Introduction
Rolipram (
1) is a compound with varied biological activity. In particular, attention has been drawn to the emetic [
1], antiinflammatory [
2], immunosupressant [
3], antidepressive [
4,
5], putative antiparkinsonian [
6], and neuroprotective [
7] effects. The best characterised biochemical activity of
1 is the selective inhibition of the cAMP-selective phosphodiesterase family known as Type IV (PDE4). Its selectivity for this subtype of phosphodiesterases is the hallmark of the classification of these enzymes [
8].
Although
1 has been often used as the racemate in biological experiments, the biological activity of the enantiomers may be widely divergent [
9,
10,
11,
12]. Therefore we considered it desirable to use this compound routinely as a single enantiomer for both
in vitro and
in vivo studies. This made it necessary to obtain sufficiently large quantities of both enantiomers. In this paper we report a new synthesis of the enantiomers of
1, and the characterisation of these compounds as inhibitors of PDE4 enzyme subtypes.
The industrial synthesis of
rac-(
1) and resolution of the enantiomers either chromatographically [
13,
14,
15] or by classical (enzymatic) resolution of an intermediate in the synthesis [
16] has been reported. Asymmetric syntheses of
1 have been recently reviewed [
17]. Additionally, several recent enantioselective syntheses of
1 [
18,
19] have been reported. For our purposes, however, we required larger quantities of both enantiomers, and the chiral chromatographic methods, although perfectly suitable for the separation of a few grams of material, were not readily suitable for the quantities required. It seemed to us that an enantiodivergent synthesis, where diastereoisomeric intermediates could be readily separated, would provide an alternative fast and efficient route to both enantiomers.
