Effects of Central Metal Ion on Binuclear Metal Phthalocyanine-Based Redox Mediator for Lithium Carbonate Decomposition



 and
and
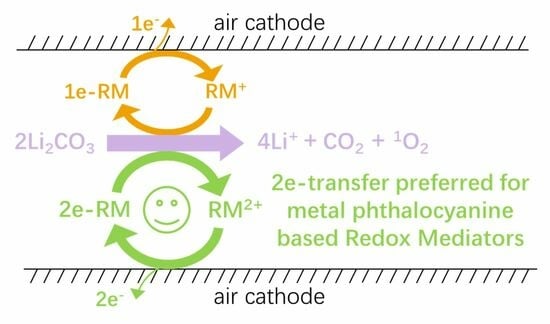
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
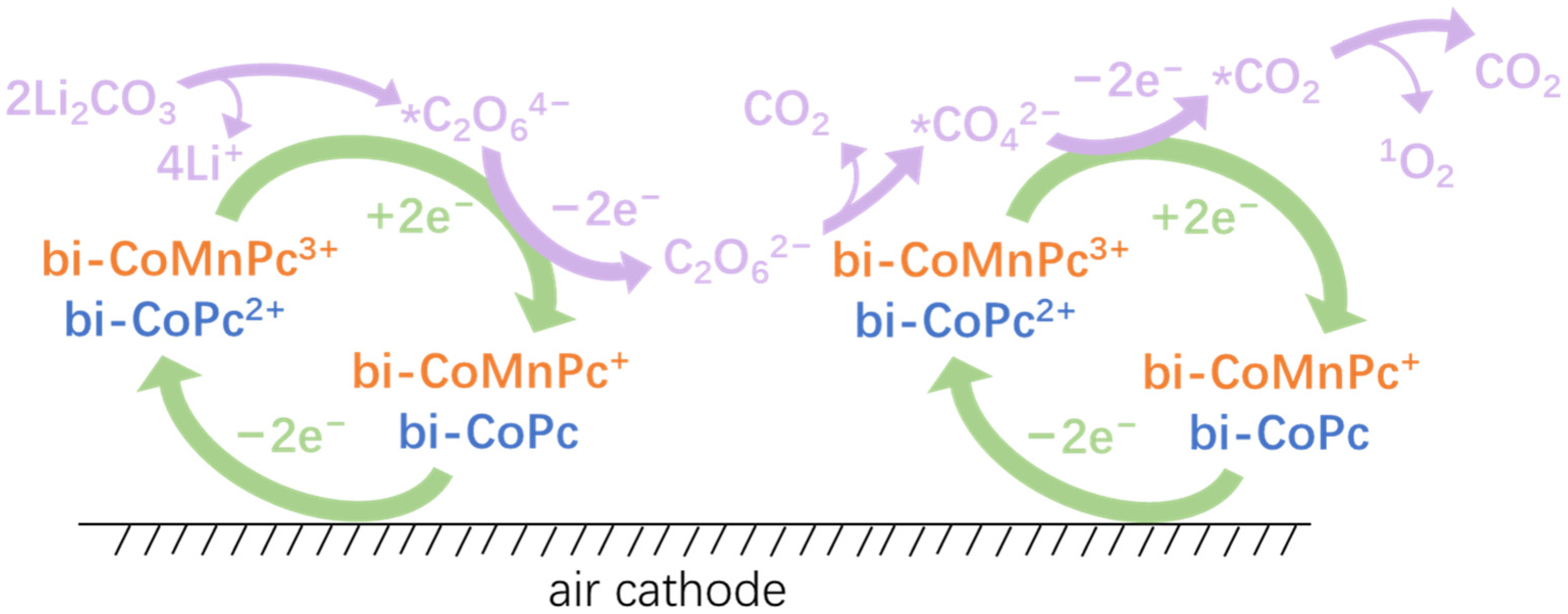
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Electrochemical Measurements
3.3. Physical Characterizations
3.4. Computation Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwak, W.-J.; Rosy; Sharon, D.; Xia, C.; Kim, H.; Johnson, L.R.; Bruce, P.G.; Nazar, L.F.; Sun, Y.-K.; Frimer, A.A.; et al. Lithium-Oxygen Batteries and Related Systems: Potential, Status, and Future. Chem. Rev. 2020, 120, 6626–6683. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Vivek, J.P.; Zhao, E.W.; Lei, J.; Garcia-Araez, N.; Grey, C.P. Current Challenges and Routes Forward for Nonaqueous Lithium–Air Batteries. Chem. Rev. 2020, 120, 6558–6625. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Yang, D.-Y.; Huang, G.; Zhang, X.-B. Lithium–Air Batteries: Air-Electrochemistry and Anode Stabilization. Acc. Chem. Res. 2021, 54, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-H.; Lee, J.; Jung, J.-W.; Park, J.; Jang, T.; Kim, H.-S.; Nam, J.-S.; Lim, H.; Yoon, K.R.; Kim, I.-D.; et al. Lithium–Air Batteries: Air-Breathing Challenges and Perspective. ACS Nano 2020, 14, 14549–14578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Huang, J.; Peng, Z. Achilles’ Heel of Lithium–Air Batteries: Lithium Carbonate. Angew. Chem. Int. Ed. 2017, 57, 3874–3886. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Jia, C.; Wan, H.; Peng, Z.; Bi, Y.; Liu, Y.; Peng, Z.; Wang, Q.; Li, H.; et al. Decomposing lithium carbonate with a mobile catalyst. Nano Energy 2017, 36, 390–397. [Google Scholar] [CrossRef]
- Wang, X.-G.; Wang, C.; Xie, Z.; Zhang, X.; Chen, Y.; Wu, D.; Zhou, Z. Improving Electrochemical Performances of Rechargeable Li−CO2 Batteries with an Electrolyte Redox Mediator. ChemElectroChem 2017, 4, 2145–2149. [Google Scholar] [CrossRef]
- Mota, F.M.; Kang, J.-H.; Jung, Y.; Park, J.; Na, M.; Kim, D.H. Mechanistic Study Revealing the Role of the Br3-/Br2 Redox Couple in CO2-Assisted Li–O2 Batteries. Adv. Energy Mater. 2020, 10, 1903486. [Google Scholar] [CrossRef]
- Chen, J.; Zou, K.; Ding, P.; Deng, J.; Zha, C.; Hu, Y.; Zhao, X.; Wu, J.; Fan, J.; Li, Y. Conjugated Cobalt Polyphthalocyanine as the Elastic and Reprocessable Catalyst for Flexible Li–CO2 Batteries. Adv. Mater. 2019, 31, 1805484. [Google Scholar] [CrossRef]
