Abstract
The photoredox-catalyzed decarboxylative cross-coupling reaction of aryl acetic acids and aryl nitriles has been achieved under an argon atmosphere in high yields. This method provides a fast way to obtain prevalent aryl acetic acids from an abundant natural source. A tentative radical mechanism has been proposed.
1. Introduction
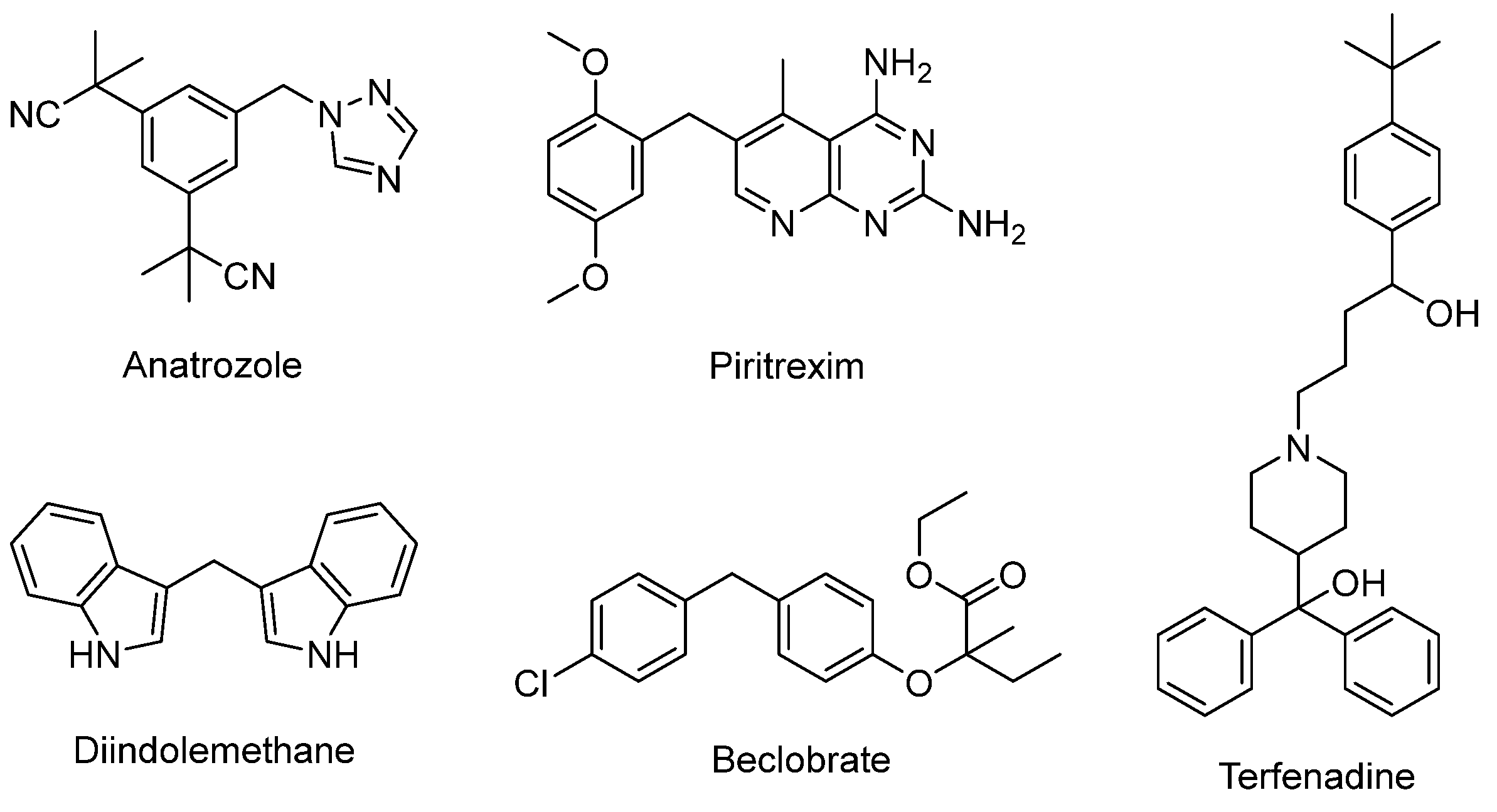
Diarylmethane motifs play a prominent role in the natural, bioactive and pharmaceutical product industries [1,2]. Some examples of anticancer agents include anastrozole [3], pitracoxin [4] and diindolemethane [5] (Figure 1). Anastrozole and letrozole are known to have pharmacological activity against breast cancer. Pharmacological experiments have shown that piritrexim has a significant inhibitory effect on dihydrofolate reductase of leukemia cells and is mainly used in the treatment of malignant tumors. Diindolemethane is present in cruciferous plants and also exhibits anticancer effects. Beclobrate [6] is a potent drug used to lower lipids and is about 20 times more effective than clofibrate. It also has some inhibition activity on platelet aggregation. Terfenadine is a second-generation antihistamine that is more selective to H1 receptors than the first generation and less irritating to the central nervous system [7]. It has been reported that some molecules containing diarylmethane skeleton may have a certain inhibitory effect on COVID-19 [8]. Carboxylic acids are widely employed as raw materials in organic synthesis because of their features of low toxicity, high stability and rich reserves. 3-indoleacetic acid (IAA) was detected by Salkowski in 1885 [9], who demonstrated that it can not only regulate the growth of plants but can also facilitate other aspects of plant development. In addition, the IAA industry is currently maintaining a stable market growth [10]. Therefore, the transformation of IAA and synthesis of diarylmethanes are highly appealing in organic chemistry due to their significant importance.
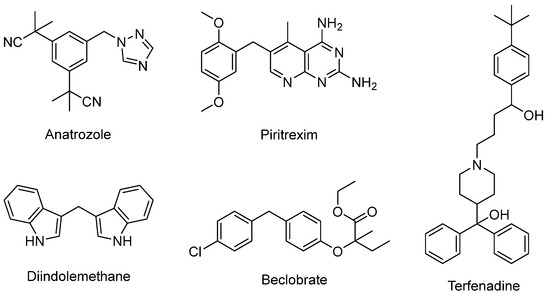
Figure 1.
The represent drugs containing diarylmethane scaffolds.