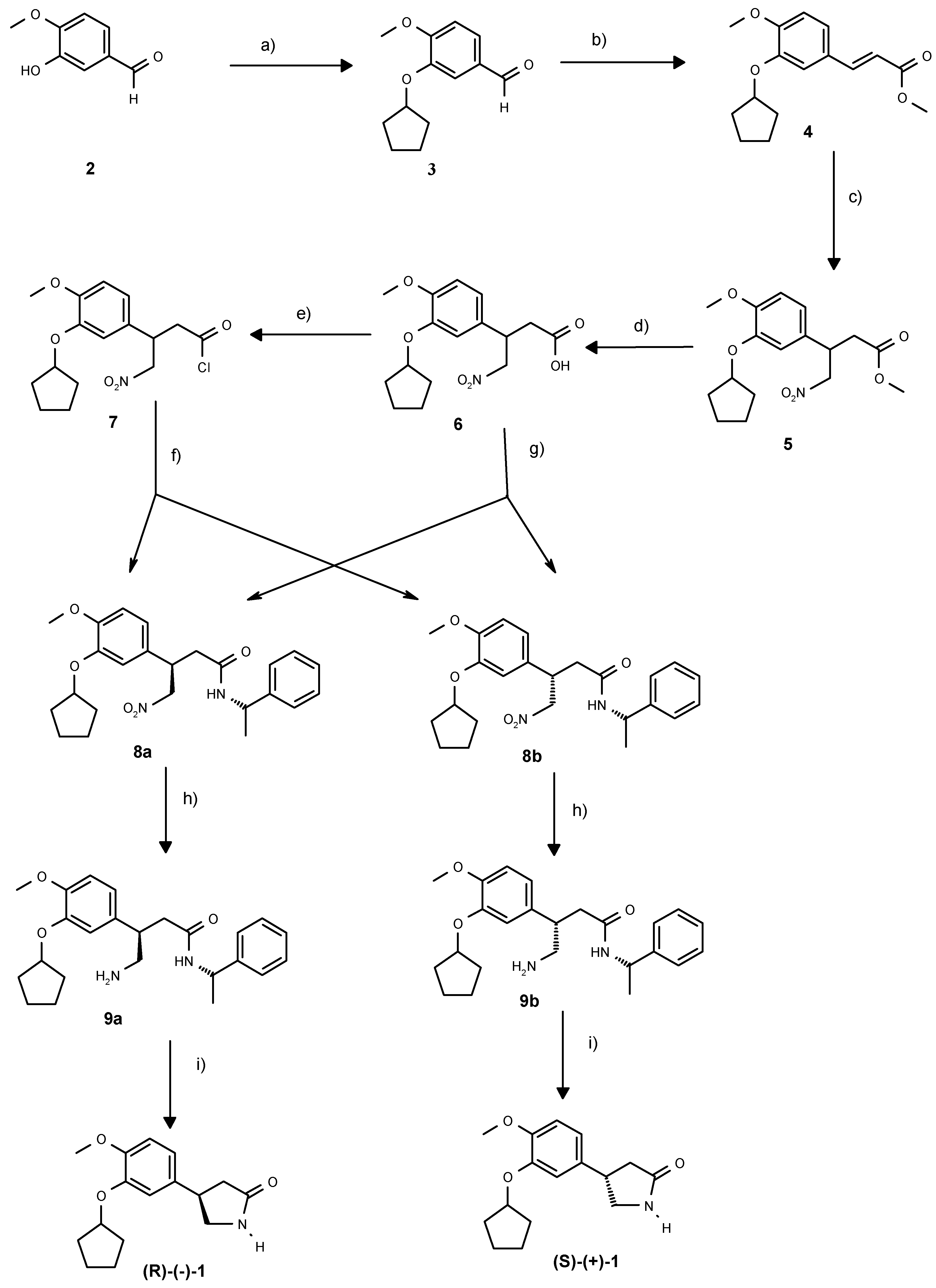
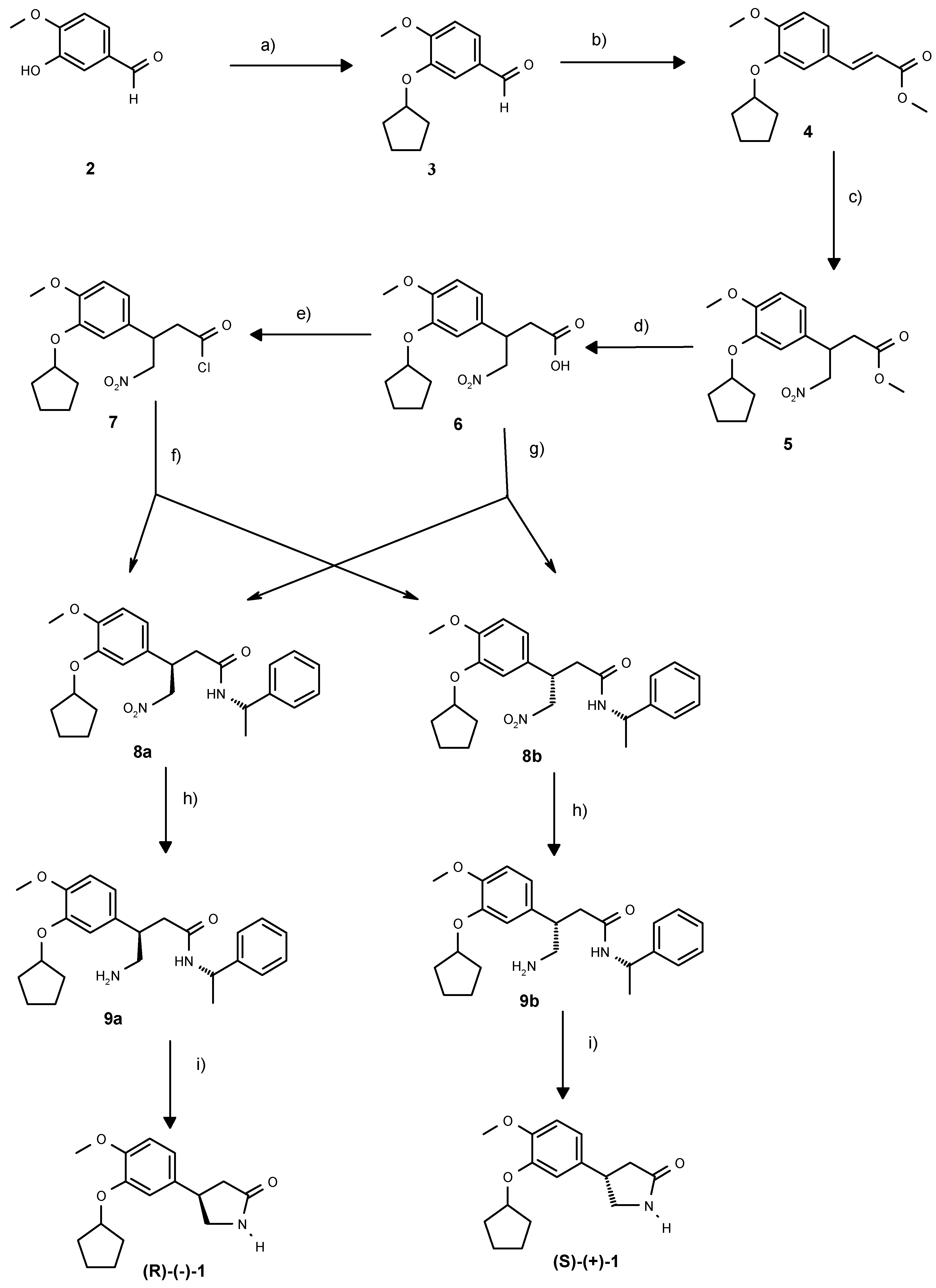
Scheme 1.
Enantiodivergent synthesis of rolipram: a) BnUb3NBr, C5H9Br, KOH, H2O, toluene, 93%. b) CH2(COOH)COOMe, aniline, piperidine, py, 85°C, 89%. c) CH3NO2, tetramethylguanidine, 75°C, 24h, 78%. d) NaOH, MeOH, 94%. e) SO2Cl2, CH2Cl2, 93%. f) (S)-phenylethylamine, CH2Cl2, reflux, 70%. g) (S)-phenylethylamine , HOBT, DMAP, EDCI, DMF, 0°C-RT, 79% h) Pd/C, 0.2 bar H2, DMF, 20 h, 100%. i). xylene, reflux 3-4h, 54%.
Scheme 1.
Enantiodivergent synthesis of rolipram: a) BnUb3NBr, C5H9Br, KOH, H2O, toluene, 93%. b) CH2(COOH)COOMe, aniline, piperidine, py, 85°C, 89%. c) CH3NO2, tetramethylguanidine, 75°C, 24h, 78%. d) NaOH, MeOH, 94%. e) SO2Cl2, CH2Cl2, 93%. f) (S)-phenylethylamine, CH2Cl2, reflux, 70%. g) (S)-phenylethylamine , HOBT, DMAP, EDCI, DMF, 0°C-RT, 79% h) Pd/C, 0.2 bar H2, DMF, 20 h, 100%. i). xylene, reflux 3-4h, 54%.
Additionally, from a previous synthesis of
rac-
1 [
20], the known racemic ester
5 [
21] was available to us in large quantities. We thus envisaged a strategy where the racemic acid could be converted to a mixture of readily separable diastereoisomeric amides, which could then be converted to the cyclic pyrrolidinones (+)-
1 and (−)-
1 respectively. In our initial experiments, the acid
6 was converted to the corresponding mixture of diastereoisomeric amides with either the Evans auxiliary, (+)-3-benzyl-oxazolidin-2-one or with (
S)- phenylethylamine. The latter mixture of diastereoisomers proved to be the easiest to separate, so we decided to use this chiral amine in the enantiodivergent step.