- Huang, S.; Li, Z.; Liu, Z.; Yan, Q.; Ma, B.; Wang, D.; Wei, F.; Chen, Z.; He, H. Surface Enrichment of Redox Mediator for Long-Cyclable Lithium−Air Batteries. Energy Fuels 2023, 37, 11465–11471. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, Y.; Shuai, L.; Tang, Y.; Qiu, M.; Xie, J.; Liu, J.; Wen, W.; Chen, H.; Nan, S.; et al. Remarkable improvement of cyclic stability in Li–O2 batteries using ruthenocene as a redox mediator. Chin. Chem. Lett. 2020, 31, 1997–2002. [Google Scholar] [CrossRef]
- Wan, H.; Sun, Y.; Yu, J.; Shi, Q.; Zhu, Y.; Qian, Y. A chiral salen-Co(II) complex as soluble redox mediator for promoting the electrochemical performance of Li-O2 batteries. Nano Res. 2022, 15, 8101–8108. [Google Scholar] [CrossRef]
- Ko, S.; Yoo, Y.; Choi, J.; Lim, H.-D.; Park, C.B.; Lee, M. Discovery of organic catalysts boosting lithium carbonate decomposition toward ambient air operational lithium–air batteries. J. Mater. Chem. A 2022, 10, 20464. [Google Scholar] [CrossRef]
- Pipes, R.; Bhargav, A.; Manthiram, A. Phenyl Disulfide Additive for Solution-Mediated Carbon Dioxide Utilization in Li–CO2 Batteries. Adv. Energy Mater. 2019, 9, 1900453. [Google Scholar] [CrossRef]
- Xiong, Q.; Huang, G.; Zhang, X.-B. High-Capacity and Stable Li-O2 Batteries Enabled by a Trifunctional Soluble Redox Mediator. Angew. Chem. Int. Ed. 2020, 59, 19311–19319. [Google Scholar] [CrossRef]
- Jiang, F.; Ma, L.; Sun, J.; Guo, L.; Peng, Z.; Cui, Z.; Li, Y.; Guo, X.; Zhang, T. Deciphering the Enigma of Li2CO3 Oxidation Using a Solid-State Li–Air Battery Configuration. ACS Appl. Mater. Interfaces 2021, 13, 14321–14326. [Google Scholar] [CrossRef]
- Yang, S.; He, P.; Zhou, H. Exploring the electrochemical reaction mechanism of carbonate oxidation in Li–air/CO2 battery through tracing missing oxygen. Energy Environ. Sci. 2016, 9, 1650–1654. [Google Scholar] [CrossRef]
- Mahne, N.; Renfrew, S.E.; McCloskey, B.D.; Freunberger, S.A. Electrochemical Oxidation of Lithium Carbonate Generates Singlet Oxygen. Angew. Chem. Int. Ed. 2018, 57, 5529–5533. [Google Scholar] [CrossRef]
- Cao, D.; Tan, C.; Chen, Y. Oxidative decomposition mechanisms of lithium carbonate on carbon substrates in lithium battery chemistries. Nat. Commun. 2022, 13, 4908. [Google Scholar] [CrossRef]
