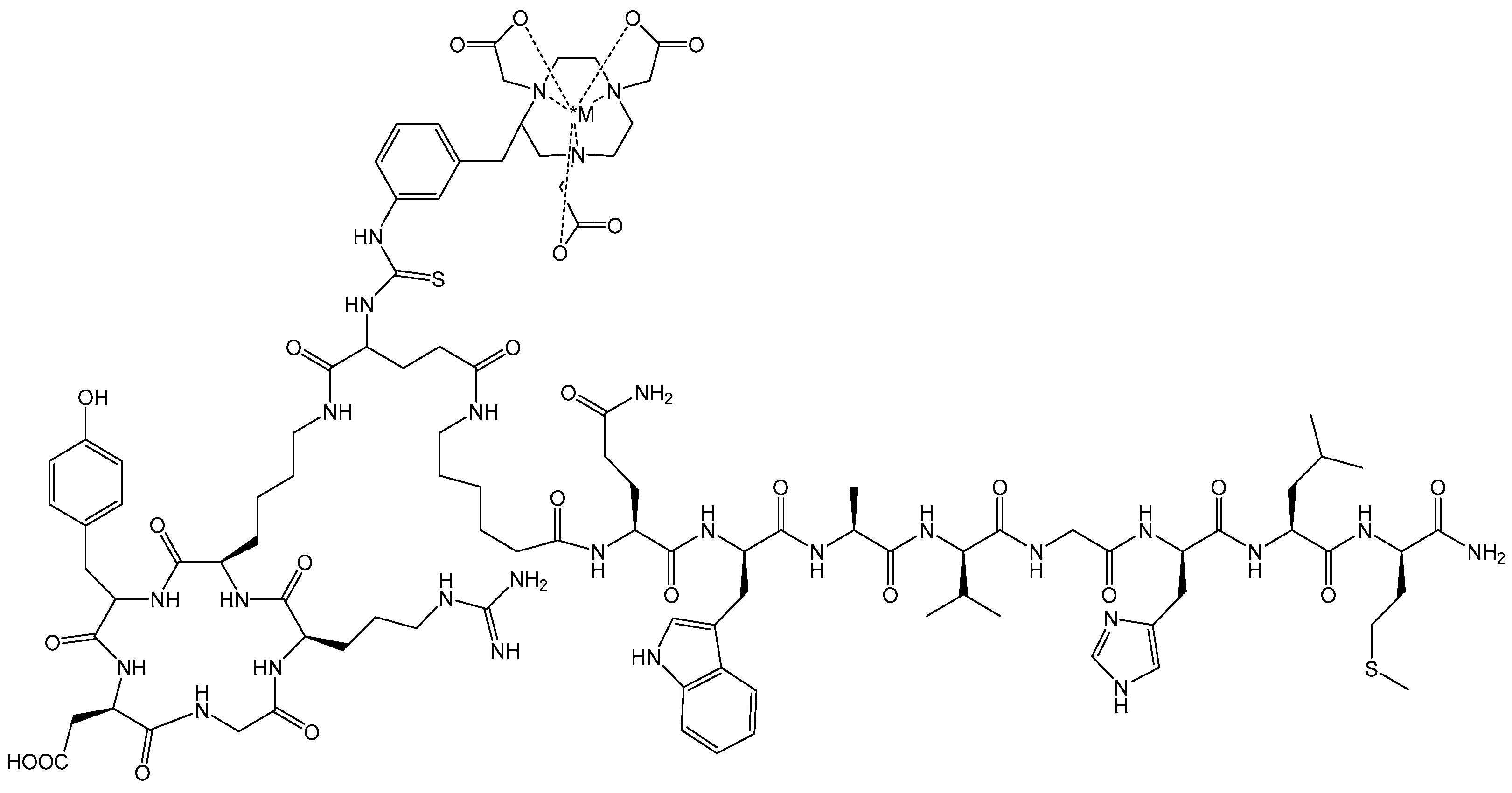

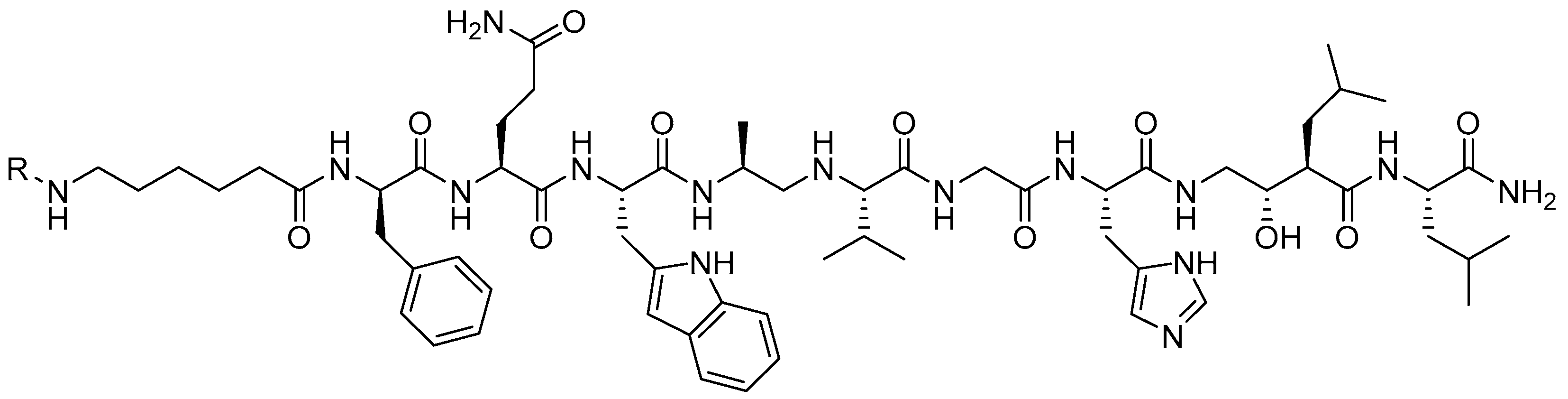
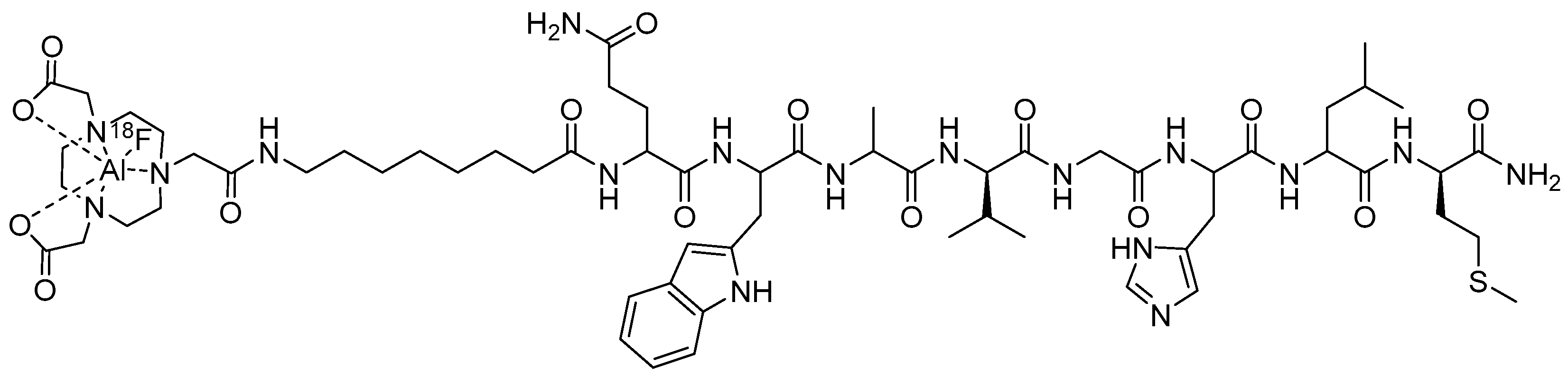
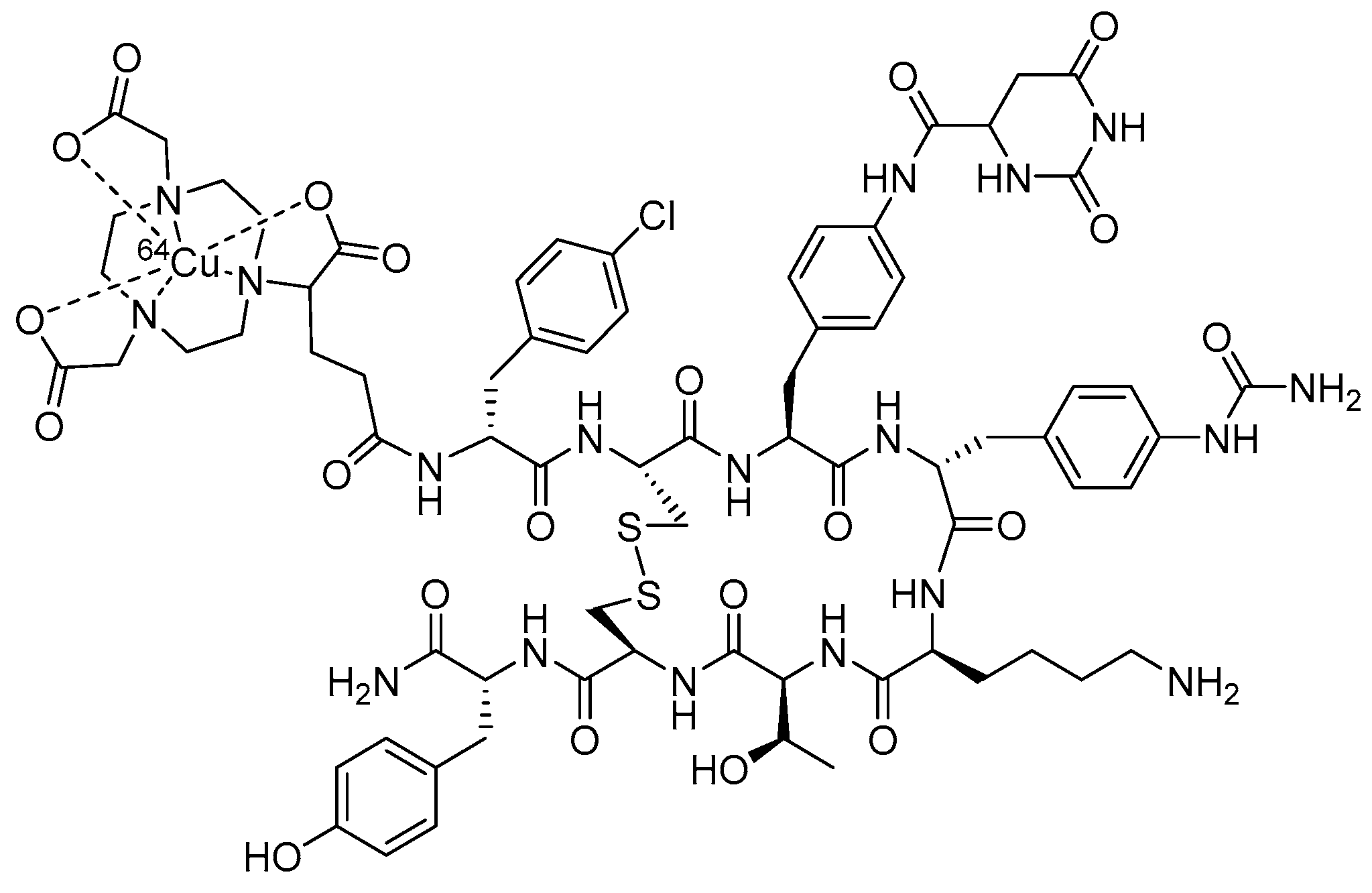




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isotope | Physical Characteristics | Availability | Specific Activity |
|---|---|---|---|
| Copper-64 | t1/2 = 12.7 h, β+—0.65 MeV, β−—0.57 MeV | Widely Available, Cyclotron-Produced | High |
| Copper-67 | t1/2 = 2.58 d, γ—93 and 185 keV, β−—0.562 MeV | Widely Available, Cyclotron-Produced | High |
| Gallium-68 | t1/2 = 1.13 h, β+—1.899 MeV | Limited, Ge-68/Ga-68 Generator | High |
| Gallium-67 | t1/2 = 78.3 h, γ—93, 185, and 300 keV | Limited, Cyclotron-Produced | High |
