

Ensemble-Based Virtual Screening Led to the Discovery of Novel Lead Molecules as Potential NMBAs

Abstract
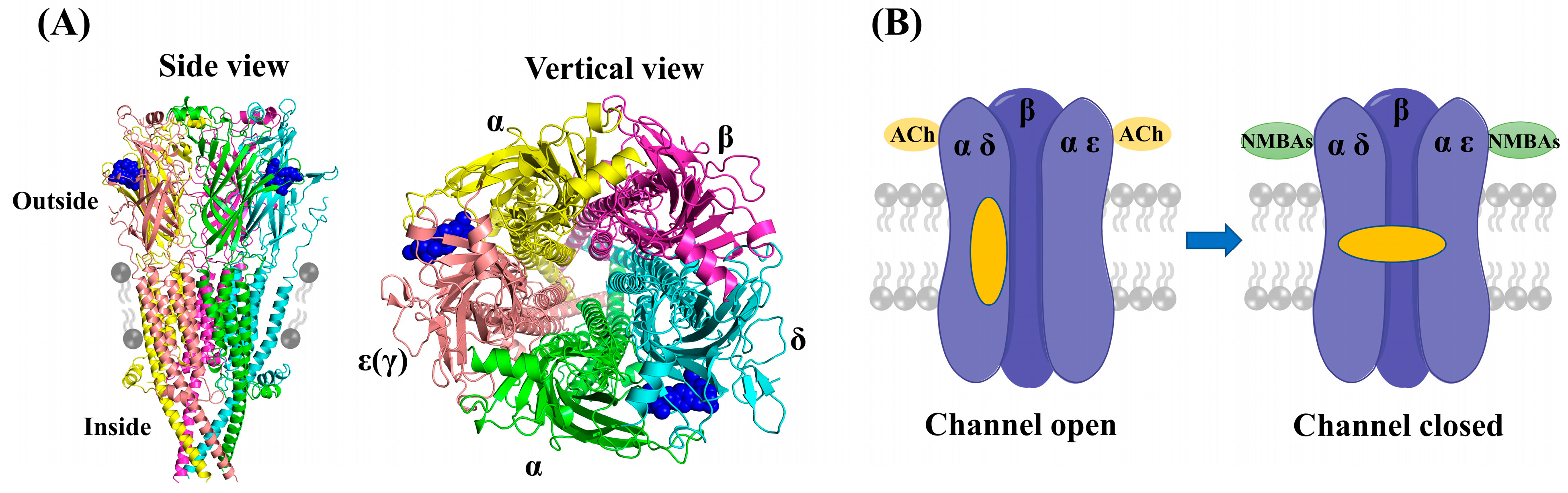
1. Introduction
2. Results
2.1. Molecular Property Filters
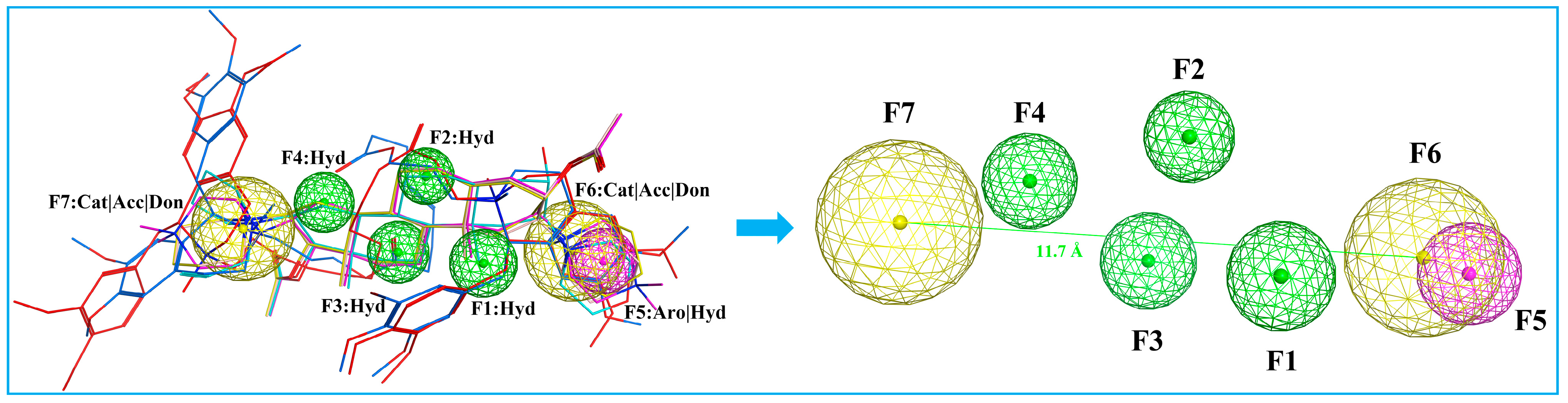
2.2. Pharmacophore Modelling
2.3. Molecular Docking
2.4. Molecular Dynamic Simulation
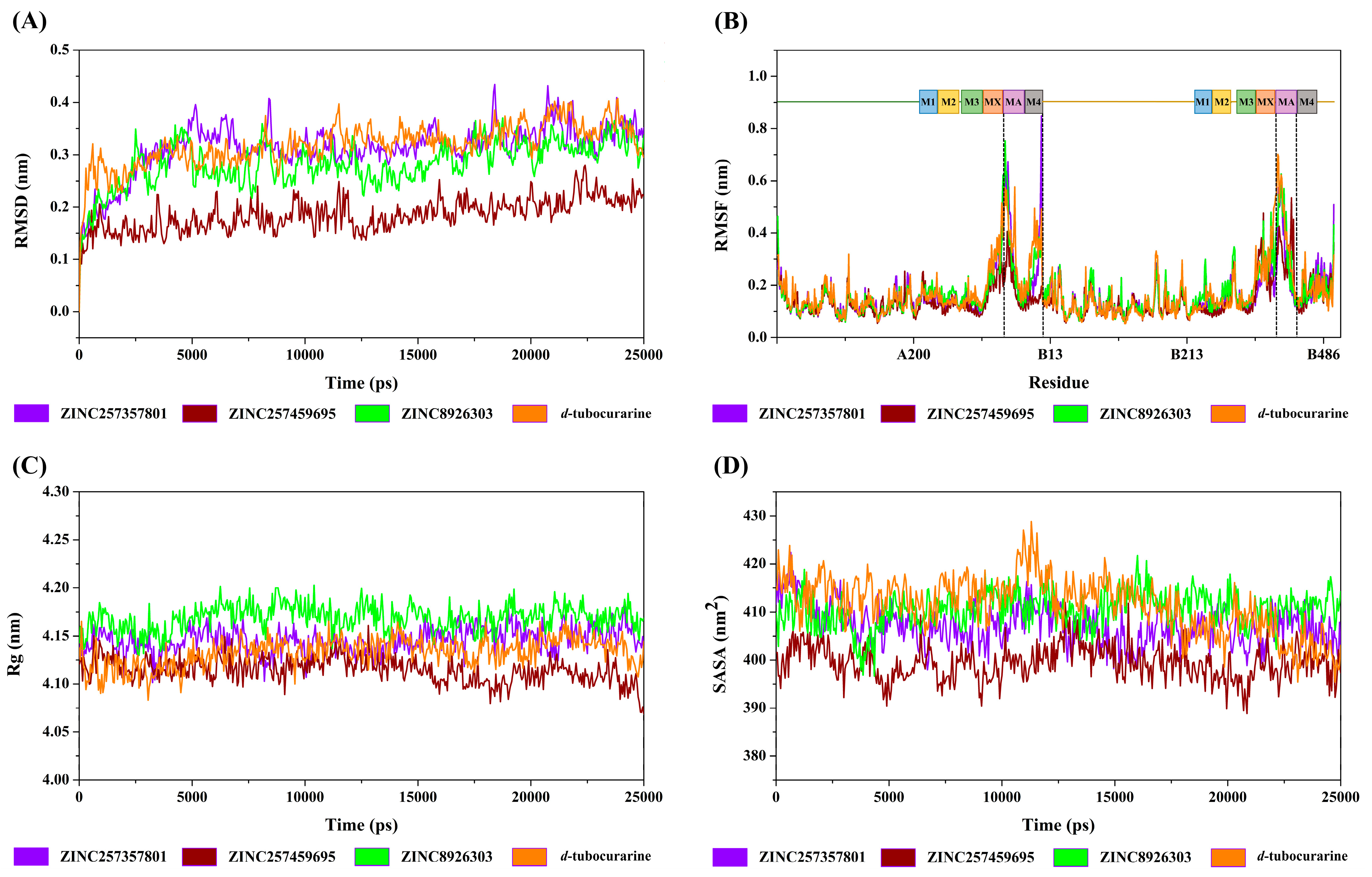
2.4.1. Structural Deviation and Compactness Analysis
2.4.2. Hydrogen Bond Analysis
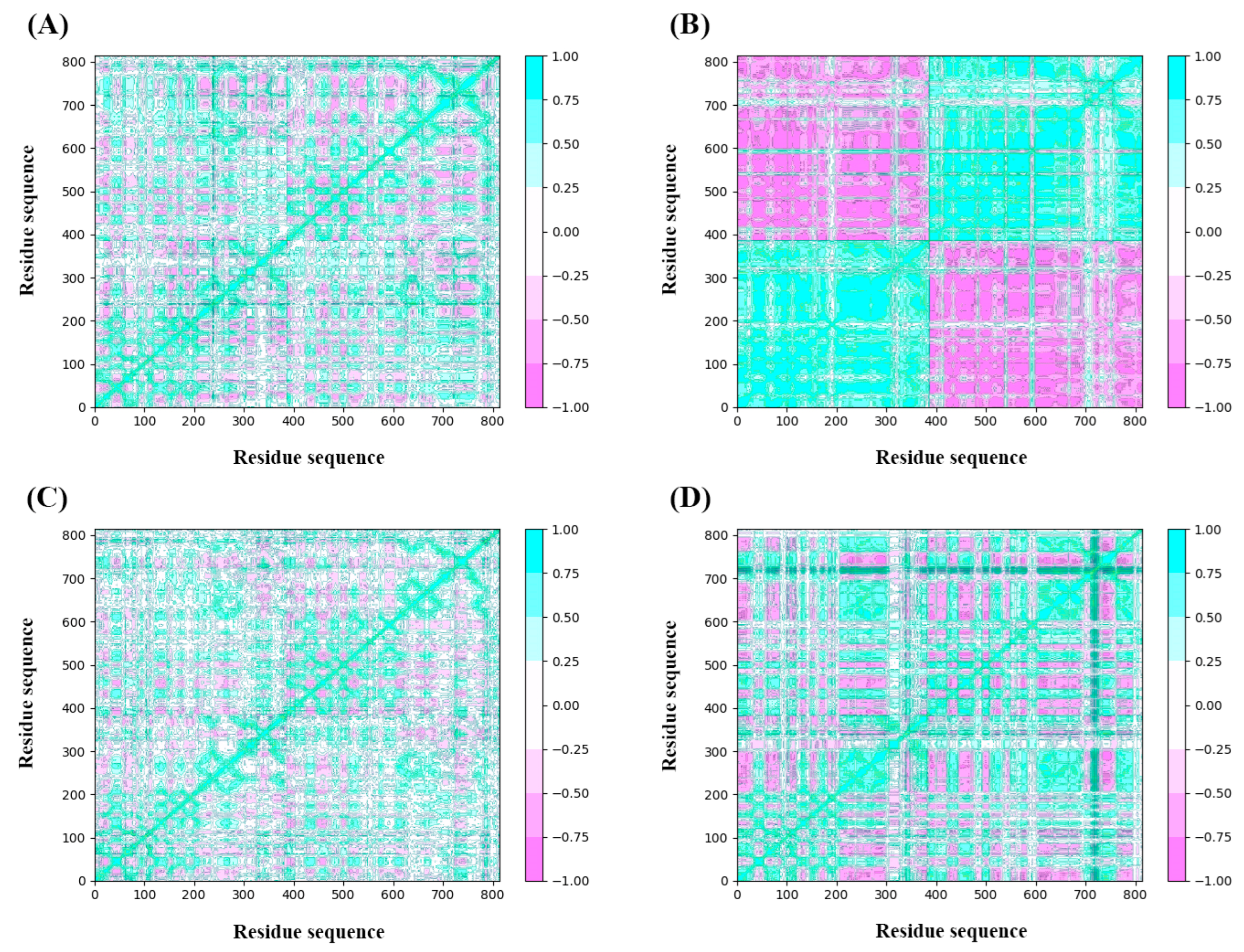
2.4.3. Dynamic Cross-Correlation Map Analysis
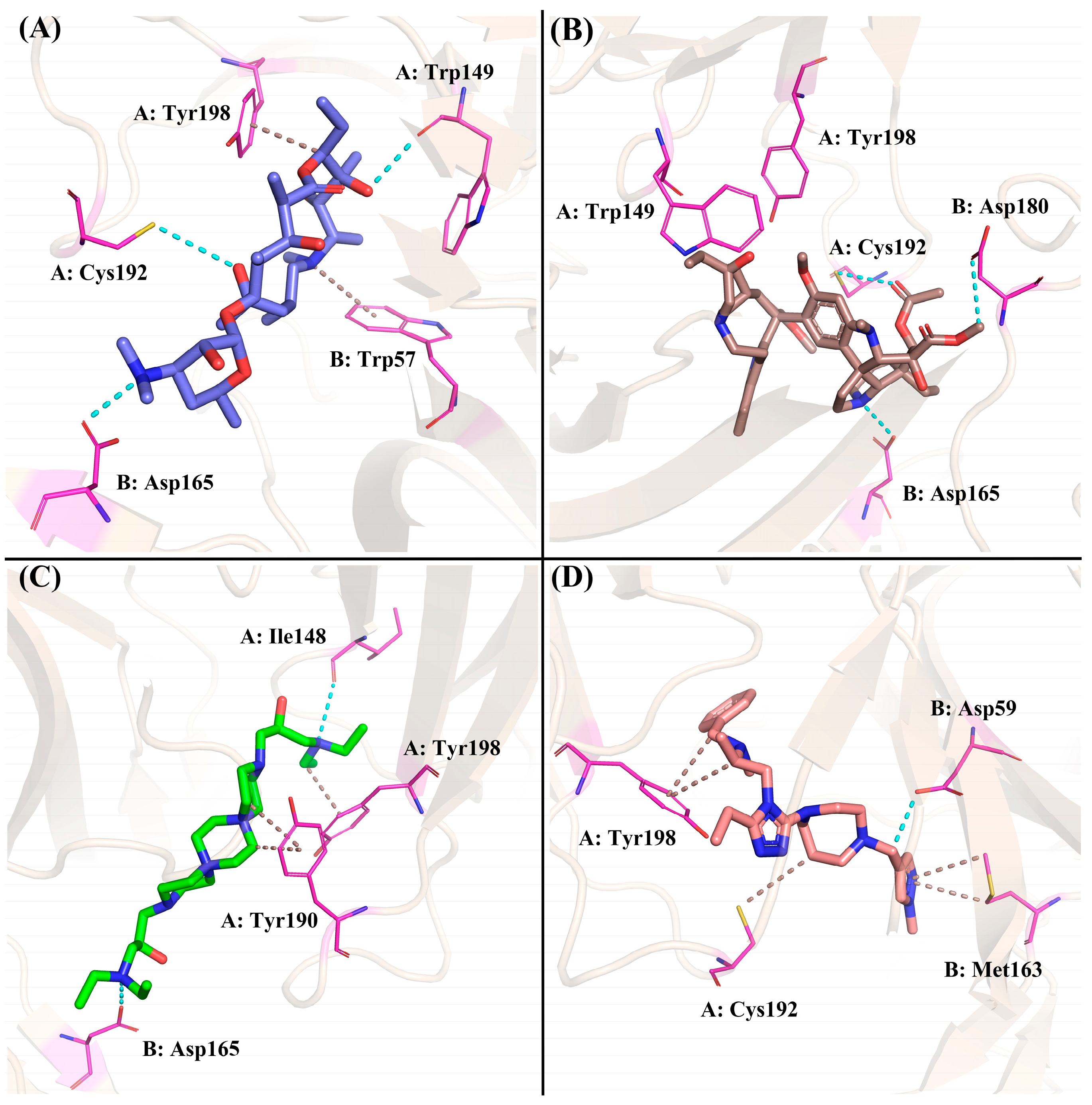
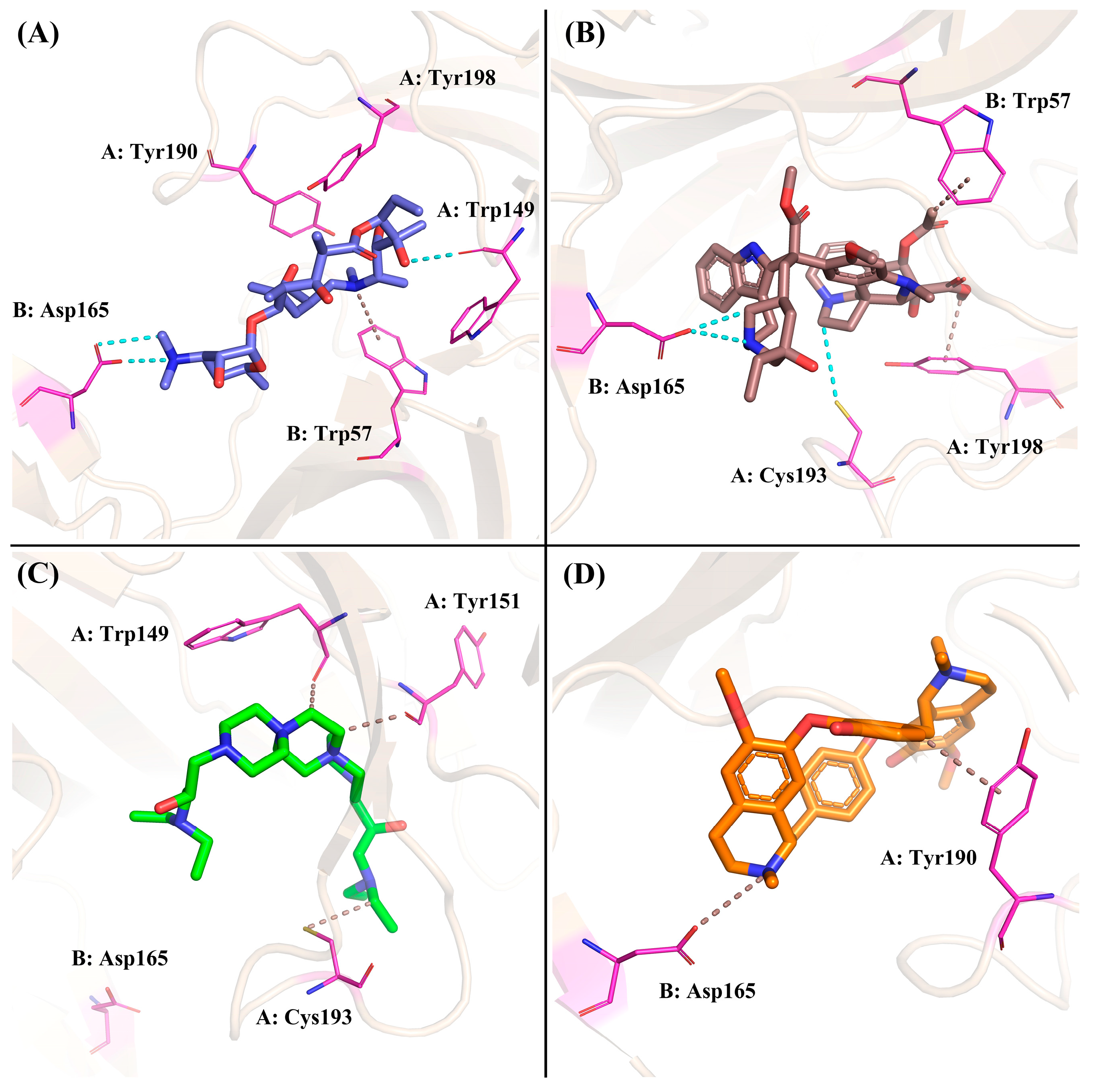
2.4.4. The Binding Modes Refined through the MD Simulations
2.4.5. Binding Free Energy Calculation by MM/GBSA Method
2.5. In Silico Pharmacokinetic Profile (ADMET)
3. Materials and Methods
3.1. Ligand-Based Pharmacophore Generation
3.2. Molecular Docking
3.3. Molecular Dynamics Simulation
3.4. Binding Free Energy Calculation
3.5. ADMET Property Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stäuble, C.G.; Blobner, M. The future of neuromuscular blocking agents. Curr. Opin. Anaesthesiol. 