
Novel Bicyclic P,S-Heterocycles via Stereoselective hetero-Diels–Alder Reactions of Thiochalcones with 1-Phenyl-4H-phosphinin-4-one 1-Oxide †

 ,
,  , , and
, , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
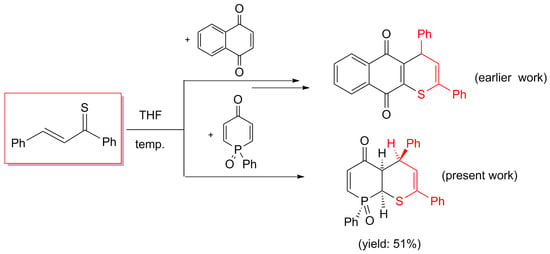
2. Results and Discussion
3. Materials and Methods
3.1. Experimental: X-ray Structure Determination of 13a and 13b
3.2. General Information
- rac-RS,8SR,9SR,10SR)-2,8-Diphenyl-4-(thiophen-2-yl)-4,4a,8a-trihydrophosphinino [2,3-b]thiopyran-5-one 8-oxide (13a). Yield = 250 mg (57%), pale yellow crystals, mp = 184 °C (decomp.) (petroleum ether/CH2Cl2).
- rac-(RS,8SR,9SR,10SR)-2,4,8-Triphenyl-4,4a,8a-trihydrophosphinino[2,3-b]thiopyran-5-one 8-oxide (13b). Yield: 220 mg (51%), beige crystals, mp = 212 °C (decomp.) (petroleum ether/CH2Cl2).
- rac-(RS,8SR,9SR,10SR)-2,8-Diphenyl-4-(4-methylphenyl)-4,4a,8a-trihydrophosphinino[2,3-b]thiopyran-5-one 8-oxide (13c). Yield: 245 mg (51%), cream-colored crystals, mp = 201 °C (decomp.) (petroleum ether/CH2Cl2).
- rac-(RS,8SR,9SR,10SR)-4-(4-Bromophenyl)-2,8-diphenyl-4,4a,8a-trihydrophosphinino[2,3-b]thiopyran-5-one 8-oxide (13d). Yield: 310 mg (61%), yellow crystals, mp = 178 °C (decomp.) (petroleum ether/CH2Cl2).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huisgen, R.; Fisera, L.; Giera, H.; Sustmann, R. Thiones as Superdipolarophiles. Rates and Equilibria of Nitrone Cycloadditions to Thioketones. J. Am. Chem. Soc. 1995, 117, 9671–9678. [Google Scholar] [CrossRef]
- Fisera, L.; Huisgen, R.; Kalwinsch, I.; Langhals, E.; Li, X.; Mlostoń, G.; Polborn, K.; Rapp, J.; Sicking, W.; Sustmann, R. New thione chemistry. Pure Appl. Chem. 1996, 68, 789–798. [Google Scholar] [CrossRef]
- Huisgen, R.; Li, X.; Giera, H.; Langhals, E. ‘Thiobenzophenone S-Methylide’ (=(Diphenylmethylidenesulfonio)methanide), and C,C Multiple Bonds: Cycloadditions and Dipolarophilic Reactivities. Helv. Chim. Acta 2001, 84, 981–999. [Google Scholar] [CrossRef]
- Breu, J.; Höcht, P.; Rohr, U.; Schatz, J.; Sauer, J. Thiocarbonyl Compounds in [4+2] Cycloadditions: Preparative Aspects. Eur. J. Org. Chem. 1998, 861–2873. [Google Scholar] [CrossRef]
- Rohr, U.; Schatz, J.; Sauer, J. Thio- and Selenocarbonyl Compounds as “Superdienophiles” in [4+2] Cycloadditions. Eur. J. Org. Chem. 1998, 2875–2883. [Google Scholar] [CrossRef]
- Karakasa, T.; Motoki, S. Chemistry of α,β-Unsaturated Thione Dimers. 1. Preparation of α,β-Unsaturated Thione Dimers and Thermolysis of These Dimers in the Presence of Acrylonitrile or Acrylamide. J. Org. Chem. 1978, 43, 4147–4150. [Google Scholar] [CrossRef]
- Karakasa, T.; Motoki, S. Chemistry of α,β-Unsaturated Thione Dimers. 2. Reactions of Thiochalcones and 2-Arylidene-1-thiotetralones with Some Olefins and the Parent Ketones of the Thiones. J. Org. Chem. 1979, 44, 4151–4155. [Google Scholar] [CrossRef]
- Motoki, S.; Saito, T.; Karakasa, T.; Kato, H.; Matsushita, T.; Hayashibe, S. Thermal and Lewis Acid-promoted Novel Asymmetric Hetero Diels-Alder Reactions of a 1-Thiabuta-1,3-diene System (Thiochalcones) with (–)-Dimenthyl fumarate. J. Chem. Soc. Perkin Trans. 1 1991, 2281–2283. [Google Scholar] [CrossRef]