Experimental Section
General
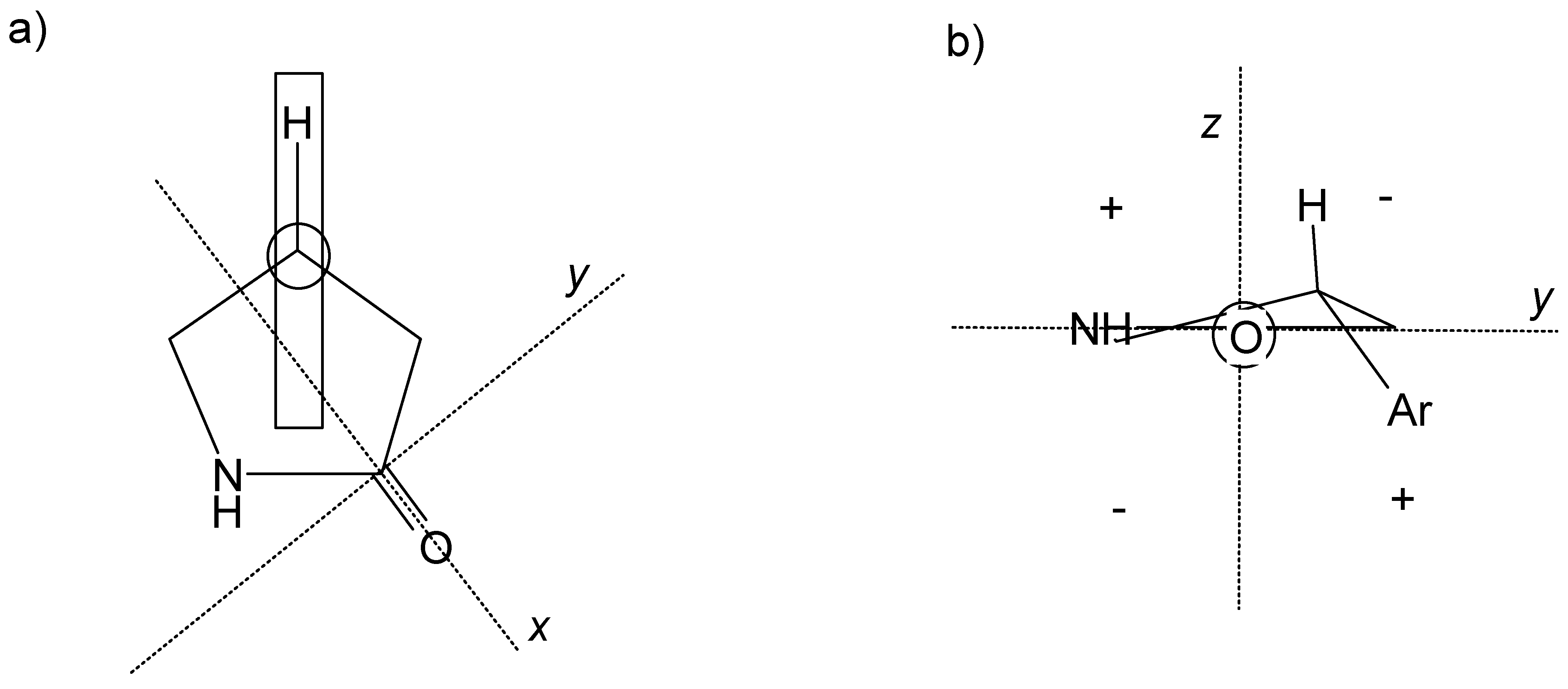
Melting points were determined on a Buechi 535 and are uncorrected. Optical rotations were determined on a Perkin-Elmer 241 polarimeter. IR spectra (ν,cm
-1) were recorded on a Bruker IFS-66 spectrometer.
1H-, and
13C- NMR spectra were recorded on a Bruker DPX 300 MHz spectrometer. Chemical shifts (δ) are given in ppm values using CDCl
3 as internal standard. Mass spectra were recorded on a VGTS-250 or aVG 70-SE spectrometer. CD spectra (λ
max (nm), [
Θ] (degrees cm² dmol
-1 )) were measured on a Jobin Yvon CD6 circular dichrograph. Solvent cutoff was defined for reference solvent absorbance = 2. Molar dichroic absorption (Δε) was calculated from the molar ellipticity according to the equation [
Θ] = 3298 Δε [
42]. Enantiomeric purity was determined by HPLC on a 5 μ Chiralcel OD column, 25 x 0.46 cm (N. Djordjevic, G. Lerch). Microanalyses were performed on a Leco CHN-800 or Leco RO-478 respectively. Silica gel chromatography was performed with columns of various lengths and diameters using Merck’s silica gel 60, particle size 0.040-0.063 mm as stationary phase. Hexane fraction boiling at 65-70 °C was used as solvent.
Cyclopentyl isovanillin (3)
Benzyltributyl-ammonium bromide (80.2 g, 0.255 mol) and bromo-cyclopentane (394 ml, 3.6 mol) were added to a rapidly stirred solution of isovanillin (288 g, 1.8 mol) in toluene (1620 ml), and this suspension was heated to 80-83 °C. To the resulting solution aqueous potassium hydroxide (225 g in 570 ml water) was added dropwise during 1 h, maintaining the solution at 80-83 °C. After 3 h the solution was cooled to room temperature, the two phases separated, and the upper toluene phase dried (MgSO4) and evaporated under vacuum. The residue was distilled to yield 3 (676 g, 93%) as a faintly green oil, b.p. 120 °C /0.1 mm. IR (neat): 1689, 1585, 1265, 810. MS (EI): 220 (M+), 152 (M-cyclopentyl). 1H-NMR (CDCl3): 1.5-2.0 (m, 8H, CH2), 3.95 (s, 1H, CH3), 4.81-4.91 (m, 1H, CH), 6.95-7.95 (m, 3H, Ar-H), 9.85 (s, 1H, CH). Anal. Calcd for C13H16O3 : C, 70.9; H, 7.3; O, 21.8. Found: C, 70.7; H, 7.3; O, 21.7.
3-cyclopentyloxy-4-methoxy-methyl cinnamate (4)
Aniline (13.6 ml) and piperidine (13.6 ml) were added to a solution of (568.9 g, 2.59 mol) 3 in pyridine (1633 ml), and the resulting solution was heated to 85°C. Monomethyl malonate (672.7g, 5.69 mol) was then added to this solution during 1 h, ensuring that the internal temperature did not exceed 90 °C. Stirring was continued for an additional 5 h at the same temperature. The mixture was cooled down to room temperature and slowly added to a mixture of 2N hydrochloric acid (13.6 l) and ice. The aqueous phase was extracted with 5.6 l EtOAc (2 x 2.8 l), and the combined organic extracts washed with brine, and dried (MgSO4). The solution was evaporated to dryness under reduced pressure, and the residue dissolved in Et2O (770 ml ), and cooled to 5 °C. Hexane (2.3 l) was added to this solution over 30 min during which a suspension formed, which was stirred for an additional 1h, filtered, washed with precooled diethyl ether/hexane (240 ml 1:3) and dried overnight to give 4 (498.2 g, 69.7 %) as colourless crystals, mp. 56.8-58.8 °C. The mother liquor was subjected to chromatography (3.8 kg Silica gel, hexane-EtOAc 3:1) to yield, after crystallisation as described above, additional 4 (136.1 g, 19.1 %), mp. 60-61 °C. IR (KBr): 1717, 1638, 1513, 1270, 1171, MS (EI): 276 (M+), 207, 198, 192, 176. 1H-NMR (CDCl3): 1.55-1.97 (m, 8H, CH2), 3.75 (s, 3H, CH3), 3.82 (s, 3H, CH3), 4.74 (m, 1H, CH), 6.24 (d, 1H, CH, J=15.9 Hz), 6.92 (dd, 2H, Ar-H, J=8.2 Hz) 7.01-7.05 (m, 1H, Ar-H), 7.57 (d, 1H, CH, J=15.9 Hz). Anal. Calcd for C16H20O4 : C, 69.6; H, 7.3; O, 23.2. Found: C, 69.0; H, 7.1; O, 23.0.