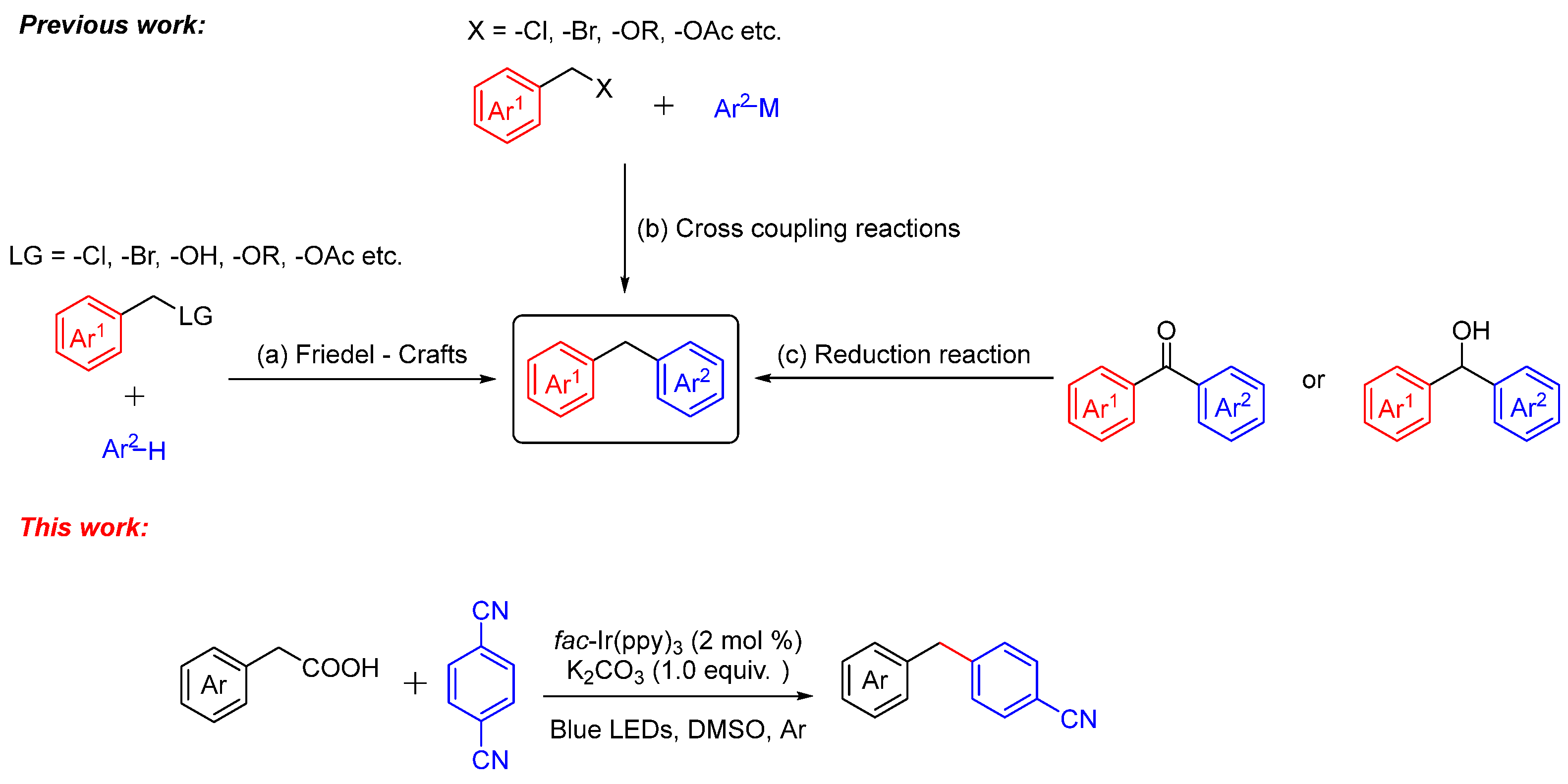
There are many ways to synthesize diarylmethane [11,12,13,14], and the conventional methods include (Scheme 1) (a) functionalization of arenes by means of a Friedel–Crafts reaction [15,16,17,18], (b) cross-coupling reaction [19,20,21,22,23,24,25,26], and (c) reduction reaction [27,28]. Among them, the typical approaches demand a high temperature and high pressure, which are generally achieved using highly demanded equipment. Recently, direct decarboxylative cross-coupling reactions have attracted researchers’ attention. In 2018, Lundgren and co-workers [29] reported the Cu-catalyzed decarboxylative cross-coupling reaction of aryl acetic acids and boronic esters to diarylmethanes. In 2021, our group [30] reported the decarboxylative cross-coupling reaction of NHPI with indoles to deliver a broad scope of diarylmethanes. Furthermore, photo-catalyzed chemistry takes over a major area in organic synthesis on account of its environmentally friendly. MacMillan [31,32,33,34] and other research groups [35,36,37,38] have explored lots of novel works on photochemical decarboxylation. Aryl nitriles were converted to their radical anion via SET reduction and underwent radical addition followed by decyanation [39,40,41]. With great efforts, we report the photoredox-catalyzed decarboxylative cross-coupling reaction of aryl acetic acids and aryl nitriles under an argon atmosphere in high yields.
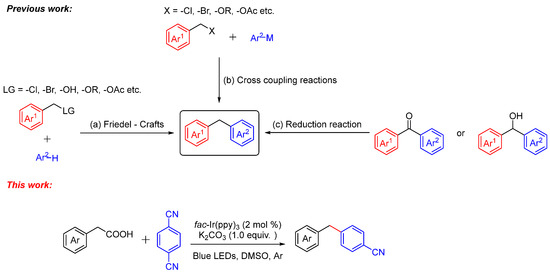
Scheme 1.
The conventional methods toward synthesis of diarylmethanes.
2. Results
Initially, our investigations focused on the blue LED irradiation of 3-indoleacetic acid (IAA) 1a and 1,4-dicyanobenzene (1,4-DCB) 2a in DMSO under argon atmosphere using 2 mol% of fac-Ir(ppy)3 in the presence of K2CO3 at room temperature. To our delight, the diarylmethane 3a was given in 82% isolated yield (Table 1, entry 1). No reaction took place when the reaction was performed without LED irradiation or without fac-Ir(ppy)3, resulting in the recovery of substrates 1a and 2a (Table 1, entries 2–3). When fac-Ir(ppy)3 as the photosensitizer was replaced by [Ir(dtbbpy)(ppy)2]PF6, the yield dropped to 53% (Table 1, entry 4). Furthermore, nearly no product was detected when the other photosensitizers including organic photocatalysts eosin Y and rose bengal were employed (Table 1, entries 5–7). The yield of the decarboxylation cross-coupling reaction dropped to 30% in the absence of a base (Table 1, entry 8). This reaction could also proceed when using other bases, such as K3PO4, NaOAc and CsF, but the yields decreased to some extent (Table 1, entries 9–11). In addition, DMSO was more optimal than other solvents for the transformation, (Table 1, entries 12–14). while the reaction did not occur when DCM was used as the solvent (Table 1, entry 12). The desired product was given with good yield in MeCN and DMA (Table 1, entries 13–14). Finally, we found that only trace amounts of diarylmethane were obtained when the reaction was performed under an air atmosphere (Table 1, entry 15). This result implies a radical involved mechanism in the reaction because of the biradical feature of the O2 molecule.

Table 1.
Screening of reaction conditions a.
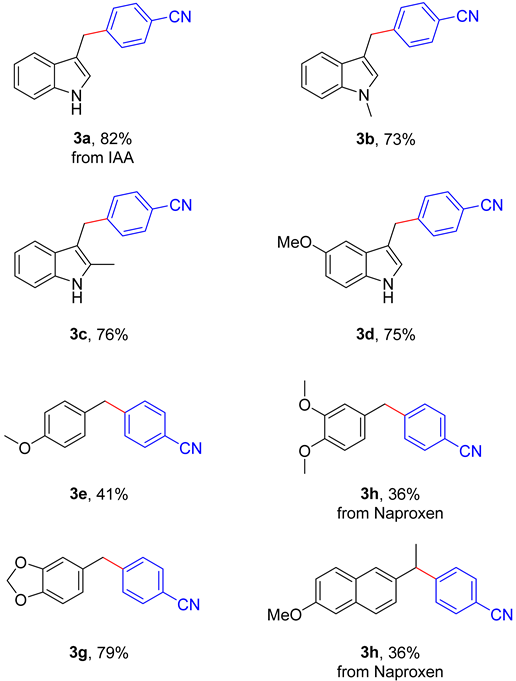
With the above optimal conditions in hand, we examined the scope of aryl acetic acids with 1,4-DCB to decarboxylative cross-coupling reaction. As shown in Table 2, all aryl acetic acid derivatives 1a–1h reacted smoothly with 2a to obtain the unsymmetrical diarylmethanes 3a–3h in good to excellent yields. First, alkyl and methoxy substituted heterocyclic acids could provide the corresponding products in yields of 73% (3b), 76% (3c) and 75% (3d), respectively. Moreover, phenyl acetic acid derivatives can also deliver this desired transformation (3e–3g). Homoveratric acid was coupled to 1,4-DCB to furnish product 3f in 83% yield. It is worth noting that the medicine molecule Naproxen was tolerated with the mild reaction conditions to obtain target product 3h in moderate yields, which is the property of non-steroidal anti-inflammatory group agents. This implies the synthetic potential of our approaches to medicinal chemistry.