- Mlostoń, G.; Grzelak, P.; Utecht, G.; Jasiński, M. First (3+2)-Cycloadditions of Thiochalcones as C=S Dipolarophiles: Efficient Synthesis of 1,3,4-Thiadiazoles via Reactions with Fluorinated Nitrile Imines. Synthesis 2017, 49, 2129–2137. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Sobiecka, M.; Heimgartner, H.; Würthwein, E.-U.; Zimmer, R.; Lentz, D.; Reissig, H.-U. Hetero-Diels-Alder Reactions of In Situ-Generated Azoalkenes with Thioketones; Experimental and Theoretical Studies. Molecules 2021, 26, e2544. [Google Scholar] [CrossRef]
- Hejmanowska, J.; Jasiński, M.; Mlostoń, G.; Albrecht, Ł. Taming of Thioketones: The First Asymmetric Thia-Diels-Alder reaction with Thioketones as Heterodienophiles. Eur. J. Org. Chem. 2017, 950–954. [Google Scholar] [CrossRef]
- Hejmanowska, J.; Jasiński, M.; Wojciechowski, J.; Mlostoń, G.; Albrecht, Ł. The first organocatalytic, ortho-regioselective inverse-electron-demand hetero-Diels-Alder reaction. Chem. Commun. 2017, 53, 11472–11475. [Google Scholar] [CrossRef] [PubMed]
- Mlostoń, G.; Grzelak, P.; Heimgartner, H. Hetero-Diels-Alder reactions of hetaryl thiochalcones with acetylenic dienophiles. J. Sulfur Chem. 2017, 38, 1–10. [Google Scholar] [CrossRef][Green Version]
- Mlostoń, G.; Hamera-Fałdyga, R.; Heimgartner, H. First synthesis of ferrocenyl-substituted thiochalcones and their [4 + 2]-cycloadditions with acetylenic dipolarophiles. J. Sulfur Chem. 2018, 39, 322–331. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Urbaniak, P.; Marko, A.; Linden, A.; Heimgartner, H. First thia-Diels-Alder reactions of thiochalcones with 1,4-quinones. Beilstein J. Org. Chem. 2018, 14, 1834–1839. [Google Scholar] [CrossRef]
- Märkl, G.; Olbricht, H. Derivates of Phospha-2,5-cyclohexadien-4-one and 4,4′-Diphosphabi-(2,5-cyclohexadienylidene). Angew. Chem. Int. Ed. 1966, 5, 588–589. [Google Scholar] [CrossRef]
- Łastawiecka, E.; Frynas, S.; Pietrusiewicz, K.M. Desymmetrization Approach to the Synthesis of Optically Active P-Stereogenic Phosphin-2-en-4-ones. J. Org. Chem. 2021, 86, 6195–6206. [Google Scholar] [CrossRef] [PubMed]
- Łastawiecka, E.; Włodarczyk, A.; Kozioł, A.E.; Małuszyńska, H.; Pietrusiewicz, K.M. Resolution of P-Stereogenic 1-Phenylphosphin-2-en-4-one 1-Oxide into Two Enantiomers by (R,R)-TADDOL and Conformational Diversity of the Phosphinenone Ring and TADDOL in the Crystal State. Molecules, 2021; 26, 6873. [Google Scholar] [CrossRef]
- Kashman, Y.; Awerbouch, O. Phosphasteroids-I. Cycloaddition reactions of phospholenes with various dienes. Tetrahedron Lett. 1973, 14, 3217–3220. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Koprowski, M.; Drzazga, Z.; Parcheta, R.; Łastawiecka, E.; Demchuk, O.M.; Justyniak, I. Efficient Oxidative Resolution of 1-Phenylphosphol-2-ene and Diels-Alder Synthesis of Enantiopure Bicyclic and Tricyclic P-Stereogenic C-P Heterocycles. Symmetry 2020, 12, 346. [Google Scholar] [CrossRef]
