
Discovery of Hydrazineyl Amide Derivative of Pseudolaric Acid B for Reprogramming Tumor-Associated Macrophages Against Tumor Growth

Abstract
1. Introduction
2. Results
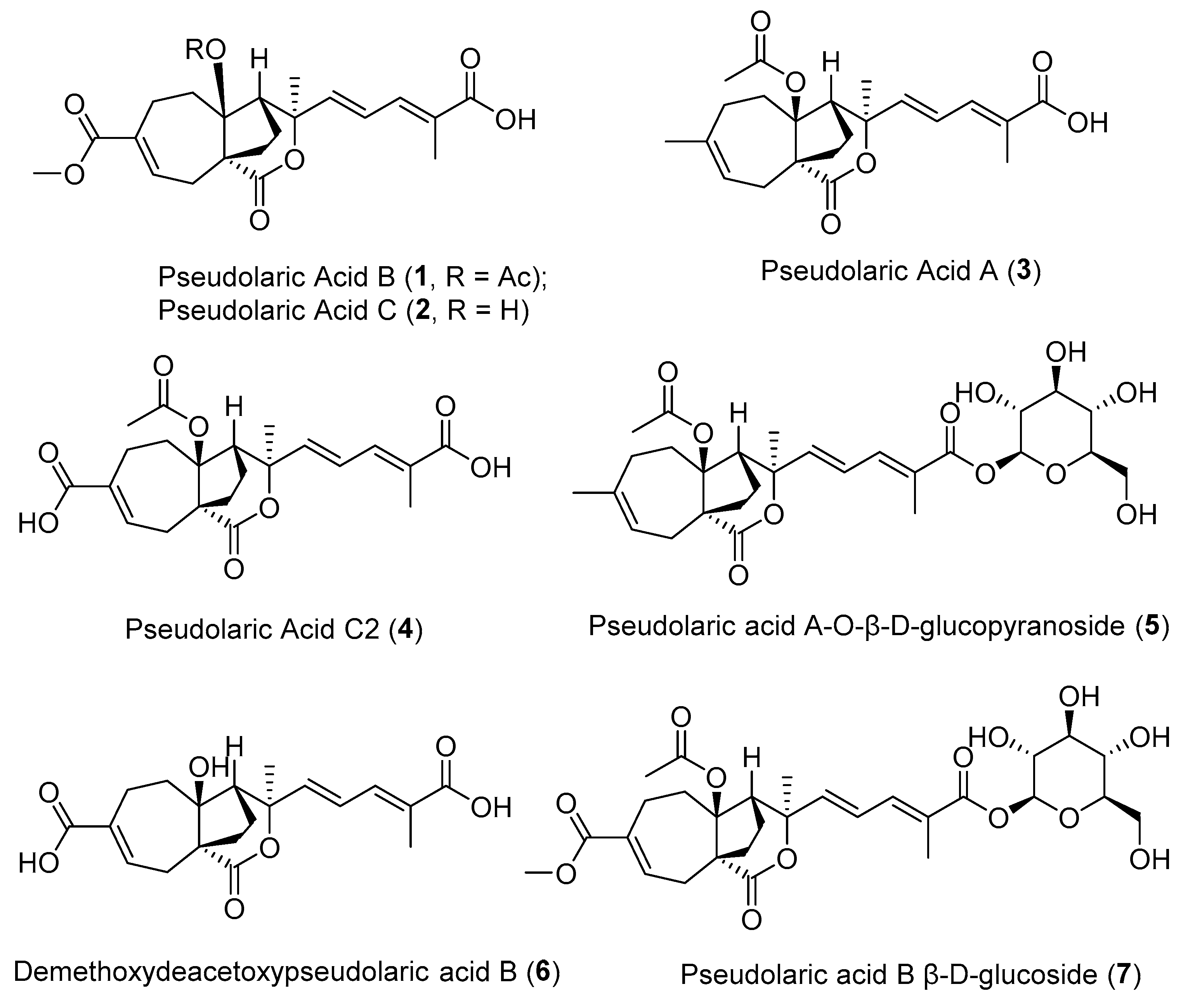
2.1. The Effects of Natural Pseudolaric Acids and Their O-Pyranosides on the Tumor-Promoting Phenotype of the RAW264.7 Cell Line
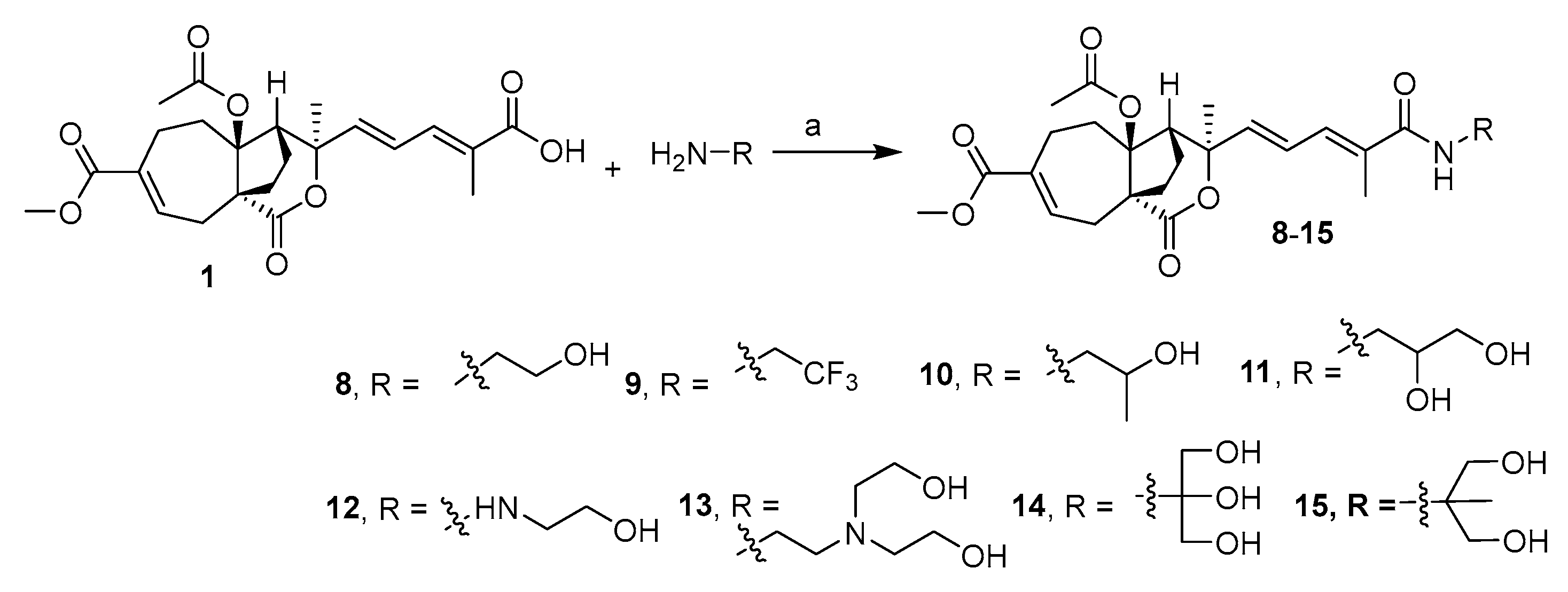
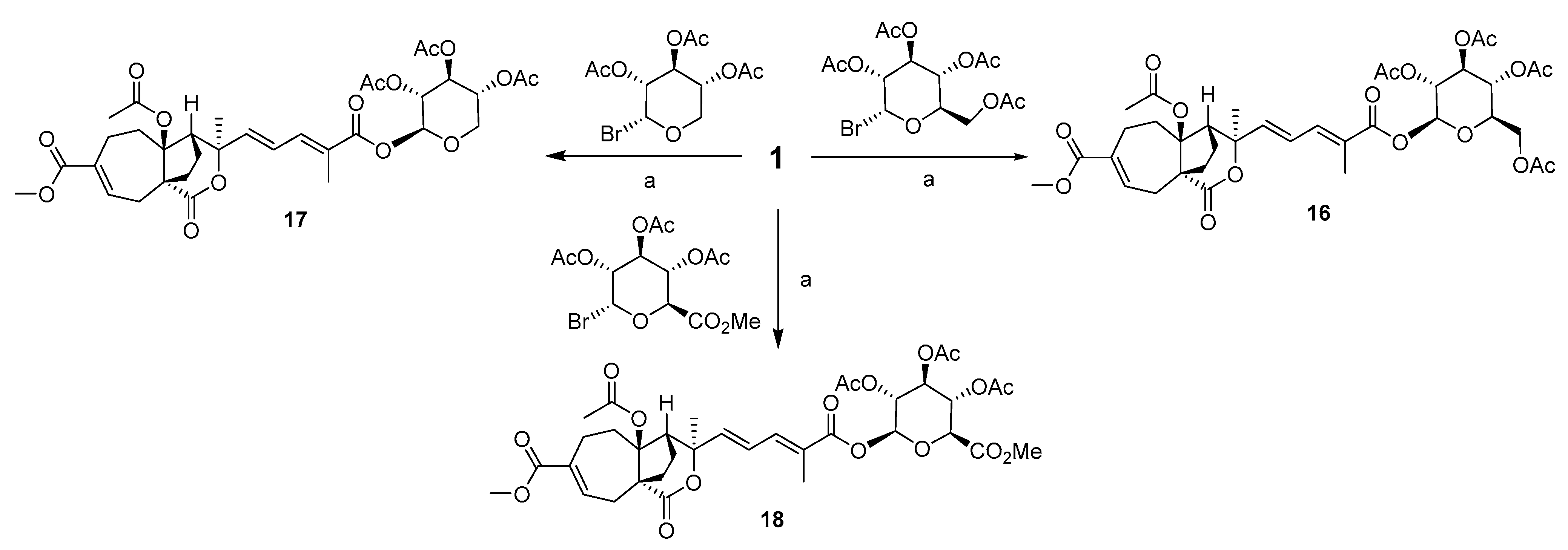
2.2. Design and Synthesis of PAB Derivatives
2.3. The Effects of the Carboxylic Acid-Modified Derivatives of PAB on the Tumor-Promoting Phenotype of the RAW264.7 Cell Line
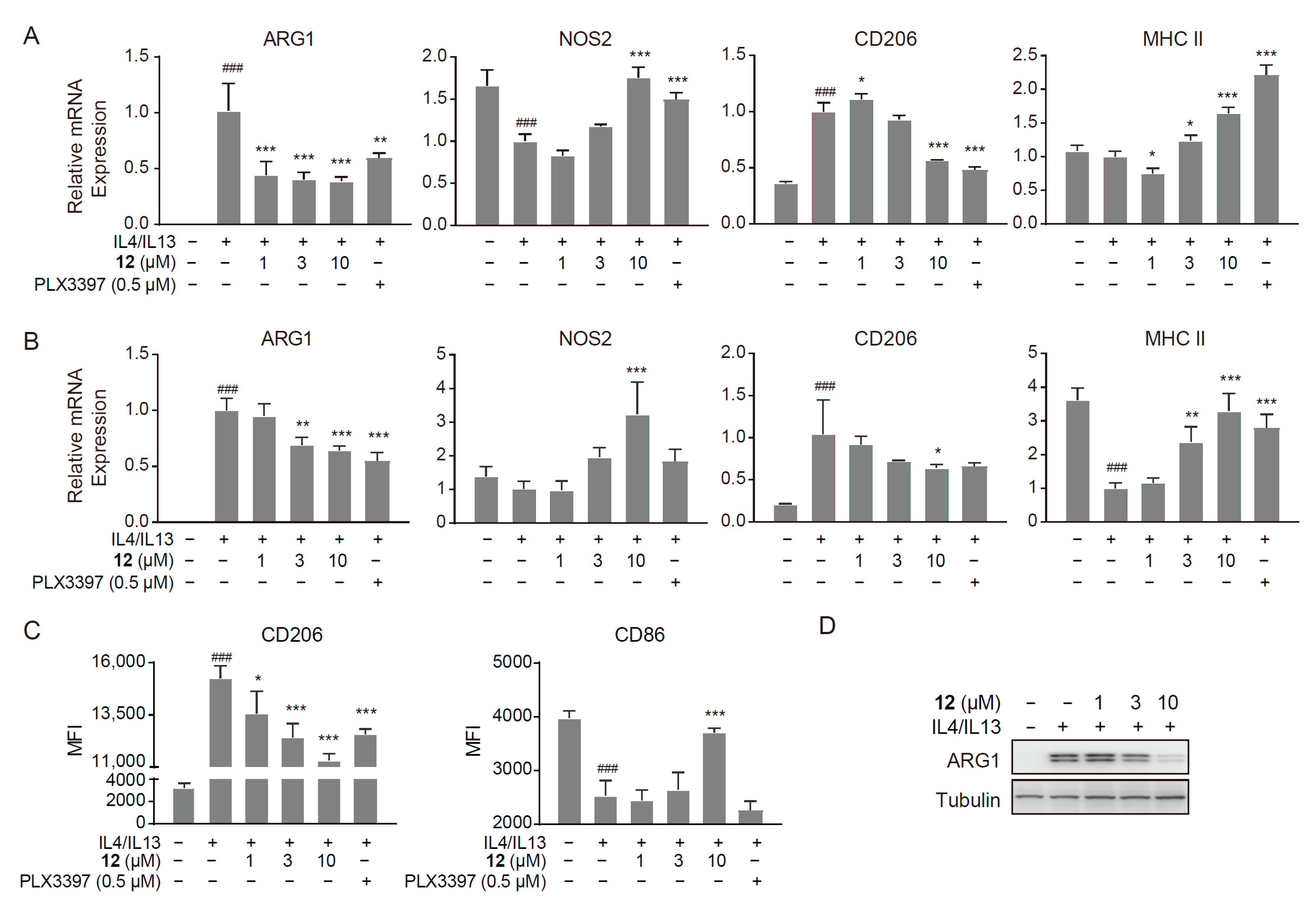
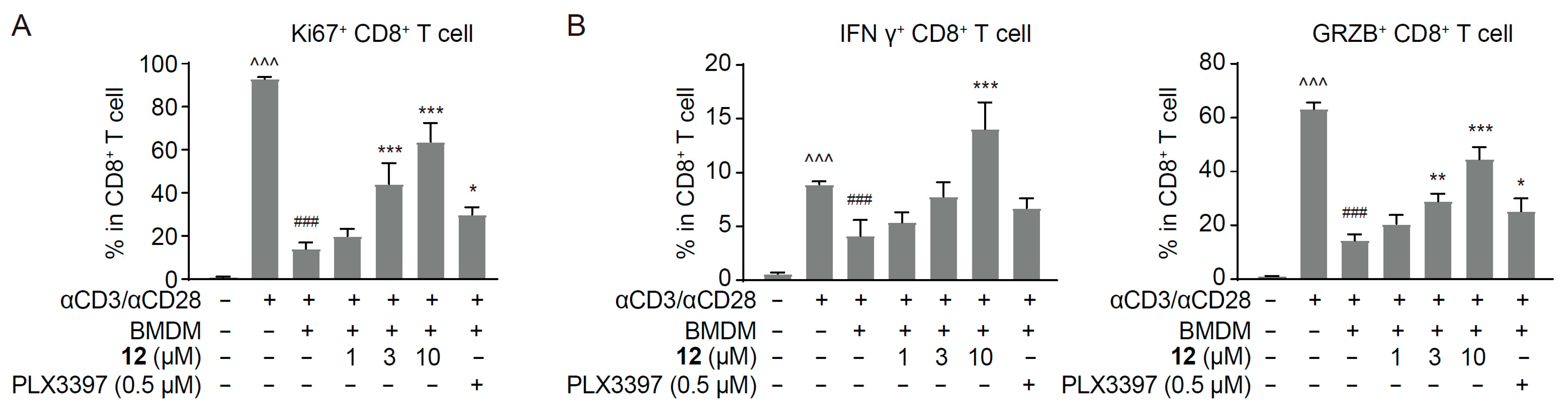
2.4. Compound 12 Inhibited the M2-like Polarized Tumor-Promoting Phenotype of Macrophages
2.5. Compound 12 Reversed Macrophage-Mediated CD8+ T Cell Suppression
2.6. Pharmacokinetic Properties of Compound 12 in Mice
2.7. Compound 12 Reshaped the Tumor Immune Microenvironment and Inhibited Tumor Growth in Immunocompetent Murine Tumor Models
2.8. Molecular Docking of Compound 12 with Hsp90
3. Materials and Methods
3.1. Chemistry
3.2. Cell Culture
3.3. Macrophage Culture and Stimulation
3.4. Quantitative Real-Time PCR (RT-PCR)
3.5. Macrophage Phenotype Analysis
3.6. Western Blot Analysis
3.7. Cell Proliferation Assay
3.8. CD8+ T Cell Suppression Assay
3.9. In Vivo Pharmacokinetic Studies in Mice
3.10. In Vivo Anti-Tumor Efficacy
3.11. Molecular Docking
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef]