- Lever, A.B.P.; Minor, P.C.; Wilshire, J.P. Electrochemistry of Manganese Phthalocyanine in Nonaqueous Media. Inorg. Chem. 1981, 20, 2550–2553. [Google Scholar] [CrossRef]
- Lin, C.-L.; Lee, C.-C.; Ho, K.-C. Spectroelectrochemical studies of manganese phthalocyanine thin films for applications in electrochromic devices. J. Electroanal. Chem. 2002, 524−525, 81–89. [Google Scholar] [CrossRef]
- Kobayashi, N.; Lam, H.; Nevin, W.A.; Janda, P.; Leznoff, C.C.; Koyama, T.; Monden, A.; Shirai, H. Synthesis, spectroscopy, electrochemistry, spectroelectrochemistry, Langmuir-Blodgett film formation, and molecular orbital calculations of planar binuclear phthalocyanines. J. Am. Chem. Soc. 1994, 116, 879–890. [Google Scholar] [CrossRef]
- Lim, H.-D.; Lee, B.; Zheng, Y.; Hong, J.; Kim, J.; Gwon, H.; Ko, Y.; Lee, M.; Cho, K.; Kang, K. Rational design of redox mediators for advanced Li–O2 batteries. Nat. Energy 2016, 1, 16066. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, X.; Johnson, L.R.; Bruce, P.G. Kinetics of lithium peroxide oxidation by redox mediators and consequences for the lithium–oxygen cell. Nat. Commun. 2018, 9, 767. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Zor, C.; Yang, S.; Lagnoni, M.; Dewar, D.; Nimmo, T.; Chau, C.; Jenkins, M.; Kibler, A.J.; Pateman, A.; et al. Why charging Li–air batteries with current low-voltage mediators is slow and singlet oxygen does not explain degradation. Nat. Chem. 2023, 15, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Yi, J.; Guo, S.; Sun, Y.; Wu, S.; Liu, X.; Yang, S.; He, P.; Zhou, H. Li2CO3-free Li–O2/CO2 battery with peroxide discharge product. Energy Environ. Sci. 2018, 11, 1211–1217. [Google Scholar] [CrossRef]
- Kim, B.; Shin, K.; Henkelman, G.; Ryu, W.-H. CO2-mediated porphyrin catalysis in reversible Li-CO2 cells. Chem. Eng. J. 2023, 477, 147141. [Google Scholar] [CrossRef]
- Lian, Z.; Lu, Y.; Ma, S.; Wang, L.; Li, Z.; Liu, Q. An integrated strategy for upgrading Li-CO2 batteries: Redox mediator and separator modification. Chem. Eng. J. 2022, 450, 138400. [Google Scholar] [CrossRef]
- Wang, L.; Lu, Y.; Ma, S.; Lian, Z.; Gu, X.; Li, J.; Li, Z.; Liu, Q. Optimizing CO2 reduction and evolution reaction mediated by o-phenylenediamine toward high performance Li-CO2 battery. Electrochim. Acta 2022, 419, 140424. [Google Scholar] [CrossRef]