2020, 33, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Tuba, Z.; Maho, S.; Vizi, E.S. Synthesis and structure-activity relationships of neuromuscular blocking agents. Curr. Med. Chem. 2002, 9, 1507–1536. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Sung, T.Y.; Yang, H.S. Factors that affect the onset of action of non-depolarizing neuromuscular blocking agents. Korean J. Anesthesiol. 2017, 70, 500–510. [Google Scholar] [CrossRef]
- Tran, D.T.; Newton, E.K.; Mount, V.A.; Lee, J.S.; Wells, G.A.; Perry, J.J. Rocuronium versus succinylcholine for rapid sequence induction intubation. Cochrane Database Syst. Rev. 2015, Cd002788. [Google Scholar] [CrossRef] [PubMed]
- Selinger, A.J.; Cavallin, N.A.; Yanai, A.; Birol, I.; Hof, F. Template-Directed Synthesis of Bivalent, Broad-Spectrum Hosts for Neuromuscular Blocking Agents. Angew. Chem. Int. Ed. Engl. 2022, 61, e202113235. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Conformation, action, and mechanism of action of neuromuscular blocking muscle relaxants. Pharmacol. Ther. 2003, 98, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Akkol, E.K.; Karatoprak, G.; Carpar, E.; Hussain, Y.; Khan, H.; Aschner, M. Effects of Natural Products on Neuromuscular Junction. Curr. Neuropharmacol. 