3(3’-cyclopentyloxy-4’-methoxy) phenyl-4-nitro methyl butyrate (5)
A solution of 4 (550.6 g, 1.99 mol) and 1,1,3,3- tetramethyl guanidine (53.9 ml) in nitromethane (1090 ml) was stirred for 24 h at 75 °C. After cooling the mixture to room temperature, EtOAc (1000 ml) was added to it. The biphasic mixture was poured into 2 N HCl (1500 ml), the organic phase separated, and the aqueous phase extracted with EtOAc (1000 ml). The combined organic phases were washed with 2 N HCl (1500 ml), followed by brine, and dried (MgSO4). Evaporation under vacuum yielded crude 5 (677 g) which was purified by chromatography (12 kg silica gel, hexane-ethyl acetate 3:1) to give 5 (523 g, 78 %) as a white solid, mp. 84.1-85.1 °C. IR (KBr): 1744, 1733, 1551, 1518, 1258, 1237. MS (EI): 337 (M+), 269, 222, 238, 222, 191, 164, 150, 131. 1H-NMR (CDCl3): 1.50-1.92 (m, 8H, CH2), 2.68 (d, 2H, CH2, J=7.3 Hz), 3.57 (s, 3H, CH3), 3.75 (s, 3H, CH3), 3.84 (m, 1H, CH), 4.59 (ddd, 2H, CH2, J=12.4 and 7.2 Hz), 4.69 (m, 1H, CH), 6.59-6.80 (m, 3H, Ar-H). Anal. Calcd for C17H23NO6: C, 60.5; H, 6.9; O, 28.5. Found: C, 60.6; H, 6.7; O, 28.0.
3(3’-cyclopentyloxy-4’-methoxy) phenyl-4-nitro butyric acid (6)
Sodium hydroxide solution (2 N, 720 ml) was added dropwise to a cold (0 °C) suspension of 5 (72 g, 0.21 mol) in methanol (1440 ml) while the internal temperature did not exceed 20 °C. After the addition was complete, the ice bath was removed and the mixture stirred for an additional 1.5 h. Methanol was removed from the reaction mixture under vacuum, and to the remaining aqueous solution EtOAc (1400 ml) was added. The mixture was acidified to pH=2 with HCl (2 N, 720 ml ). The two layers were separated and the aqueous layer extracted twice with EtOAc (2 x 700 ml). The combined organic extracts were dried (MgSO4) and evaporated to yield 6 (68.5 g, 99.3%) as beige crystals. The obtained crude material was almost pure and could be used directly for the subsequent step. Recrystallisation from EtOAc-hexane (1:1, 140 ml) proceeded to give 6 (64.5, 93.5 %) mp 119-121 °C. IR (KBr): 1703, 1590, 1553, 1260. MS (FAB): 323 (M+), 263, 255. 1H-NMR (CDCl3): 1.54-1.98 (m, 8H, CH2), 2.72 (ddd, 2H, CH2, J=16.6 and 7.5 Hz), 3.74 (s, 3H, CH3), 3.81 (m, 1H, CH), 4.56 (ddd, 2H, CH2, J=12.5 and 7.7 Hz), 4.68 (m, 1H, CH), 6.65-6.76 (m, 1H, Ar-H), 6.71 (dd, 2H, Ar-H, J=8.5 Hz). Anal. Calcd for C16H21NO6: C, 59.4; H, 6.6; N, 4.3. Found: C, 59.6; H, 6.4; N, 4.2.
3(3’-Cyclopentyloxy-4’-methoxy) phenyl-4-nitro butyryl chloride (7)
Thionyl chloride (24 ml, 0.33 mol) was added over 15 min. to a suspension of 6 (64.7 g, 0.2 mol) in CH2Cl2 (470 ml) at room temperature. After complete addition the suspension was heated to reflux for 3 h. The yellow solution was evaporated to give 7 (69.5 g) as a brown oil which solidified upon cooling. This crude material was taken up in Et2O (100 ml) and CH2Cl2 (50 ml), and crystallised by slow addition of the hexane fraction (500 ml) giving rise to 7 (63.7 g, 93.2 %) as beige crystals, mp 75-76 °C. IR (KBr): 1788, 1551, 1260. MS (EI): 341 (M+), 305, 273. 1H-NMR (CDCl3): 1.56-1.88 (m, 8H, CH2), 3.28 (ddd, 2H, CH2, J=17.5 and 7.9 Hz), 3.76 (s, 3H, CH3), 3.87 (m, 1H, CH), 4.55 (ddd, 2H, CH2, J=12.7 and 7.2 Hz), 4.69 (m, 1H, CH), 6.64-6.77 (m, 1H, Ar-H), 6.71 (dd, 2H, Ar-H, J=8 Hz). Anal. calcd for C16H20NO5Cl: C, 56.2; H, 5.9; N, 4.1. Found: C, 56.6; H, 5.9; N, 4.2.
(3R)-(3’-Cyclopentyloxy-4’-methoxy) phenyl-4-nitro butyryl - (S)-(−)- phenyl-ethylamide (8a) and (3S)-(3’- Cyclopentyloxy-4’-methoxy) phenyl-4-nitro butyryl -(S)- (−)- phenyl-ethylamide (8b)
Via carboxylic acid 6
4-Dimethylaminopyridine (116 g, 0.95 mol), solid hydroxybenzotriazole (HOBT, 73.2 g, 0.54 mol), and (146g, 0.45 mol) solid 6 were added in sequence to a cooled (0°C), stirred solution of (S)-(−)-1-phenyl ethylamine (54.7 g, 0.45 mol) in DMF (2700 ml). After stirring for 15 min, N-(3-dimethylaminopropyl)-N’-ethyl- carbodiimide hydrochloride (EDCI, 97.9 g, 0.51 mol) was added in portions over 15 min. Stirring was continued at 0°C for 1 h. The ice bath was removed, and the solution stirred overnight. After cooling the mixture to 5-10 °C, EtOAc (2000 ml) was added and the solution washed with 2N HCl (4500 ml). The aqueous phase was extracted with EtOAc (1800 ml) and the combined organic extracts were dried (MgSO4). Filtration and evaporation under reduced pressure yielded a mixture of diastereoisomers 8a and 8b. Purification by chromatography as described below gave 8a (51.8 g, 26.8 %), 8b (57 g, 29.5 %) and a mixed fraction (45 g 23 %) which was not further purified. For analytical data see below.