Table 2.
Scope of the decarboxylative cross-coupling reaction of various aryl acetic acids with 1,4-DCB a,b.
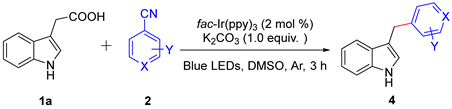
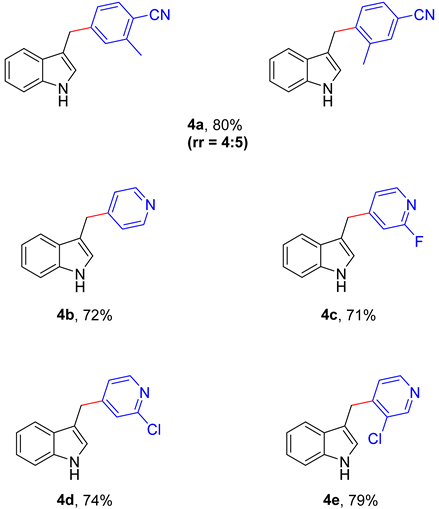
Next, a range of cyanoaromatics and cyano-heteroaromatics were capable of participating decarboxylative cross-coupling reaction with excellent yields (4a–4f, 71–80%) in Table 3. 2-methylterephthalonitrile was successfully converted into the corresponding product in 80% yield (4a). In addition, isonicotinonitrile was tolerated under the standard conditions (4b). Finally, it is worth noting that isonicotinonitrile with halogen substituents were all compatible reaction partners (4c–4e).
Table 3.
Scope of the decarboxylative cross-coupling reaction of various aryl nitriles with 3-indoleacetic acid a,b.
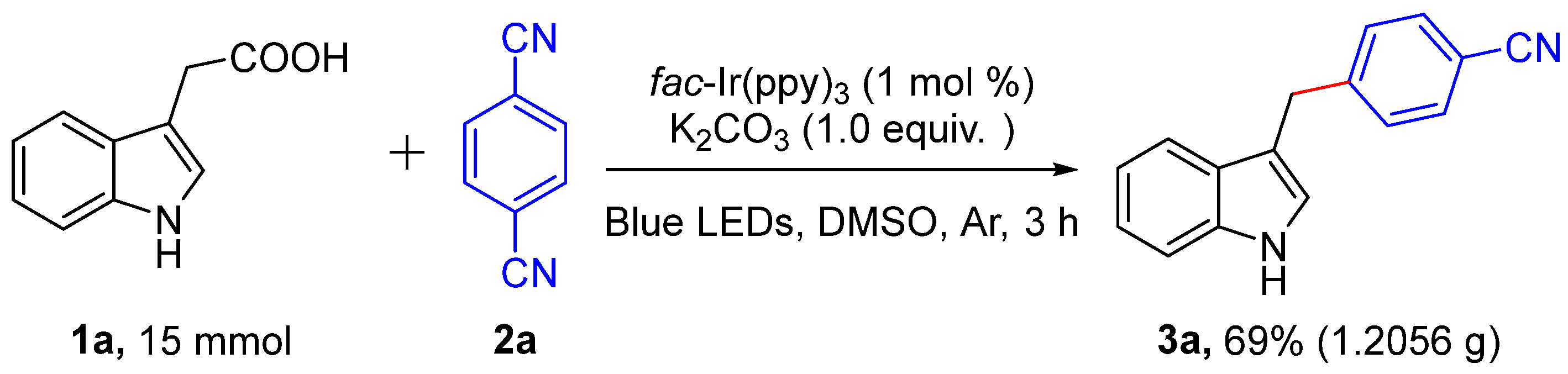
In addition, the practicality of this decarboxylative cross-coupling reaction was further proved by a gram-scale reaction (Scheme 2). When the reaction was carried out under photoredox-catalyzed method, the objected unsymmetrical diarylmethanes 3a could be gained in high isolated yield (69%).
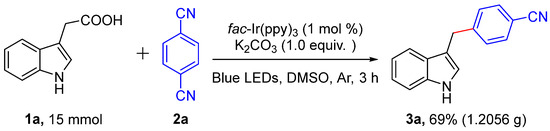
Scheme 2.
Gram-scale synthesis.
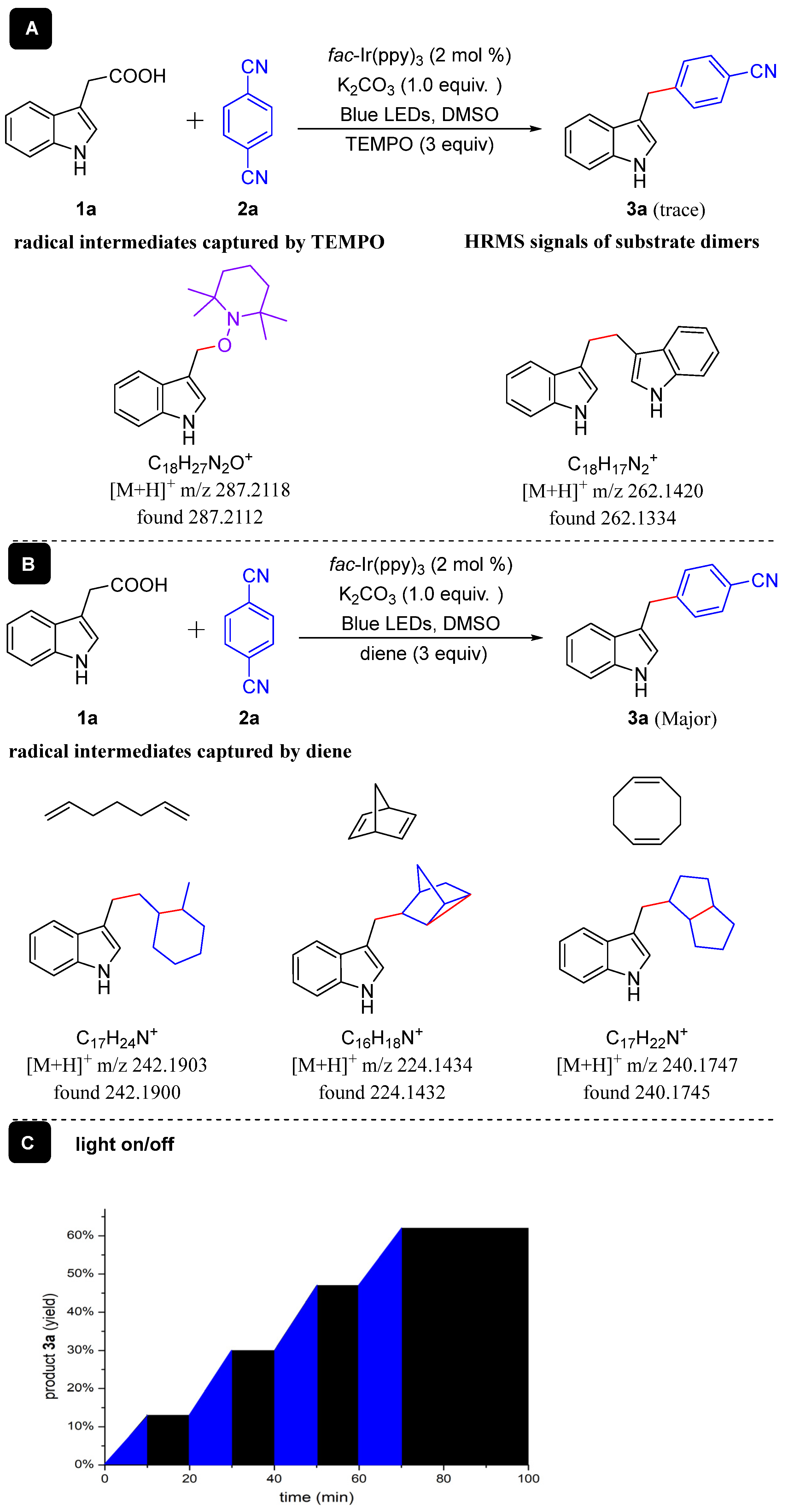
A series of control experiments were achieved to gain more information about the decarboxylative cross-coupling reaction mechanism between aryl acetic acids and aryl nitriles (Scheme 3). Initially, the reaction of 3-indoleacetic acid (1a) with 1,4-DCB (2a) was conducted in the presence of radical scavenger TEMPO (2,2,6,6-tetramethylpiperidine) under standard reaction conditions (Scheme 3A), and only a trace amount of compound 3a was perceived, and TEMPO-trapping compounds could be discerned by HRMS (Scheme 3A), and homocoupling dimer could be detected by HRMS too (Scheme 3A). Then, the radical intermediate in the reaction was compromised by some cyclizations [42] (Scheme 3B). Dienes caused addition/cyclization compatible with intramolecular radical addition based upon HRMS analysis. Finally, the light on/off experiment (Scheme 3C) also verifies that the reaction involves a radical process [43]. These control experiments reveal that this reaction involved radical intermediates.