- Cao, D.; Liu, X.; Yuan, X.; Yu, F.; Chen, Y. Redox Mediator-Enhanced Performance and Generation of Singlet Oxygen in Li−CO2 Batteries. ACS Appl. Mater. Interfaces 2021, 13, 39341–39346. [Google Scholar] [CrossRef]
- Sun, D.; Shen, Y.; Zhang, W.; Yu, L.; Yi, Z.; Yin, W.; Wang, D.; Huang, Y.; Wang, J.; Wang, D.; et al. A Solution-Phase Bifunctional Catalyst for Lithium–Oxygen Batteries. J. Am. Chem. Soc. 2014, 136, 8941–8946. [Google Scholar] [CrossRef] [PubMed]
- Ottakam Thotiyl, M.M.; Freunberger, S.A.; Peng, Z.; Bruce, P.G. The Carbon Electrode in Nonaqueous Li–O2 Cells. J. Am. Chem. Soc. 2013, 135, 494–500. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Lu, T. Gau_Xtb: A Gaussian Interface for Xtb Code. Available online: https://sobereva.com/soft/gau_xtb (accessed on 15 August 2023).
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Weinhold, F.J. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J. Mol. Struct. Theochem. 1988, 46, 41–62. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F.J. Natural Atomic Orbitals and Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F.J. Natural Localized Molecular Orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F.J. Natural Bond Orbital Analysis of Near-Hartree-Fock Water Dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F.J. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Q.; Yan, L.; Huang, H.; Chen, Z.; Liu, Z.; Zhou, S.; He, H. Effects of Central Metal Ion on Binuclear Metal Phthalocyanine-Based Redox Mediator for Lithium Carbonate Decomposition. Molecules 2024, 29, 2034. https://doi.org/10.3390/molecules29092034
Yan Q, Yan L, Huang H, Chen Z, Liu Z, Zhou S, He H. Effects of Central Metal Ion on Binuclear Metal Phthalocyanine-Based Redox Mediator for Lithium Carbonate Decomposition. Molecules. 2024; 29(9):2034. https://doi.org/10.3390/molecules29092034
Chicago/Turabian StyleYan, Qinghui, Linghui Yan, Haoshen Huang, Zhengfei Chen, Zixuan Liu, Shaodong Zhou, and Haiyong He. 2024. "Effects of Central Metal Ion on Binuclear Metal Phthalocyanine-Based Redox Mediator for Lithium Carbonate Decomposition" Molecules 29, no. 9: 2034. https://doi.org/10.3390/molecules29092034
APA StyleYan, Q., Yan, L., Huang, H., Chen, Z., Liu, Z., Zhou, S., & He, H. (2024). Effects of Central Metal Ion on Binuclear Metal Phthalocyanine-Based Redox Mediator for Lithium Carbonate Decomposition. Molecules, 29(9), 2034. https://doi.org/10.3390/molecules29092034