Via acid chloride 7
A solution of (S)-(-)-1-phenyl-ethylamine (49 ml, 0.39 mol) in CH2Cl2 (60 ml) was added dropwise over 15 min to a refluxing solution of 7 (60 g, 0.175 mol) in CH2Cl2 (660 ml). After maintaining reflux for 2 h, the reaction was complete. The solution was cooled to room temperature and washed successively with 2N HCl (240 ml), 2N NaHCO3 (240 ml), and brine (240 ml). The united aqueous phases were extracted twice with CH2Cl2 (240 ml and 120 ml). The combined organic phases were dried (MgSO4), and evaporated under vacuum to furnish a diastereomeric mixture of 8a and 8b (76.2 g). The mixture was dissolved in CH2Cl2 (500 ml) and subjected to chromatography (silica gel EtOAc-hexane fraction- CH2Cl2 50:30:20) yielding 8a (28.9 g, 38.7 %) and 8b (31 g, 41.5 %).
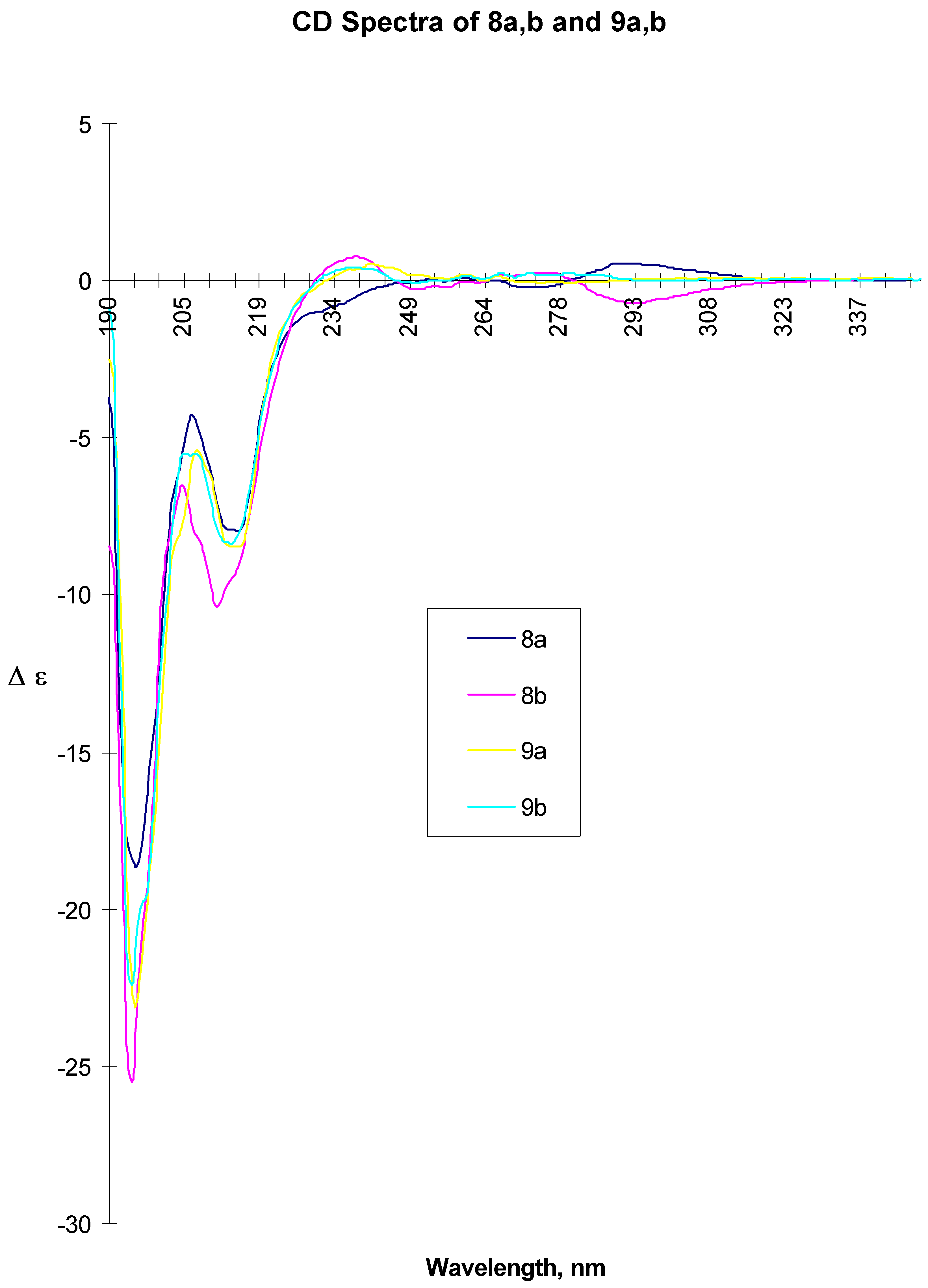
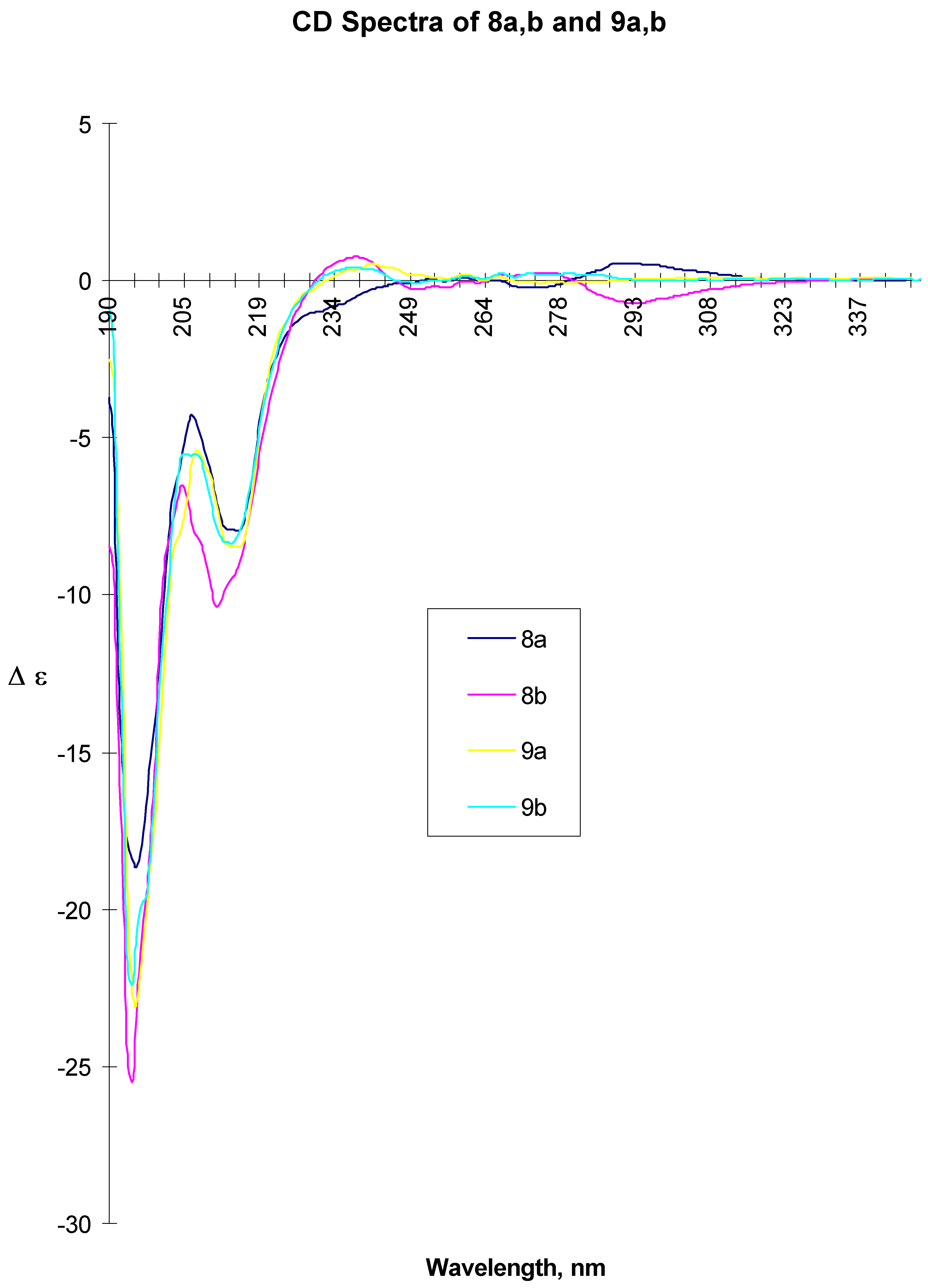
8a: white beige crystals, mp 143-144 °C, [α] D20 = +7.15 (c=0.47 acetone). CD ( c= 2.00 mg/ml, MeOH, l = 0.01 cm) 194.5 (-18.387), 206.5 (-4.276), 211 (sh, ca. 7.9), 215 (-7.952), 275.5 (-0.225), 294.5 (0.551). CD ( c= 2.00 mg/ml, MeOH, l = 1.00 cm) 277.5 (-0.203), 296.5 (0.717). IR (KBr): 3336, 1643, 1550. MS (EI): 427 (MH+), 323 , 281. 13C-NMR (CDCl3): 21.81, 24.30, 33.04, 40.45, 40.77, 49.17, 56.34, 79.87, 80.56, 112.59, 114.75, 119.55, 126.16, 126.32, 127.75, 128.97, 131.07, 143.00, 148.23, 150.03, 169.03. 1H-NMR (CDCl3): 1.27 (d, 3H, CH3, J=6.9 Hz), 1.53-1.83 (m, 8H, CH2), 2.50 (ddd, 2H, CH2, J=14.6 and 7.3 Hz), 3.75 (s, 3H, CH3), 3.82 (m, 1H, CH), 4.61 (ddd, 2H, CH2, J=12.3 and 8 Hz), 4.65 (m, 1H, CH), 4.96 (m, 1H, CH), 5.49 (d, 1H, NH, J=8 Hz), 6.65-6.73 (m, 1H, Ar-H), 6.67 (dd, 2H, Ar-H, J=8.9 Hz), 7.10-7.26 (m, 5H, Ar-H). Anal. Calcd for C24H30N2O5: C, 67.6; H, 7.1; N, 6.6. Found: C, 67.5; H, 7.2; N, 6.6.