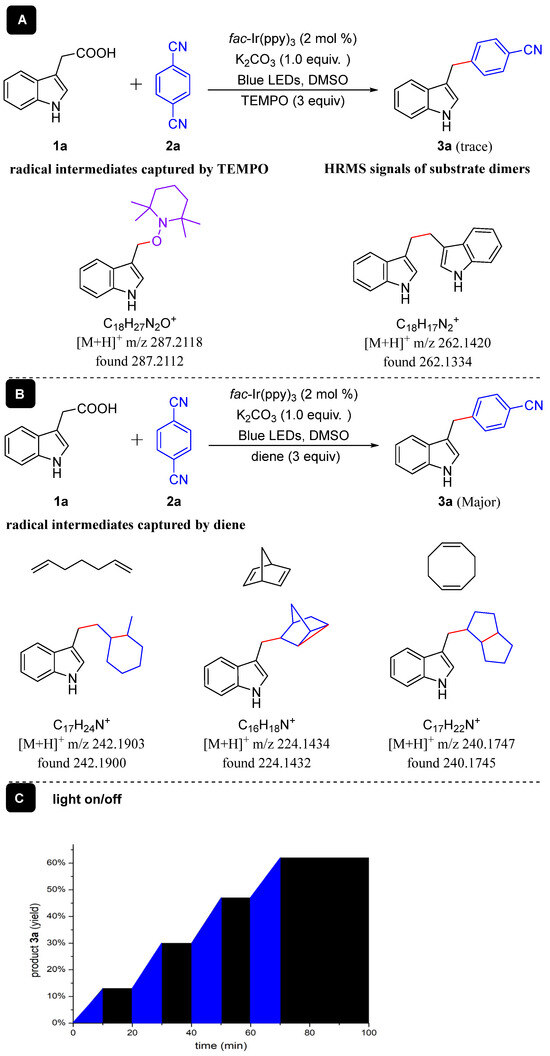
Scheme 3.
Control experiments.
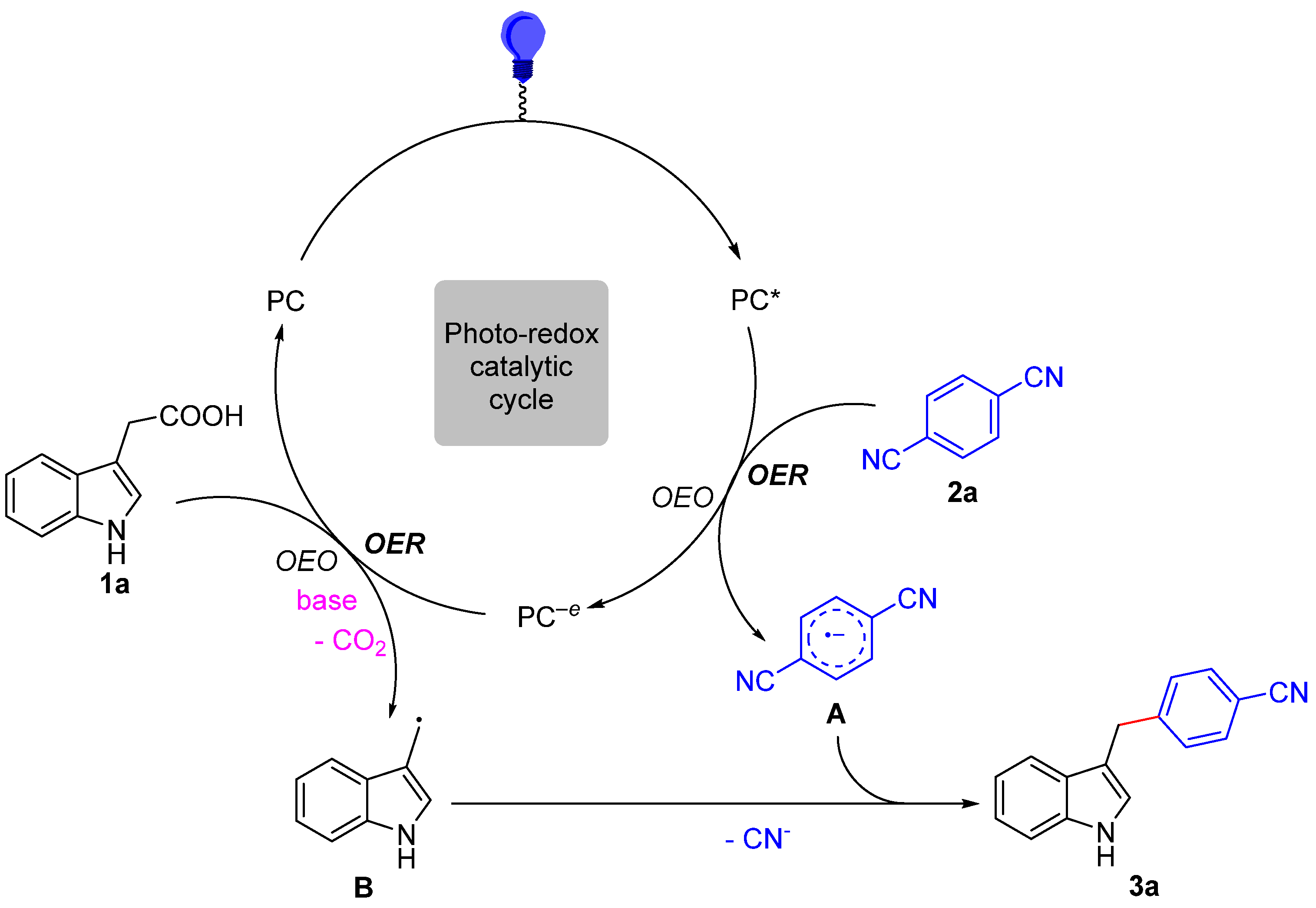
Guided by the above experimental results and previous literature [44,45,46,47,48,49], a plausible mechanism is presented in Scheme 4 for a photoredox-catalyzed decarboxylative cross-coupling reaction. In the first place, the irradiation of photocatalyst PC [Ir(ppy)3] with visible light produces a photoexcited state PC*[*Ir(ppy)3], which is a strong reductant (E1/2red [IrIV/*IrIII] = −1.73 V vs. SCE) [50]. We assumed that the reduction of 1,4-DCB (E1/2red = −1.61 V vs. SCE) [51] by single electron transfer to form the corresponding radical anion intermediate A and oxidized photocatalyst PC−e [IrIV(ppy)3] would be easy. Then, the PC−e [IrIV(ppy)3] is a strong oxidant (E1/2red = +0.77 V vs. SCE) and could undergo a single electron transfer with 3-indoleacetic acid substrate [52] to deliver the radical intermediate B with the elimination of CO2, together with regenerating PC [IrIII(ppy)3] and finishing the photoredox cycle. Finally, the radical–radical coupling of intermediate A with B and then aromatization with loss of CN− would produce the diarylmethane.