- Conte, E. Targeting monocytes/macrophages in fibrosis and cancer diseases: Therapeutic approaches. Pharmacol. Ther. 2022, 234, 108031. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Li, X.L.; Liu, R.; Su, X.; Pan, Y.S.; Han, X.F.; Shao, C.S.; Shi, Y.F. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef]
- Cheng, N.; Bai, X.X.; Shu, Y.X.; Ahmad, O.; Shen, P.P. Targeting tumor-associated macrophages as an antitumor strategy. Biochem. Pharmacol. 2021, 183, 114354. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, D.J.; Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 2023, 186, 1627–1651. [Google Scholar] [CrossRef] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Luo, Z.W.; Yin, F.C.; Wang, X.B.; Kong, L.Y. Progress in approved drugs from natural product resources. Chin. J. Nat. Med. 2024, 22, 195–211. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Zhou, B.N.; Ying, B.P.; Song, G.Q.; Chen, Z.X.; Han, J.; Yan, Y.F. Pseudolaric Acids from Pseudolarix-Kaempferi. Planta Med. 1983, 47, 35–38. [Google Scholar] [CrossRef]
- Chiu, P.; Leung, L.T.; Ko, B.C.B. Pseudolaric acids: Isolation, bioactivity and synthetic studies. Nat. Prod. Rep. 2010, 27, 1066–1083. [Google Scholar] [CrossRef]
- Liu, M.L.; Sun, D.; Li, T.; Chen, H. A Systematic Review of the Immune-Regulating and Anticancer Activities of Pseudolaric Acid B. Front. Pharmacol. 2017, 8, 394. [Google Scholar] [CrossRef]
- Yin, M.; Li, N.; Zhang, L.N.; Lin, J.; Wang, Q.; Gu, L.W.; Zheng, H.R.; Zhao, G.Q.; Li, C. Pseudolaric Acid B Ameliorates Fungal Keratitis Progression by Suppressing Inflammation and Reducing Fungal Load. ACS Infect. Dis. 2023, 9, 1196–1205. [Google Scholar] [CrossRef]