8b: white beige crystals, mp 158-159 °C, [α] D20 = - 38.3 (c=0.52 acetone). CD ( c= 2.00 mg/ml, MeOH, l = 0.01 cm) 194.5 (-25.552), 204.5 (-6.524), 211.5 (-10.373), 215 (sh, ca. 10), 239.5 (0.750), 295.5 (-0.733). ). CD (c= 2.00 mg/ml, MeOH, l = 1.00 cm) 272.5 (0.541), 302.5 (-0.561). IR (KBr): 3368, 1648, 1637, 1555,1521. MS (EI): 427 (MH+), 323, 282. 13C-NMR (CDCl3): 21.77, 24.24, 32.98, 40.63, 40.78, 48.97, 56.24, 79.93, 80.71, 112.50, 114.76, 119.33, 126.11, 127.55, 128.81, 130.91, 142.79, 148.19, 149.95, 168.90. 1H-NMR (CDCl3): 1.34 (d, 3H, CH3, J=6.9 Hz), 1.50-1.97 (m, 8H, CH2), 2.50 (ddd, 2H, CH2, J=14.4 and 7.7 Hz), 3.75 (s, 3H, CH3), 384 (m, 1H, CH), 4.60 (ddd, 2H, CH2, J=12.4 and 8 Hz), 4.59 (m, 1H, CH), 4.97 (m, 1H, CH), 5.45 (d, 1H, NH, J=8Hz), 6.62-6.73 (m, 1H, Ar-H), 6.67 (dd, 2H, Ar-H, J=8.8 Hz), 6.96-7.19 (m, 5H, Ar-H). Anal. Calcd for C24H30N2O5: C, 67.6; H, 7.1; N, 6.6. Found: C, 67.5; H, 7.1; N, 6.6.
(3R)(3’-Cyclopentyloxy-4’-methoxy) phenyl-4-amino butyryl - (S)-(-)- phenyl-ethylamide (9a)
A suspension of 8a (38.7 g, 0.09 mol) and 10% Pd/C (10 g) in isopropanol (2900 ml) and DMF (580 ml) was hydrogenated at room temperature and 0.2 bar pressure overnight (20 h). The reaction mixture was filtered over celite, washed with isopropanol and concentrated under vacuum to dryness yielding crude 9a (40 g) as off-white solid, which was used without further purification for the subsequent step. For analytical data, a sample was purified from a different experiment (Silica gel, EtOAc-MeOH-NH4OH 80:20:2) Rf = 0.4. mp 94-95 °C, [α]rtD = -59.2° (c = 0.54, MeOH), CD ( c= 2.00 mg/ml, MeOH, l = 0.01 cm) 195.5 (-23.081), 208 (-5.479), 215 (-8.435), 242 (0.533)). CD ( c= 10.00 mg/ml, MeOH, l = 0.01 cm) 245 (0.330), 261 (0.213), 267 (0.176). IR (KBr): 3326, 2925, 1641, 1538, 1520. MS (EI): 397 (MH+). 13C-NMR (CDCl3): 21.94, 24.37, 33.16, 41.84, 45.98, 47.69, 48.97, 56.53, 80.89, 112.77, 115.22, 120.11, 126.46, 127.61, 128.94, 134.86, 143.51, 148.29, 149.46, 170.99. 1H-NMR (CDCl3): 1.20 (d, 3H, CH3, J=6.9 Hz), 1.52-1.93 (m, 8H, CH2), 2.40 (ddd, 2H, CH2, J=14.0 and 6.5 Hz), 2.70-2.89 (m, 2H, CH2), 2.94-3.05 (m, 1H, CH), 3.75 (s, 3H, CH3), 4.66 (m, 1H, CH), 4.93 (m, 1H, CH), 5.68 (d, 1H, NH, J=7.8Hz), 6.66-6.76 (m, 1H, Ar-H), 6.70 (dd, 2H, Ar-H, J=8.8 Hz), 7.08-7.45 (m, 5H, Ar-H).