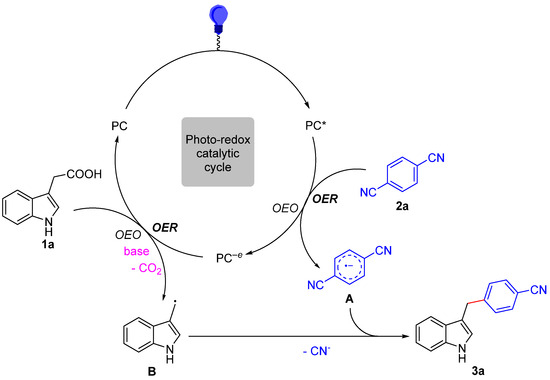
Scheme 4.
Proposed mechanism.
3. Materials and Methods
3.1. General Information
All materials, reagents and solvents were purchased from commercial suppliers and were used without further purification. Analytical TLC was performed with silica gel GF254 plates, and the products were visualized by UV detection. Flash chromatography was carried out using silica gel 200–300. The 1H NMR (400 or 600 MHz) and 13C NMR (151 MHz) spectra were measured with CDCl3 as solvent. All chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz. High-resolution mass spectra (HR-MS) were recorded under electrospray ionization (ESI) conditions.
3.2. General Procedure for Light-Promoted Decarboxylative Cross-Coupling Reaction between Aryl Acetic Acids and 1,4-Dicyanobenzene
Aryl acetic acids (1, 0.2 mmol), 1,4-dicyanobenzene (2a, 0.1 mmol), K2CO3 (1 equiv.) and fac-Ir(ppy)3 (2 mol%) were successively added to a dried reaction tube (10 mL) with a magnetic stirring bar. Air was then withdrawn and backfilled with argon 3 times. Subsequently, degassed DMSO (1 mL) was injected into the tube by syringe. Then, the resulting reaction mixture was performed at room temperature under blue LED (6 W) irradiation for 3–24 h. The reaction progress was monitored by TLC. After the reactions were completed, the reaction mixture was diluted with water (10 mL) and washed with EA (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography to afford the desired compounds 3 (ethylacetate/n-Hexane = 1:8 to 1:100).
4-((1H-indol-3-yl)methyl)benzonitrile (3a): The desired pure product was obtained in 82% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.60–7.52 (m, 2H), 7.47–7.32 (m, 4H), 7.25–7.16 (m, 1H), 7.14–7.04 (m, 1H), 6.98 (d, J = 2.3 Hz, 1H), 4.18 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 147.0, 136. 5, 132.2, 129.3, 127.1, 122.6, 122.3, 119.6, 119.1, 118.8, 113.9, 111.3, 109.7, 31.8. HRMS (ESI) exact mass calcd for C16H14N2 [M+H]+ m/z 233.1073, found 233.1083.
4-((1-methyl-1H-indol-3-yl)methyl)benzonitrile (3b): The desired pure product was obtained in 73% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 1H), 7.23–7.16 (m, 1H), 7.10–7.01 (m, 1H), 6.78 (s, 1H), 4.12 (s, 2H), 3.71 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 147.3, 137.3, 132.2, 129.4, 127.6, 127.4, 121.9, 119.2, 119.1, 119.0, 112.4, 109.7, 109.4, 32.7, 31.7. HRMS (ESI) exact mass calcd for C17H15N2 [M+H]+ m/z 247.1235, found 247.1240.
4-((2-methyl-1H-indol-3-yl)methyl)benzonitrile (3c): The desired pure product was obtained in 76% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.31–7.21 (m, 4H), 7.10 (t, J = 7.5 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 4.06 (s, 2H), 2.31 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 147.5, 135.4, 132.2, 132.2, 129.1, 128.5, 121.3, 119.5, 119.3, 118.0, 110.5, 109.4, 108.9, 30.4, 11.7. HRMS (ESI) exact mass calcd for C17H15N2 [M+H]+ m/z 247.1235, found 247.1229.
4-((5-methoxy-1H-indol-3-yl)methyl)benzonitrile (3d): The desired pure product was obtained in 75% yield as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.98 (s, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.8 Hz, 1H), 6.93 (d, J = 0.9 Hz, 1H), 6.89–6.85 (m, 1H), 6.84 (d, J = 2.3 Hz, 1H), 4.13 (s, 2H), 3.79 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 154.1, 146.9, 132.2, 131.6, 129.3, 127.5, 123.4, 119.1, 113.6, 112.3, 112.0, 109.7, 100.9, 55.9, 31.8. HRMS (ESI) exact mass calcd for C17H15N2O [M+H]+ m/z 263.1184, found 263.1185.
4-(4-methoxybenzyl)benzonitrile (3e): The desired pure product was obtained in 41% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.0 Hz, 2H), 7.28 (s, 2H), 7.08 (d, J = 8.3 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 3.97 (s, 2H), 3.79 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 158.3, 147.2, 132.2, 131.4, 129.9, 129.5, 119.0, 114.1, 109.9, 55.3, 41.1. HRMS (ESI) exact mass calcd for C15H14NO [M+H]+ m/z 224.1075, found 224.1079.
4-(3,4-dimethoxybenzyl)benzonitrile (3f): The desired pure product was obtained in 83% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 7.8 Hz, 2H), 6.82 (d, J = 8.1 Hz, 1H), 6.72–6.69 (m, 1H), 6.66 (s, 1H), 3.98 (s, 2H), 3.86 (s, 3H), 3.83 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 149.1, 147.8, 147.0, 132.2, 131.8, 129.5, 121.0, 119.0, 112.2, 111.4, 110.0, 55.9, 55.9, 41.5. HRMS (ESI) exact mass calcd for C16H16NO2 [M+H]+ m/z 254.1181, found 254.1187.
4-(benzo[d][1,3]dioxol-5-ylmethyl)benzonitrile (3g): The desired pure product was obtained in 79% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 7.9 Hz, 2H), 7.26 (d, J = 7.8 Hz, 2H), 6.75 (d, J = 7.8 Hz, 1H), 6.62 (d, J = 11.4 Hz, 2H), 5.92 (s, 2H), 3.93 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 147.9, 146.9, 146.3, 133.0, 132.3, 129.5, 121.9, 119.0, 110.1, 109.3, 108.4, 101.0, 41.6. HRMS (ESI) exact mass calcd for C15H12NO2 [M+H]+ m/z 238.0868, found 238.0859.