- Wong, V.K.W.; Chiu, P.; Chung, S.S.M.; Chow, L.M.C.; Zhao, Y.Z.; Yang, B.B.; Ko, B.C.B. Pseudolaric acid B, a novel microtubule-destabilizing agent that circumvents multidrug resistance phenotype and exhibits antitumor activity. Clin. Cancer Res. 2005, 11, 6002–6011. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Cai, H.R.; Wang, Z.Q.; Jiang, Y.Q. Pseudolaric Acid B Inhibits Proliferation, Invasion, and Angiogenesis in Esophageal Squamous Cell Carcinoma Through Regulating CD147. Drug Des. Dev. Ther. 2020, 14, 4561–4573. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Li, T.; Chen, H.; Mei, X.; Cao, B.; Zhang, Y.Y. The Immunosuppressive Activity of Pseudolaric Acid B on T lymphocytes. Phytother. Res. 2013, 27, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, H.; Yang, Z.; Wang, W.; Wang, Y.T.; Zhang, L.M.; Zhao, J.H.; Zhou, X.; Li, Y.M. A novel Pseudolaric acid B derivative, Hexahydropseudolaric acid B, exterts an immunomodulatory effect in vitro/in vivo evaluation. Eur. J. Pharmacol. 2014, 745, 10–18. [Google Scholar] [CrossRef]
- Li, T.; Chen, H.; Yang, Z.; Wei, N.; Zhang, S.; Mei, X.; Bai, S.F. Topical application of Pseudolaric acid B improve DNFB-induced contact hypersensitivity via regulating the balance of Th1/Th17/Treg cell subsets. Eur. J. Pharm. Sci. 2012, 45, 668–676. [Google Scholar] [CrossRef]
- Li, T.; Wong, V.K.W.; Yi, X.Q.; Wong, Y.F.; Zhou, H.; Liu, L. Pseudolaric Acid B Suppresses T Lymphocyte Activation Through Inhibition of NF-κB Signaling Pathway and p38 Phosphorylation. J. Cell. Biochem. 2009, 108, 87–95. [Google Scholar] [CrossRef]