2022, 20, 594–610. [Google Scholar] [CrossRef]
- Karlin, A. Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 2002, 3, 102–114. [Google Scholar] [CrossRef]
- Unwin, N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: Insights from Torpedo postsynaptic membranes. Q. Rev. Biophys. 2013, 46, 283–322. [Google Scholar] [CrossRef]
- Liu, Y.; Sugiura, Y.; Padgett, D.; Lin, W. Postsynaptic development of the neuromuscular junction in mice lacking the gamma-subunit of muscle nicotinic acetylcholine receptor. J. Mol. Neurosci. 2010, 40, 21–26. [Google Scholar] [CrossRef][Green Version]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef] [PubMed]
- Gyermek, L. Development of ultra short-acting muscle relaxant agents: History, research strategies, and challenges. Med. Res. Rev. 2005, 25, 610–654. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.; Changeux, J.P. The nicotinic acetylcholine receptor and its prokaryotic homologues: Structure, conformational transitions & allosteric modulation. Neuropharmacology 2015, 96, 137–149. [Google Scholar] [CrossRef] [PubMed]
- El-Subbagh, H.I.; El-Azab, A.S.; Hassan, G.S.; El-Messery, S.M.; Abdel-Aziz, A.A.; El-Taher, K.E.H. Thiadiazolodiazepine analogues as a new class of neuromuscular blocking agents: Synthesis, biological evaluation and molecular modeling study. Eur. J. Med. Chem. 2017, 126, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Rao, Z.; Xu, J.; Zhu, Q.; Altenbach, H.J.; Chen, H.; Zhou, D.; Xiao, Y.; Ke, X.; Guo, H.; et al. 16-morpholino quaternary ammonium steroidal derivatives as neuromuscular blocking agents: Synthesis, biological evaluation and in silico probe of ligand-receptor interaction. Eur. J. Med. Chem. 2012, 56, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.S.; Vender, J.S. Neuromuscular-blocking drugs. Use and misuse in the intensive care unit. Crit. Care Clin. 2001, 17, 925–942. [Google Scholar] [CrossRef]
- Plaud, B.; Baillard, C.; Bourgain, J.L.; Bouroche, G.; Desplanque, L.; Devys, J.M.; Fletcher, D.; Fuchs-Buder, T.; Lebuffe, G.; Meistelman, C.; et al. Guidelines on muscle relaxants and reversal in anaesthesia. Anaesth. Crit. Care