4-(1-(6-methoxynaphthalen-2-yl)ethyl)benzonitrile (3h): The desired pure product was obtained in 36% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.9 Hz, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.60 (s, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 8.9 Hz, 1H), 7.17–7.13 (m, 1H), 7.11 (s, 1H), 4.33 (q, J = 7.2 Hz, 1H), 3.91 (s, 3H), 1.72 (d, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 157.6, 152.0, 139.7, 133.3, 132.2, 129.2, 128.9, 128.5, 127.2, 126.8, 125.4, 119.0, 109.9, 105.6, 55.3, 44.8, 21.4. HRMS (ESI) exact mass calcd for C20H18NO [M+H]+ m/z 288.1388, found 288.1382.
3.3. General Procedure for Light-Promoted Decarboxylative Cross-Coupling Reaction between 3-Indoleacetic Acid and Nitriles
3-indoleacetic acid (1a, 0.2 mmol), nitriles (2, 0.1 mmol), K2CO3 (1 equiv.) and fac-Ir(ppy)3 (2 mol%) were successively added to a dried reaction tube (10 mL) with a magnetic stirring bar. Air was then withdrawn and backfilled with argon 3 times. Subsequently, degassed DMSO (1 mL) was injected into the tube by syringe. Then, the resulting reaction mixture was performed at room temperature under blue LED (6 W) irradiation for 3 h. The reaction progress was monitored by TLC. After the reactions were completed, the reaction mixture was diluted with water (10 mL) and washed with EA (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography to afford the desired compounds 4 (ethylacetate/n-Hexane = 1:3 to 1:10).
4-((1H-indol-3-yl)methyl)-2-methylbenzonitrile (4a) and 4-((1H-indol-3-yl)methyl)-3-methylbenzonitrile (4a’): The desired pure product was obtained in 80% yield as a white solid (rr = 4:5). 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H), 7.53–7.39 (m, 2H), 7.36 (d, J = 3.6 Hz, 1H), 7.26–7.11 (m, 3H), 7.11–7.02 (m, 1H), 6.94 (s, 0.4H), 6.75 (s, 0.5H), 4.10 (d, J = 6.7 Hz, 2H), 2.46 (s, 1.3H), 2.35 (s, 1.7H). 13C NMR (151 MHz, CDCl3) δ 146.9, 145.1, 141.9, 137.9, 136.5, 133.4, 132.5, 130.5, 129.9, 129.8, 127.2, 127.2, 126.6, 122.6, 122.3, 122.3, 119.6, 119.4, 118.9, 118.8, 118.5, 114.0, 113.2, 111.3, 111.3, 110.1, 109.8, 31.7, 29.5, 20.5, 19.4. HRMS (ESI) exact mass calcd for C17H15N2 [M+H]+ m/z 247.1235, found 247.1240.
3-(pyridin-4-ylmethyl)-1H-indole (4b): The desired pure product was obtained in 72% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.47 (d, J = 5.0 Hz, 2H), 7.43 (d, J = 7.8 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.18 (t, J = 6.7 Hz, 3H), 7.07 (t, J = 7.4 Hz, 1H), 6.96 (s, 1H), 4.10 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 150.8, 149.4, 136.6, 127.2, 124.2, 122.9, 122.2, 119.5, 118.8, 113.0, 111.4, 31.1. HRMS (ESI) exact mass calcd for C14H13N2 [M+H]+ m/z 209.1079, found 209.1082.
3-((2-fluoropyridin-4-yl)methyl)-1H-indole (4c): The desired pure product was obtained in 71% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 8.08 (d, J = 5.1 Hz, 1H), 7.46–7.31 (m, 2H), 7.24–7.19 (m, 1H), 7.14–7.06 (m, 2H), 7.02 (s, 1H), 6.80 (s, 1H), 4.14 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 164.2 (d, J = 238.2 Hz), 156.6 (d, J = 7.6 Hz), 147.2 (d, J = 15.1 Hz), 136.4, 127.0, 122.7, 122.5, 121.7 (d, J = 3.9 Hz), 119.8, 118.7, 112.6, 111.3, 109.2 (d, J = 37.0 Hz), 30.9, 30.9. HRMS (ESI) exact mass calcd for C14H12FN2 [M+H]+ m/z 227.0985, found 227.0989.
3-((2-chloropyridin-4-yl)methyl)-1H-indole (4d): The desired pure product was obtained in 74% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.24 (d, J = 5.1 Hz, 1H), 7.47–7.36 (m, 2H), 7.22 (t, J = 7.5 Hz, 2H), 7.15–7.06 (m, 2H), 7.00 (s, 1H), 4.09 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 154.0, 151.6, 149.4, 136.4, 126.9, 124.2, 122.8, 122.8, 122. 5, 119.8, 118.7, 112.5, 111.3, 30.8. HRMS (ESI) exact mass calcd for C14H12ClN2 [M+H]+ m/z 243.0689, found 243.0683.
3-((3-chloropyridin-4-yl)methyl)-1H-indole (4e): The desired pure product was obtained in 79% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 8.55 (d, J = 2.3 Hz, 1H), 8.30 (d, J = 5.0 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.22 (d, J = 7.2 Hz, 1H), 7.16–7.10 (m, 1H), 7.08 (d, J = 5.0 Hz, 1H), 7.03 (d, J = 2.1 Hz, 1H), 4.23 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 148.9, 148.0, 147.5, 136.4, 132.1, 127.1, 125.0, 123.2, 122.4, 119.7, 118.8, 111.4, 111.4, 28.5. HRMS (ESI) exact mass calcd for C14H12ClN2 [M+H]+ m/z 243.0689, found 243.0692.
3.4. Scale-Up Experiment
3-Indoleacetic acid (1a, 15 mmol), 1,4-dicyanobenzene (2a, 7.5 mmol), K2CO3 (7.5 mmol) and fac-Ir(ppy)3 (1 mol%) were successively added to a dried reaction tube (100 mL) with a magnetic stirring bar. Air was then withdrawn and backfilled with argon 3 times. Subsequently, degassed DMSO (25 mL) was injected into the tube by syringe. Then, the resulting reaction mixture was performed at room temperature under blue LED (6 W) irradiation for 15 h. After the reaction was completed, the reaction mixture was diluted with water (50 mL) and washed with EA (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (ethylacetate/n-Hexane = 1:8) to afford the desired compound 3a (1.2056 g, 69%).
3.5. The Light On/Off Experiment
Following the literature procedure [53], to a dried reaction tube (10 mL) with a magnetic stirring bar were added 3-Indoleacetic acid (1a, 0.2 mmol), 1,4-dicyanobenzene (2a, 0.1 mmol), K2CO3 (1 equiv.) and fac-Ir(ppy)3 (2 mol%) successively. Air was then withdrawn and backfilled with argon 3 times. Subsequently, degassed DMSO (1 mL) was injected into the tube by syringe. Then, the resulting reaction mixture was performed at room temperature under blue LED (6 W) irradiation. eight identical reactions were carried out simultaneously and yield was determined by 1H NMR of the crude mixture using 1,3,5-trimethylbenzene (13.9 μL, 0.10 mmol, 0.5 equiv.) as internal standard. The experiments with continuous intervals of irradiation and dark periods led to total interruption of the reaction proceed in the absence of light, and reactivity is restored under further light (The details are in the Supplementary Materials). These results indicated that light is an essential component of the reaction.
4. Conclusions
In summary, we have described a photoredox-catalyzed decarboxylative cross-coupling reaction of aryl acetic acids and aryl nitriles achieved under an argon atmosphere in high yields. A tentative radical mechanism has been proposed. Further studies of the detailed reaction mechanism and application are underway in our laboratory.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29092156/s1, Part 1, Experimental Section; Part 2, Scale-up experiment; Part 3, The light on/off experiment; Part 4, Characterizations of compounds; Part 5, NMR spectra of the products.