- Li, Y.; Li, T.; Ji, W.; Li, X.; Ma, Y.; Zhao, J.; Zhou, X.; Li, Y. Inhibitory effects of pseudolaric acid B on inflammatory response and M1 phenotype polarization in RAW264.7 macrophages induced by lipopolysaccharide. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi Chin. J. Cell. Mol. Immunol. 2016, 32, 625–629. [Google Scholar]
- Liu, Y.; Xu, X.; Chen, H.; Ke, Y.; Bai, S.F. Synthesis of pseudolaric acid B derivatives and their antitumor activity. Med. J. Chin. People’s Armed Police Forces 2007, 9, 660–663. [Google Scholar]
- Yang, S.P.; Cai, Y.J.; Zhang, B.L.; Tong, L.J.; Xie, H.; Wu, Y.; Lin, L.P.; Ding, J.; Yue, J.M. Structural modification of an angiogenesis inhibitor discovered from traditional Chinese medicine and a structure—Activity relationship study. J. Med. Chem. 2008, 51, 77–85. [Google Scholar] [CrossRef]
- Deng, H.; Xu, Q.; Li, X.T.; Huang, X.; Liu, J.Y.; Yan, R.; Quan, Z.S.; Shen, Q.K.; Guo, H.Y. Design, synthesis, and evaluation of antitumor activity in Pseudolaric acid B Azole derivatives: Novel and potent angiogenesis inhibitor via regulation of the PI3K/AKT and MAPK mediated HIF-1/VEGF signaling pathway. Eur. J. Med. Chem. 2024, 278, 116813. [Google Scholar] [CrossRef]
- Xu, Q.; Deng, H.; Huang, X.; Liu, J.Y.; Chen, G.Q.; Shen, Q.K.; Quan, Z.S.; Guo, H.Y.; Yin, X.M. Design, synthesis, and biological screening of pseudolaric acid B derivatives as potential anti-tumor agents. Bioorganic Chem. 2024, 151, 107670. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.H.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 339–340. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Lafta, H.A.; AbdulHussein, A.H.; Al-Shalah, S.A.J.; Alnassar, Y.S.; Mohammed, N.M.; Akram, S.M.; Qasim, M.T.; Najafi, M. Tumor-associated Macrophages (TAMs) in Cancer Resistance; Modulation by Natural Products. Curr. Top. Med. Chem. 2023, 23, 1104–1122. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Hong, L.; Tanaka, M.; Yasui, M.; Hara-Chikuma, M. HSP90 promotes tumor associated macrophage differentiation during triple-negative breast cancer progression. Sci. Rep. 2024, 14, 22541. [Google Scholar] [CrossRef]
- Fan, C.S.; Chen, C.C.; Chen, L.L.; Chua, K.V.; Hung, H.C.; Hsu, J.T.; Huang, T.S. Extracellular HSP90α Induces MyD88-IRAK Complex-Associated IKKα/β-NF-κB/IRF3 and JAK2/TYK2-STAT-3 Signaling in Macrophages for Tumor-Promoting M2-Polarization. Cells 2022, 11, 229. [Google Scholar] [CrossRef]
- Yoshimura, K.; Zou, G.; Fan, Y.; Yamashita, K.; Wang, L.; Wu, J.; Wang, R.; Shao, S.; Scott, A.W.; Jin, J.; et al. HSP90 inhibitor AUY922 suppresses tumor growth and modulates immune response through YAP1-TEAD pathway inhibition in gastric cancer. Cancer Lett. 2025, 610, 217354. [Google Scholar] [CrossRef]
- Klemke, L.; De Oliveira, T.; Witt, D.; Winkler, N.; Bohnenberger, H.; Bucala, R.; Conradi, L.C.; Schulz-Heddergott, R. Hsp90-stabilized MIF supports tumor progression via macrophage recruitment and angiogenesis in colorectal cancer. Cell Death Dis. 2021, 12, 155. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, L.; Zhou, Y.; Zhou, P.; Yan, Q.; Chen, X.; Ding, S.; Zhu, F. CKLF1 Enhances Inflammation-Mediated Carcinogenesis and Prevents Doxorubicin-Induced Apoptosis via IL6/STAT3 Signaling in HCC. Clin. Cancer Res. 2019, 25, 4141–4154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [PubMed]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.F.; He, D.H.; Zhang, S.B.; Lu, Y.Z.; Ye, X.W.; Fan, X.Z.; Wang, H.; Wang, Q.; Liu, Y.Q. Identification of pseudolaric acid B as a novel Hedgehog pathway inhibitor in medulloblastoma. Biochem. Pharmacol. 2021, 190, 114593. [Google Scholar] [CrossRef]
- Liu, J.X.; Wu, X.D.; Li, W.Y.; Yuan, Z.F.; Yang, K.; Zhao, Q.S. Discovery of pseudolaric acid A as a new Hsp90 inhibitor uncovers its potential anticancer mechanism. Bioorganic Chem. 2021, 112, 104963. [Google Scholar] [CrossRef]
- Zhou, Y.Q.; Di, Z.A.; Li, X.M.; Shan, Y.H.; Li, W.; Zhang, H.C.; Xiao, Y.L. Chemical proteomics reveal CD147 as a functional target of pseudolaric acid B in human cancer cells. Chem. Commun. 2017, 53, 8671–8674. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mRNA Expression Level | ||||
|---|---|---|---|---|
| ARG1 | NOS2 | |||
| Relative Expression (%) | p-Value | Relative Expression (%) | p-Value | |
| Vehicle (M0) | 0.5 ± 0.2 | / | 157.6 ± 11.9 | / |
| IL4/IL13 treatment (M2) | 100.4 ± 8.8 | ### | 100.2 ± 6.1 | ### |
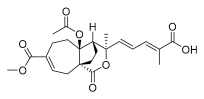 1 (10 μM) | 9.9 ± 0.8 | ** | 135.0 ± 19.5 | * |
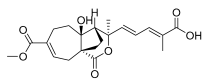 2 (10 μM) | 17.3 ± 9.1 | ** | 80.1 ± 15.6 | ns |
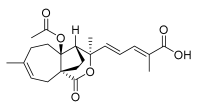 3 (10 μM) | 342.6 ± 27.9 | *** | 297.4 ± 6.8 | *** |
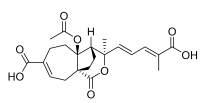 4 (10 μM) | 87.4 ± 25.4 | ns | 131.2 ± 14.5 | ns |