(3S)(3’-Cyclopentyloxy-4’-methoxy) phenyl-4-amino butyryl - (S)-(-)- phenyl-ethylamide (9b)
A suspension of 8b (57.1 g, 0.13 mol) and 10% Pd/C (14.3 g) in isopropanol (4300 ml) and DMF (850 ml) was hydrogenated as described above to give crude 9b (57.9g, > 100%) as a viscous oil, which was of sufficient purity for the subsequent step. A sample of this oil obtained in a separate experiment was purified by chromatography (Silica gel, EtOAc-MeOH-NH4OH 80:20:2; Rf = 0.1) for chiroptical measurements. [α]rtD = -20.3° (c=1.3, MeOH), CD ( c = 2.00 mg/ml, MeOH, l = 0.01 cm) 194.5 (-22.422), 205.5 (-5.540), 214 (-8.365), 239 (0.419). CD (c = 10.00 mg/ml, MeOH, l = 0.01 cm) 239.5 (0.482), 251 (-0.123), 268 (0.242). IR (KBr): 3291, 2961, 1641, 1515, 1259. MS (EI): 397 (MH+), 367, 299, 194. 13C-NMR (CDCl3): 22.05, 24.35, 33.11, 41.74, 45.95, 47.74, 48.68, 56.41, 80.65, 112.70, 115.40, 119.94, 126.24, 127.15, 128.72, 134.75, 143.45, 148.17, 149.33, 171.08. 1H-NMR (CDCl3): 1.38 (d, 3H, CH3, J=6.9 Hz), 1.45-1.84 (m, 8H, CH2), 2.48 (ddd, 2H, CH2, J=13.9 and 8.7 Hz), 2.84-2.97 (m, 2H, CH2), 3.07-3.11 (m, 1H, CH), 3.83 (s, 3H, CH3), 4.70 (m, 1H, CH), 5.02 (m, 1H, CH), 5.89 (d, 1H, NH, J=7.9Hz), 6.65-6.83 (m, 1H, Ar-H), 6.74 (dd, 2H, Ar-H, J=8.7 Hz), 6.98-7.23 (m, 5H, Ar-H).
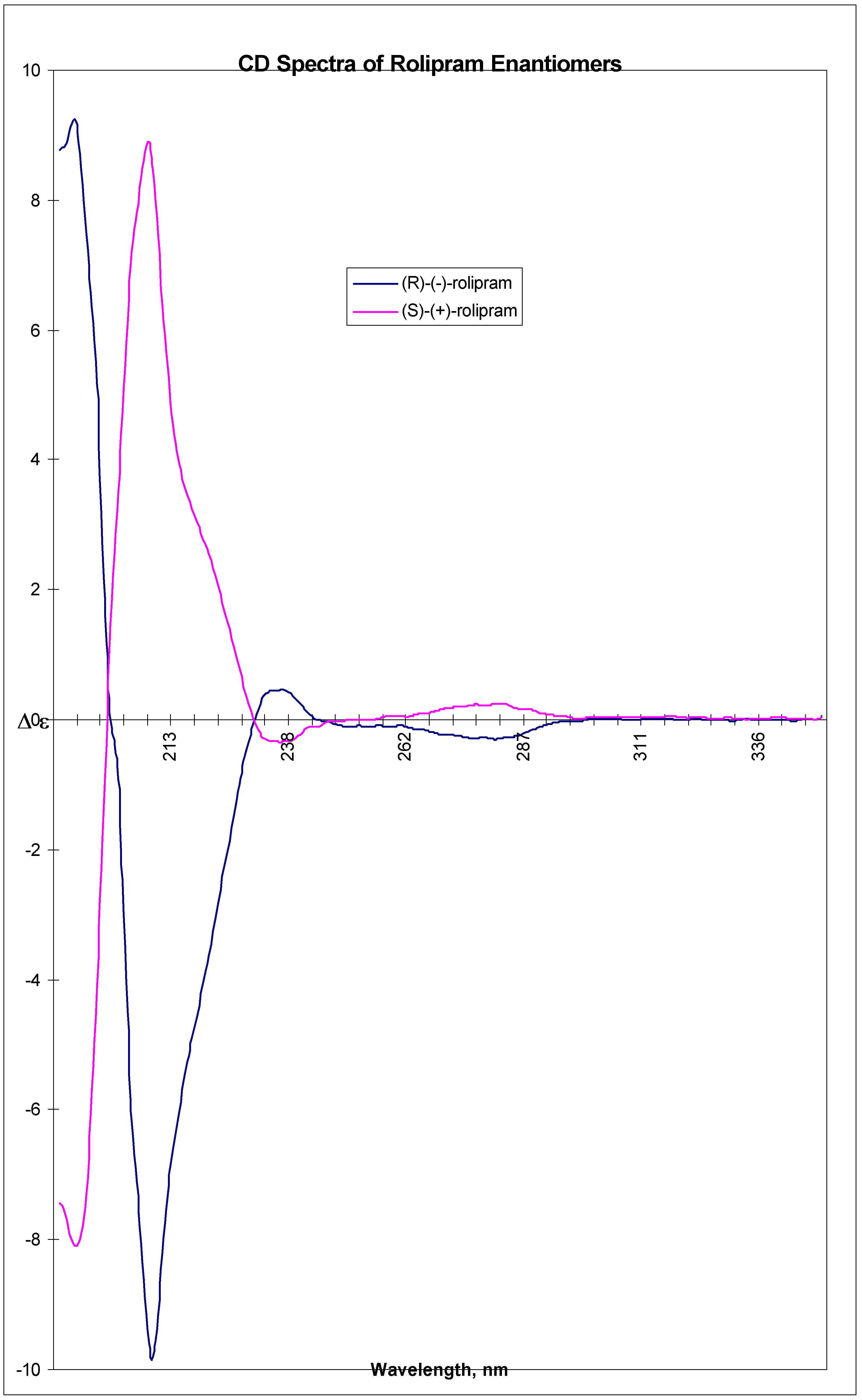
(4R)-(3’-Cyclopentyloxy-4’-methoxy)phenyl-2-pyrrolidone ((R)-(-)-1)
A suspension of 9a (38.3 g, 96.5 mmol) in xylene (2300 ml, isomeric mixture) was heated to reflux for 3-4 h. The clear yellowish solution was evaporated to dryness under vacuum. The resulting solid was suspended in t-butyl methyl ether (500 ml) and stirred overnight at room temperature. Finally the suspension was cooled to -5 °C , filtered, washed with precooled (-5 °C) t-butyl methyl ether (400 ml), and dried overnight at 50 °C, 2 mbar to give (R)-(-)-1 (14.3 g, 54 %) as off white crystals, mp 127-130 °C, [α]rtD = -35° (c = 0.5, MeOH), Enantiomeric purity (5 μ Chiralcel OD column, 25 x 0.46 cm, mobile phase: CH3CN 0.6 ml min-1, retention time 10.3 min): e.e. 99.70% CD ( c= 2.00 mg/ml, MeOH, l = 0.01 cm)192.5 (9.186), 209.5 (-9.857), 237 (0.461), 282.5 (-0.299). CD ( c= 10.00 mg/ml, MeOH, l = 0.01 cm) 236 (0.478), 280 (-0.242). IR (KBr): 3196, 3094, 1709, 1689, 1517. MS (EI): 275 (M+), 207, 177, 150, 135, 107. 13C-NMR (CDCl3): 26.02, 34.82, 40.15, 41.99, 51.79, 58.16, 82.62, 114.25, 115.88, 120.81, 136.59, 149.92, 151.21, 179.82. 1H-NMR (CDCl3): 1.48-1.93 (m, 8H, CH2), 2.52 (ddd, 2H, CH2, J=16.9 and 8.9 Hz), 3.50 (ddd, 2H, CH2, J=9.2 and 7.3 Hz), 3.55 (m, 1H, CH), 3.76 (s, 3H, CH3), 4.70 (m, 1H, CH), 6.57 (s, 1H, NH), 6.69-6.77 (m, 3H, Ar-H). Anal. Calcd for C16H21NO3: C, 69.8; H, 7.7; N,5.1. Found: C, 69.7; H, 7.5; N, 5.1.
(4S)-(3’-Cyclopentyloxy-4’-methoxy)phenyl-2-pyrrolidone ((S)-(+)-1)