- Goti, A.; Cicchi, S.; Brandi, A.; Pietrusiewicz, K.M. Nitrone Cycloadditions to 2,3-Dihydro-1-phenyl-1H-phosphole 1-Oxide. Double Asymmetric Reaction and Kinetic Resolution by a Chiral Nitrone. Tetrahedron: Asymmetry 1991, 2, 1371–1378. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Hołody, W.; Koprowski, M.; Cicchi, S.; Goti, A.; Brandi, A. Asymmetric and Doubly Asymmetric 1,3-Dipolar Cycloadditions in the Synthesis of Enantiopure Organophosphorus Compounds. Phosphorus Sulfur Silicon Relat. Elem. 1999, 144, 389–392. [Google Scholar] [CrossRef]
- Märkl, G.; Potthast, R. l-Phenyl-phosphol. Tetrahedron Lett. 1968, 9, 1755–1758. [Google Scholar] [CrossRef]
- Quin, L.D.; Mesch, K.A.; Bodalski, R.; Pietrusiewicz, K.M. 13C NMR spectra of phosphole–P(IV) dimers; ABX and AA′X 13C–31P coupling in some derived structures. Org. Magn. Reson. 1982, 20, 83–91. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, J.; Körtvélyesi, T.; Parlagh, G.; Imre, T.; Ludányi, K.; Hegedűs, L.; Hanusz, M.; Simon, K.; Márton, A.; et al. Novel 2-phosphabicyclo [2.2.2]oct-5-ene derivatives and their use in phosphinylations. Heteroat. Chem. 2004, 15, 97–106. [Google Scholar] [CrossRef]
- Allen, F.H.; Baalham, C.A.; Lommerse, J.P.M.; Raithby, P.R. Carbonyl–Carbonyl Interactions can be Competitive with Hydrogen Bonds. Acta Cryst. 1998, B54, 320–329. [Google Scholar] [CrossRef]
- Breugst, M.; Reissig, H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef] [PubMed]
- Jasiński, R.; Dresler, E. On the Question of Zwitterionic Intermediates in the [3+2] Cycloaddition Reactions: A Critical Review. Organics 2020, 1, 49–69. [Google Scholar] [CrossRef]
- Houk, K.N.; Liu, F.; Yang, Z.; Seeman, J.I. Evolution of the Diels-Alder Reaction Mechanism since the 1930s: Woodward, Houk with Woodward, and the Influence of Computational Chemistry on Understanding Cycloadditions. Angew. Chem. Int. Ed. 2021, 60, 12660–12681. [Google Scholar] [CrossRef]
- Firestone, R.A. The Low Energy of Concert in Many Symmetry-Allowed Cycloadditions Supports a Stepwise-Diradical Mechanism. Int. J. Chem. Kinet. 2013, 45, 415–428. [Google Scholar] [CrossRef]
- Sustmann, R.; Tappanchai, S.; Bandmann, H. (E)-1-Methoxy-1,3-butadiene and 1,1-Dimethoxy-1,3-butadiene in (4 + 2) Cycloadditions. A Mechanistic Comparison. J. Am. Chem. Soc. 1996, 118, 12555–12561. [Google Scholar] [CrossRef]
- CrysAlisPRO Software System; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2015.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Emerson, W.S.; Patrick, R.T.M. The preparation of 2-thiophenealdehyde and some of its derivatives. J. Org. Chem. 1949, 14, 790–797. [Google Scholar] [CrossRef]
- Hayat, F.; Salahuddin, A.; Umar, S.; Azam, A. Synthesis, characterization, antiamoebic activity and cytotoxicity of novel series of pyrazoline derivatives bearing quinoline tail. Eur. J. Med. Chem. 2010, 45, 4669–4675. [Google Scholar] [CrossRef]