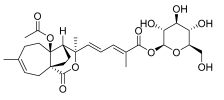 5 (10 μM) | 16.2 ± 9.2 | ** | 158.1 ± 17.2 | ns |
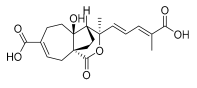 6 (10 μM) | 126.3 ± 43.8 | ns | 119.0 ± 25.0 | ns |
 7 (10 μM) | 21.2 ± 13.0 | ** | 306.6 ± 36.2 | *** |
| b PLX3397@0.5 μM | 3.2 ± 2.4 | *** | 193.3 ± 10.2 | *** |
| mRNA Expression Level | ||||
|---|---|---|---|---|
| ARG1 | NOS2 | |||
| Relative Expression (%) | p-Value | Relative Expression (%) | p-Value | |
| Vehicle (M0) | 0.5 ± 0.2 | / | 157.6 ± 11.9 | / |
| IL4/IL13 treatment (M2) | 100.4 ± 8.8 | ### | 100.2 ± 6.1 | ### |
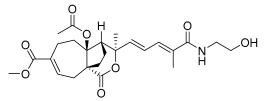 8 (10 μM) | 78.7 ± 18.4 | ns | 74.0 ± 5.5 | ns |
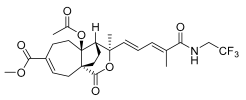 9 (10 μM) | 7.4 ± 7.9 | *** | 148.7 ± 14.1 | ** |
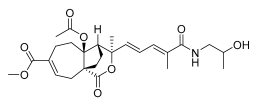 10 (10 μM) | 7.5 ± 5.9 | *** | 102.6 ± 7.2 | ns |
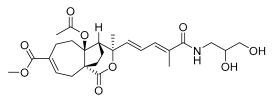 11 (10 μM) | 17.5 ± 17.1 | *** | 105.1 ± 7.5 | ns |
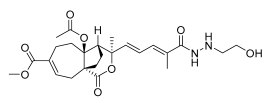 12 (10 μM) | 1.4 ± 1.1 | *** | 182.2 ± 5.3 | *** |
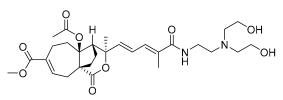 13 (10 μM) | 72.2 ± 10.2 | ns | 133.5 ± 5.0 | * |
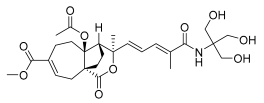 14 (10 μM) | 42.2 ± 9.5 | ** | 136.1 ± 8.0 | * |
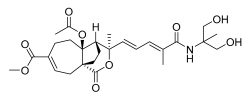 15 (10 μM) | 18.7 ± 18.0 | *** | 130.3 ± 5.8 | ns |
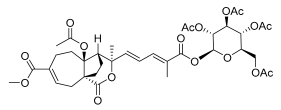 16 (10 μM) | 80.0 ± 27.5 | ns | 95.2 ± 23.0 | ns |
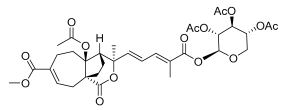 17 (10 μM) | 32.3 ± 19.2 | ** | 91.3 ± 6.9 | ns |
 18 (10 μM) | 42.6 ± 30.2 | ** | 58.7 ± 4.5 | ** |
| 1 (10 μM) | 9.9 ± 0.8 | ** | 135.0 ± 19.5 | * |
| PLX3397 (0.5 μM) | 3.2 ± 2.4 | *** | 193.3 ± 10.2 | *** |