Pain Med. 2020, 39, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Klucka, J.; Kosinova, M.; Zacharowski, K.; De Hert, S.; Kratochvil, M.; Toukalkova, M.; Stoudek, R.; Zelinkova, H.; Stourac, P. Rapid sequence induction: An international survey. Eur. J. Anaesthesiol. 2020, 37, 435–442. [Google Scholar] [CrossRef]
- Schlaich, N.; Mertzlufft, F.; Soltész, S.; Fuchs-Buder, T. Remifentanil and propofol without muscle relaxants or with different doses of rocuronium for tracheal intubation in outpatient anaesthesia. Acta Anaesthesiol. Scand. 2000, 44, 720–726. [Google Scholar] [CrossRef]
- Hraiech, S.; Yoshida, T.; Annane, D.; Duggal, A.; Fanelli, V.; Gacouin, A.; Heunks, L.; Jaber, S.; Sottile, P.D.; Papazian, L. Myorelaxants in ARDS patients. Intensive Care Med. 2020, 46, 2357–2372. [Google Scholar] [CrossRef]
- McCall, M.; Jeejeebhoy, K.; Pencharz, P.; Moulton, R. Effect of neuromuscular blockade on energy expenditure in patients with severe head injury. JPEN J. Parenter Enter. Nutr. 2003, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Lascarrou, J.B.; Le