A solution of 9b (57.8 g, 146 mmol) in xylene (3000 ml) was subjected to the same procedure as described above for (R)-(-)-1 giving (S)-(+)-1 (21.5 g, 54 %) as off-white crystals, mp 131-134 °C, [α]rtD = +31° (c = 0.6, MeOH) Enantiomeric purity: (5 μ Chiralcel OD column, 25 x 0.46 cm, mobile phase: CH3CN 0.6 ml min-1, retention time 9.5 min): 98.95% e.e. CD ( c= 2.00 mg/ml, MeOH, l = 0.01 cm) 193.5 (-8.105), 208.5 (8.893), 237.5 (-0.336), 283.5 (0.248). CD ( c= 10.00 mg/ml, MeOH, l = 0.01 cm) 237.5 (-0.351), 280.5 (0.270). IR (KBr): 3196, 3094, 1709, 1688, 1517. MS (EI): 276 (MH+), 208, 150, 107. 13C-NMR (CDCl3): 26.36, 35.16, 40.53, 42.32, 52.16, 58.50, 82.96, 114.60, 116.24, 121.16, 136.95, 150.26, 151.54, 180.24. 1H-NMR (CDCl3): 1.52-1.92 (m, 8H, CH2), 2.52 (ddd, 2H, CH2, J=16.9 and 8.8 Hz), 3.50 (ddd, 2H, CH2, J=9.2 and 7.3 Hz), 3.55 (m, 1H, CH), 3.76 (s, 3H, CH3), 4.70 (m, 1H, CH), 6.43 (s, 1H, NH), 6.69-6.77 (m, 3H, Ar-H). Anal. calcd for C16H21NO3: C, 69.8; H, 7.7; N,5.1. Found: C, 69.7; H, 7.8; N, 5.1.
(S)-Phenylethylamine acetamide (10)
Acetic anhydride (10.2 g, 0.1 mmol) was added dropwise to a solution of (
S)-(-)-1-phenyl-ethylamine (12.11 g, 0.1 mmol) in toluene (20 ml) at such a rate that slow reflux was maintained. When the addition was complete, the solution was allowed to cool to rt. And hexane (20 ml) added with stirring. After leaving for 8 h at 4 °C, the precipitate was filtered off, washed with hexane (10 ml) and dried (50 °C, 0,4 mbar, 8h) to give crystals (8.66 g, 53.5%) which were of high purity, and were used directly for CD measurements. mp. 103 °C [Lit [Ref.
26] 102-103 °C], [α]
rtD = -152.8° (c = 10.9, EtOH). CD (
c= 2.00 mg/ml, MeOH,
l = 0.01 cm) (λ
max nm, (Δε)): 215(-12.862), 211(-13.332), 197.5(-28.522) CD (
c= 20.00 mg/ml, MeOH,
l = 0.01 cm) (λ
max nm, (Δε)): 267.5 (0.224), 260.5 (0.231), 254.5 (0.126), 248 (0.047), 234 (0.037)..
Inhibition of cAMP phosphodiesterase (PDE4) from human neutrophil homogenate
Phosphodiesterase was prepared from human neutrophils by ultrasonic homogenisation. Activity was assayed by the column method of Thompson [
43]. Inhibitors were dissolved in DMSO and diluted to the required concentration in buffer T (MgCl
2 5mM, mercaptoethanol 3.6 mM, bovine serum albumin 1mg/ml, tris-hydroxymethyl-aminomethane 40mM, pH 8.0) containing 10% DMSO.
Inhibition of Human cAMP-specific phosphodiesterase (PDE4) isoenzymes
PDE4 activity was assessed as previously described [
40]. With the exception of PDE4B (rat; expressed in
S. cerevisae), all isoenzyme preparations were from human sources: PDE4A, PDE4B, PDE4D expressed in
S. cerevisae.
Inhibition of rolipram binding to rat brain membranes
Binding of [
3H]-rolipram to rat brain membranes was performed according to published procedures [
40] adapted for use in 96-well microtitre plates.


{kind=link}
{kind=link}
{kind=link}
{kind=link}