- Urbaniak, K.; Mlostoń, G.; Gulea, M.; Masson, S.; Linden, A.; Heimgartner, H. Thio- and Dithioesters as Dipolarophiles in Reactions with Thiocarbonyl Ylides. Eur. J. Org. Chem. 2005, 1604–1612. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Gulea, M.; Masson, S.; Linden, A.; Heimgartner, H. [2+3]-Cycloadditions of Phosphonodithioformate S-Methanides with C=S, N=N, and C=C Dipolarophiles. Helv. Chim. Acta 2005, 88, 2582–2592. [Google Scholar] [CrossRef]
- Munasinghe, D.S.; Kasper, M.-A.; Jasiński, R.; Kula, K.; Palusiak, M.; Celeda, M.; Mlostoń, G.; Hackenberger, C.P.R. (3+2)-Cyclization Reactions of Unsaturated Phosphonites with Aldehydes and Thioketones. Chem. Eur. J. 2023, 29, e202300806. [Google Scholar] [CrossRef]
- McReynolds, M.D.; Dougherty, J.M.; Hanson, P.R. Synthesis of Phosphorus and Sulfur Heterocycles via Ring-Closing Olefin Metathesis. Chem. Rev. 2004, 104, 2239–2258. [Google Scholar] [CrossRef]
- Jun, J.H.; Javed, S.; Ndi, C.N.; Hanson, P.R. Synthesis of P-, S-, Si-, B-, and Se-Heterocycles via Ring-Closing Metathesis. In Synthesis of Heterocycles by Metathesis Reactions. Topics in Heterocyclic Chemistry; Springer: Cham, Switzerland, 2017; Volume 47, pp. 319–379. [Google Scholar] [CrossRef]
- Laxmikeshav, K.; Kumari, P.; Shankaraiah, N. Expedition of sulfur-containing heterocyclic derivatives as cytotoxic agents in medicinal chemistry: A decade update. Med. Res. Rev. 2022, 42, 513–575. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Smaligo, A.J.; Song, X.-R.; Kwon, O. Phosphorus-Based Catalysis. ACS Cent. Sci. 2021, 7, 536–558. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, D.; Van Elslande, E.; Masson, G. Isothiourea-Catalyzed Enantioselective Synthesis of trans-3,4-Dihydrothiopyranones: Harnessing Thiochalcones as Original Michael Acceptors. Org. Lett. 2023, 25, 5395–5399. [Google Scholar] [CrossRef] [PubMed]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlostoń, G.; Urbaniak, K.; Palusiak, M.; Łastawiecka, E.; Frynas, S.; Pietrusiewicz, K.M.; Heimgartner, H. Novel Bicyclic P,S-Heterocycles via Stereoselective hetero-Diels–Alder Reactions of Thiochalcones with 1-Phenyl-4H-phosphinin-4-one 1-Oxide. Molecules 2024, 29, 2036. https://doi.org/10.3390/molecules29092036
Mlostoń G, Urbaniak K, Palusiak M, Łastawiecka E, Frynas S, Pietrusiewicz KM, Heimgartner H. Novel Bicyclic P,S-Heterocycles via Stereoselective hetero-Diels–Alder Reactions of Thiochalcones with 1-Phenyl-4H-phosphinin-4-one 1-Oxide. Molecules. 2024; 29(9):2036. https://doi.org/10.3390/molecules29092036
Chicago/Turabian StyleMlostoń, Grzegorz, Katarzyna Urbaniak, Marcin Palusiak, Elżbieta Łastawiecka, Sławomir Frynas, Kazimierz Michał Pietrusiewicz, and Heinz Heimgartner. 2024. "Novel Bicyclic P,S-Heterocycles via Stereoselective hetero-Diels–Alder Reactions of Thiochalcones with 1-Phenyl-4H-phosphinin-4-one 1-Oxide" Molecules 29, no. 9: 2036. https://doi.org/10.3390/molecules29092036
APA StyleMlostoń, G., Urbaniak, K., Palusiak, M., Łastawiecka, E., Frynas, S., Pietrusiewicz, K. M., & Heimgartner, H. (2024). Novel Bicyclic P,S-Heterocycles via Stereoselective hetero-Diels–Alder Reactions of Thiochalcones with 1-Phenyl-4H-phosphinin-4-one 1-Oxide. Molecules, 29(9), 2036. https://doi.org/10.3390/molecules29092036