Gouge, A.; Dimet, J.; Lacherade, J.C.; Martin-Lefèvre, L.; Fiancette, M.; Vinatier, I.; Lebert, C.; Bachoumas, K.; Yehia, A.; et al. Neuromuscular blockade during therapeutic hypothermia after cardiac arrest: Observational study of neurological and infectious outcomes. Resuscitation 2014, 85, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Savarese, J.J.; Sunaga, H.; McGilvra, J.D.; Belmont, M.R.; Murrell, M.T.; Jeannotte, E.; Cooke, F.E.; Wastila, W.B.; Heerdt, P.M. Preclinical Pharmacology in the Rhesus Monkey of CW 1759-50, a New Ultra-short Acting Nondepolarizing Neuromuscular Blocking Agent, Degraded and Antagonized by L-Cysteine. Anesthesiology 2018, 129, 970–988. [Google Scholar] [CrossRef]
- Herrera-Arozamena, C.; Estrada-Valencia, M.; Martí-Marí, O.; Pérez, C.; de la Fuente Revenga, M.; Villalba-Galea, C.A.; Rodríguez-Franco, M.I. Optical control of muscular nicotinic channels with azocuroniums, photoswitchable azobenzenes bearing two N-methyl-N-carbocyclic quaternary ammonium groups. Eur. J. Med. Chem. 2020, 200, 112403. [Google Scholar] [CrossRef] [PubMed]