Author Contributions
Conceptualization, G.G. and Y.Z.; methodology, G.G. and Y.Z.; formal analysis, Z.L.; resources, Y.L.; data curation, Y.L.; writing—original draft preparation, G.G.; writing—review and editing, Z.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Doctor Funding Project of Longdong University (XYBYZK2224), the Natural Science Foundation of Gansu Province (23JRRM0754) and the Gansu Key Laboratory of Efficient Utilization of Oil and Gas Resources for financial support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kc, S.; Dhungana, R.K.; Khanal, N.; Giri, R. Nickel-Catalyzed α-Carbonylalkylarylation of Vinylarenes: Expedient Access to γ,γ-Diarylcarbonyl and Aryltetralone Derivatives. Angew. Chem. Int. Ed. 2020, 59, 8047–8051. [Google Scholar] [CrossRef] [PubMed]
- Buzdar, A.; Jonat, W.; Howell, A.; Jones, S.E.; Blomqvist, C.; Vogel, C.L.; Eiermann, W.; Wolter, J.M.; Azab, M.; Webster, A.; et al. Anastrozole, a Potent and Selective Aromatase Inhibitor, Versus Megestrol Acetate in Postmenopausal Women with Advanced Breast Cancer: Results of Overview Analysis of Two Phase III Trials. Arimidex Study Group. J. Clin. Oncol. 1996, 14, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Lassiter, L.K.; Tummala, M.K.; Hussain, M.H.; Stadler, W.M.; Petrylak, D.P.; Carducci, M.A. Phase II Open-Label Study of Oral Piritrexim in Patients with Advanced Carcinoma of the Urothelium Who Have Experienced Failure with Standard Chemotherapy. Clin. Genitourin. Cancer. 2008, 6, 31–35. [Google Scholar] [CrossRef] [PubMed]
- McGuire, K.P.; Ngoubilly, N.; Neavyn, M.; Lanza-Jacoby, S. 3,3′-Diindolylmethane and Paclitaxel Act Synergistically to Promote Apoptosis in HER2/Neu Human Breast Cancer Cells. J. Surg. Res. 2006, 132, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Wieland, H.; Schollmeyer, P.; Hörl, W.H. Beclobrate: Pharmacodynamic Properties and Therapeutic Use in Hyperlipidemia. Eur. J. Clin. Pharmacol. 1991, 40, S85–S89. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.W.; Zechnich, A.D.; Haxby, D.G. Second-Generation Antihistamines. Drugs 1999, 57, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Matsumoto, A.; Kano, T.; Maruoka, K. Cu-Catalyzed Enantioselective Alkylarylation of Vinylarenes Enabled by Chiral Binaphthyl–BOX Hybrid Ligands. J. Am. Chem. Soc. 2020, 142, 19017–19022. [Google Scholar] [CrossRef] [PubMed]
- Gulati, U.; Gandhi, R.; Laha, J.K. Benzylic Methylene Functionalizations of Diarylmethanes. Asian J. Org. 2020, 15, 3135–3161. [Google Scholar] [CrossRef] [PubMed]
- Keswani, C.; Singh, S.P.; Cueto, L.; García-Estrada, C.; Mezaache-Aichour, S.; Glare, T.R.; Borriss, R.; Singh, S.P.; Blázquez, M.A.; Sansinenea, E. Auxins of microbial origin and their use in agriculture. Appl. Microbiol. Biotechnol. 2020, 104, 8549–8565. [Google Scholar] [CrossRef]
- Duca, D.R.; Glick, B.R. Indole-3-acetic acid biosynthesis and its regulation in plant-associated bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 8607–8619. [Google Scholar] [CrossRef]
- Cheng, X.; Lu, H.; Lu, Z. Enantioselective benzylic C–H arylation via photoredox and nickel dual catalysis. Nat. Commun. 2019, 10, 3549. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zheng, P.; Xu, T. Dual Nickel- and Photoredox-Catalyzed Reductive Cross-Coupling of Aryl Halides with Dichloromethane via a Radical Process. Org. Lett. 2020, 22, 8643–8647. [Google Scholar] [CrossRef]
- Struwe, J.; Korvorapun, K.; Zangarelli, A.; Ackermann, L. Photo-Induced Ruthenium-Catalyzed C–H Benzylations and Allylations at Room Temperature. Chem. Eur. J. 2021, 27, 16237–16241. [Google Scholar] [CrossRef] [PubMed]
- Geniller, L.; Taillefer, M.; Jaroschik, F.; Prieto, A. Nickel Metallaphotoredox Catalysis Enabling Desulfurative Cross Coupling Reactions. Adv. Synth. Catal. 2022, 364, 4249–4254. [Google Scholar] [CrossRef]
- Huston, R.C.; Friedemann, T.E. Action of Aromatic Alcohols On Aromatic Compounds in The Presence of Aluminum Chloride. J. Am. Chem. Soc. 1916, 38, 2527–2533. [Google Scholar] [CrossRef]
- Huo, C.; Wang, C.; Sun, C.; Jia, X.; Wang, X.; Chang, W.; Wu, M. Triarylaminium Salt-Initiated Aerobic Double Friedel–Crafts Reaction of Glycine Derivatives with Indoles. Adv. Synth. Catal. 2013, 355, 1911–1916. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Zhou, H.; Li, S.; Zhu, Y.; Li, Y. Visible light-induced aerobic oxidative cross-coupling of glycine derivatives with indoles: A facile access to 3,3′ bisindolylmethanes. Org. Chem. Front. 2018, 5, 2120–2125. [Google Scholar] [CrossRef]
- Tanemura, K. Acceleration under solvent-drop grinding: Synthesis of bis(indolyl)methanes using small amounts of organic solvents or ionic liquids. Tetrahedron Lett. 2021, 82, 153391. [Google Scholar] [CrossRef]
- Maity, P.; Shacklady-McAtee, D.M.; Yap, G.P.A.; Sirianni, E.R.; Watson, M.P. Nickel-Catalyzed Cross Couplings of Benzylic Ammonium Salts and Boronic Acids: Stereospecific Formation of Diarylethanes via C–N Bond Activation. J. Am. Chem. Soc. 2013, 135, 280–285. [Google Scholar] [CrossRef]
- Rubial, B.; Collins, B.S.L.; Bigler, R.; Aichhorn, S.; Noble, A.; Aggarwal, V.K. Enantiospecific Synthesis of ortho-Substituted 1,1-Diarylalkanes by a 1,2-Metalate Rearrangement/anti-SN2′ Elimination/Rearomatizing Allylic Suzuki–Miyaura Reaction Sequence. Angew. Chem. Int. Ed. 2019, 58, 1366–1370. [Google Scholar] [CrossRef]