| Indium-111 | t1/2 = 2.80 d, γ—171 and 245 keV | Widely Available, Cyclotron-Produced | High |
| Technetium-99m | t1/2 = 6.04 h, γ—140 keV | Widely Available, Generator-Produced | High |
| Fluorine-18 | t1/2 = 110 min, β+—0.635 MeV | Widely Available, Cyclotron-Produced | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chambers, C.G.; Wang, J.; Sakr, T.M.; Miao, Y.; Smith, C.J. NOTA and NODAGA Radionuclide Complexing Agents: Versatile Approaches for Advancements in Radiochemistry. Molecules 2025, 30, 2095. https://doi.org/10.3390/molecules30102095
Chambers CG, Wang J, Sakr TM, Miao Y, Smith CJ. NOTA and NODAGA Radionuclide Complexing Agents: Versatile Approaches for Advancements in Radiochemistry. Molecules. 2025; 30(10):2095. https://doi.org/10.3390/molecules30102095
Chicago/Turabian StyleChambers, Claudia G., Jing Wang, Tamer M. Sakr, Yubin Miao, and Charles J. Smith. 2025. "NOTA and NODAGA Radionuclide Complexing Agents: Versatile Approaches for Advancements in Radiochemistry" Molecules 30, no. 10: 2095. https://doi.org/10.3390/molecules30102095
APA StyleChambers, C. G., Wang, J., Sakr, T. M., Miao, Y., & Smith, C. J. (2025). NOTA and NODAGA Radionuclide Complexing Agents: Versatile Approaches for Advancements in Radiochemistry. Molecules, 30(10), 2095. https://doi.org/10.3390/molecules30102095