| Compd. | Dosage (mg/kg) | T1/2 (hr) | Tmax (hr) | Cmax (ng/mL) | AUC0–∞ (hr × ng/mL) | Cl (mL/hr/kg) | MRT0–∞ (hr) |
|---|---|---|---|---|---|---|---|
| 12 | 1 (i.v.) | 40.2 ± 6.88 | 0.0833 ± 0.000 | 32.1 ± 11.7 | 363 ± 29.4 | 2766 ± 234.8 | 63.1 ± 7.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, X.; Yu, S.; Xu, L.; Wang, Q.; Yang, L.; Su, Y.; Xiong, Z.; Shao, M.; Geng, M.; Zhang, A.; et al. Discovery of Hydrazineyl Amide Derivative of Pseudolaric Acid B for Reprogramming Tumor-Associated Macrophages Against Tumor Growth. Molecules 2025, 30, 2088. https://doi.org/10.3390/molecules30102088
Peng X, Yu S, Xu L, Wang Q, Yang L, Su Y, Xiong Z, Shao M, Geng M, Zhang A, et al. Discovery of Hydrazineyl Amide Derivative of Pseudolaric Acid B for Reprogramming Tumor-Associated Macrophages Against Tumor Growth. Molecules. 2025; 30(10):2088. https://doi.org/10.3390/molecules30102088
Chicago/Turabian StylePeng, Xia, Siqi Yu, Lin Xu, Qinghua Wang, Lin Yang, Yi Su, Zhirou Xiong, Mengjie Shao, Meiyu Geng, Ao Zhang, and et al. 2025. "Discovery of Hydrazineyl Amide Derivative of Pseudolaric Acid B for Reprogramming Tumor-Associated Macrophages Against Tumor Growth" Molecules 30, no. 10: 2088. https://doi.org/10.3390/molecules30102088
APA StylePeng, X., Yu, S., Xu, L., Wang, Q., Yang, L., Su, Y., Xiong, Z., Shao, M., Geng, M., Zhang, A., Zhang, L., Ai, J., & Ding, C. (2025). Discovery of Hydrazineyl Amide Derivative of Pseudolaric Acid B for Reprogramming Tumor-Associated Macrophages Against Tumor Growth. Molecules, 30(10), 2088. https://doi.org/10.3390/molecules30102088