- Jiang, S.-P.; Dong, X.-Y.; Gu, Q.-S.; Ye, L.; Li, Z.-L.; Liu, X.-Y. Copper-Catalyzed Enantioconvergent Radical Suzuki–Miyaura C(sp3)–C(sp2) Cross-Coupling. J. Am. Chem. Soc. 2020, 142, 19652–19659. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Ren, Q.; Bein, T.; Knochel, P. Transition-Metal-Free Synthesis of Polyfunctional Triarylmethanes and 1,1-Diarylalkanes by Sequential Cross-Coupling of Benzal Diacetates with Organozinc Reagents. Angew. Chem. Int. Ed. 2021, 60, 10409–10414. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Wang, K.; Jin, W.-J.; Xie, H.; Qi, L.; Liu, X.-Y.; Shu, X.-Z. Dynamic Kinetic Cross-Electrophile Arylation of Benzyl Alcohols by Nickel Catalysis. J. Am. Chem. Soc. 2021, 143, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, L.; Chen, P.; Liu, G. Enantioselective Arylation of Benzylic C–H Bonds by Copper-Catalyzed Radical Relay. Angew. Chem. Int. Ed. 2019, 58, 6425–6429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, P.; Liu, G. Copper-Catalyzed Arylation of Benzylic C–H bonds with Alkylarenes as the Limiting Reagents. J. Am. Chem. Soc. 2017, 139, 7709–7712. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulos, A.; Zultanski, S.L.; Stahl, S.S. Feedstocks to Pharmacophores: Cu-Catalyzed Oxidative Arylation of Inexpensive Alkylarenes Enabling Direct Access to Diarylalkanes. J. Am. Chem. Soc. 2017, 139, 7705–7708. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Holthausen, M.H.; Mallov, I.; Pérez, M.; Qu, Z.-W.; Grimme, S.; Stephan, D.W. Catalytic Ketone Hydrodeoxygenation Mediated by Highly Electrophilic Phosphonium Cations. Angew. Chem. Int. Ed. 2015, 54, 8250–8254. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Goicoechea, J.M. Nitrenium Salts in Lewis Acid Catalysis. Angew. Chem. Int. Ed. 2020, 59, 2715–2719. [Google Scholar] [CrossRef]
- Moon, P.J.; Fahandej-Sadi, A.; Qian, W.; Lundgren, R.J. Decarboxylative Benzylation of Aryl and Alkenyl Boronic Esters. Angew. Chem. Int. Ed. 2018, 57, 4612–4616. [Google Scholar] [CrossRef]
- Guo, G.; Yuan, Y.; Bao, X.; Cao, X.; Sang, T.; Wang, J.; Huo, C. Photocatalytic Redox-Neutral Approach to Diarylmethanes. Org. Lett. 2021, 23, 6936–6940. [Google Scholar] [CrossRef]
- Chu, L.; Ohta, C.; Zuo, Z.; MacMillan, D.W.C. Carboxylic Acids as A Traceless Activation Group for Conjugate Additions: A Three-Step Synthesis of (±)-Pregabalin. J. Am. Chem. Soc. 2014, 136, 10886–10889. [Google Scholar] [CrossRef] [PubMed]
- Qvortrup, K.; Rankic, D.A.; MacMillan, D.W.C. A General Strategy for Organocatalytic Activation of C–H Bonds via Photoredox Catalysis: Direct Arylation of Benzylic Ethers. J. Am. Chem. Soc. 2014, 136, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Noble, A.; McCarver, S.J.; MacMillan, D.W.C. Merging Photoredox and Nickel Catalysis: Decarboxylative Cross-Coupling of Carboxylic Acids with Vinyl Halides. J. Am. Chem. Soc. 2015, 137, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Lipshultz, J.M.; MacMillan, D.W.C. Merging Photoredox and Nickel Catalysis: The Direct Synthesis of Ketones by the Decarboxylative Arylation of α-Oxo Acids. Angew. Chem. Int. Ed. 2015, 54, 7929–7933. [Google Scholar] [CrossRef] [PubMed]
- Cassani, C.; Bergonzini, G.; Wallentin, C.-J. Photocatalytic Decarboxylative Reduction of Carboxylic Acids and Its Application in Asymmetric Synthesis. Org. Lett. 2014, 16, 4228–4231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.-Q.; Guo, W.; Ding, W.; Wu, X.; Chen, X.; Lu, L.-Q.; Xiao, W.-J. Decarboxylative Alkynylation and Carbonylative Alkynylation of Carboxylic Acids Enabled by Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2015, 54, 11196–11199. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jia, K.; Chen, Y. Hypervalent Iodine Reagents Enable Chemoselective Deboronative/Decarboxylative Alkenylation by Photoredox Catalysis. Angew. Chem. Int. Ed. 2015, 54, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Jin, C.; Liu, J.; Zhang, H.; Yuan, M.; Li, G. Acylation of indoles via photoredox catalysis: A route to 3-acylindoles. Green Chem. 2016, 18, 1201–1205. [Google Scholar] [CrossRef]
- McNally, A.; Prier, C.K.; MacMillan, D.W.C. Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334, 1114–1117. [Google Scholar] [CrossRef]
- Wang, T.; Yu, W.; Lan, J.; Wang, H.; Jiang, Z.; Li, Y.; Fu, J. Visible-light-driven three-component reductive 1,2-diarylation of alkenes. Chem. Catal. 2023, 3, 100619. [Google Scholar] [CrossRef]
- Venditto, N.J.; Boerth, J.A. Deoxy-Arylation of Amides via a Tandem Hydrosilylation/Radical– Radical Coupling Sequence. Org. Lett. 2024, 26, 3617–3621. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Ready, J.M. Hydroesterification and Difunctionalization of Olefins with N-Hydroxyphthalimide Esters. ACS Catal. 2021, 11, 13714–13720. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Bai, Z.-Q.; Yuan, P.-F.; Wu, L.-Z.; Liu, Q. Highly Efficient Iridium-Based Photosensitizers for Thia-Paternò–Büchi Reaction and Aza-Photocyclization. ACS Catal. 2021, 11, 446–455. [Google Scholar] [CrossRef]
- Li, W.; Pohl, T.; Rost, J.M.; Rittenhouse Seth, T.; Sadeghpour, H.R.; Nipper, J.; Butscher, B.; Balewski, J.B.; Bendkowsky, V.; Löw, R.; et al. A Homonuclear Molecule with a Permanent Electric Dipole Moment. Science 2011, 334, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, A.; Zhang, J.; Linic, S. Tuning Selectivity in Propylene Epoxidation by Plasmon Mediated Photo-Switching of Cu Oxidation State. Science 2013, 339, 1590–1593. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Gong, Y.; Zhou, D. Transition Metal-Free Oxidative Radical Decarboxylation/Cyclization for the Construction of 6-Alkyl/Aryl Phenanthridines. J. Org. Chem. 2015, 80, 9336–9341. [Google Scholar] [CrossRef] [PubMed]
- Lipp, B.; Nauth, A.M.; Opatz, T. Transition-Metal-Free Decarboxylative Photoredox Coupling of Carboxylic Acids and Alcohols with Aromatic Nitriles. J. Org. Chem. 2016, 81, 6875–6882. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, P.; Wang, Y. 3-Amino-fluorene-2,4-dicarbonitriles (AFDCs) as Photocatalysts for the Decarboxylative Arylation of α-Amino Acids and α-Oxy Acids with Arylnitriles. Org. Lett. 2019, 21, 2130–2133. [Google Scholar] [CrossRef]
- Shi, J.; Yuan, T.; Zheng, M.; Wang, X. Metal-Free Heterogeneous Semiconductor for Visible-Light Photocatalytic Decarboxylation of Carboxylic Acids. ACS Catal. 2021, 11, 3040–3047. [Google Scholar] [CrossRef]
- Zuo, Z.; MacMillan, D.W.C. Decarboxylative Arylation of α-Amino Acids via Photoredox Catalysis: A One-Step Conversion of Biomass to Drug Pharmacophore. J. Am. Chem. Soc. 2014, 136, 5257–5260. [Google Scholar] [CrossRef]
- Sang, T.; Li, K.; Ouyang, X.; Song, R.; Li, J. Recent advances in the radical-mediated decyanative alkylation of cyano(hetero)arene. Green Synth. Catal. 2021, 2, 145–155. [Google Scholar]
- Roth, H.G.; Romero, N.A.; Nicewicz, D.A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. Visible-Light-Induced Chemoselective Deboronative Alkynylation under Biomolecule-Compatible Conditions. J. Am. Chem. Soc. 2014, 136, 2280–2283. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).