- Goswami, L.N.; Olds, T.J.; Monk, T.G.; Johnson, Q.L.; Dilger, J.P.; Shanawaz, M.A.; Jalisatgi, S.S.; Hawthorne, M.F.; Kracke, G.R. Isomeric Carborane Neuromuscular Blocking Agents. ChemMedChem 2019, 14, 1108–1114. [Google Scholar] [CrossRef]
- Blanes-Mira, C.; Fernández-Aguado, P.; de Andrés-López, J.; Fernández-Carvajal, A.; Ferrer-Montiel, A.; Fernández-Ballester, G. Comprehensive Survey of Consensus Docking for High-Throughput Virtual Screening. Molecules 2022, 28, 175. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Tropsha, A.; Golbraikh, A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Basta, T.; Teng, J.; Lee, M.; Worrell, B.T.; Stowell, M.H.B.; Hibbs, R.E. Structural mechanism of muscle nicotinic receptor desensitization and block by curare. Nat. Struct. Mol. Biol. 2022, 29, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Erkan, S. Activity of the rocuronium molecule and its derivatives: A theoretical calculation. J. Mol. Struct. 2019, 1189, 257–264. [Google Scholar] [CrossRef]
- Udayakumar, M.; Kumar, P.S.; Hemavathi, K.; Shanmugapriya, P.; Seenivasagam, R. Receptor-based pharmacophore tool for design and development of next-generation drugs. Int. J. Bioinform. Res. Appl. 2013, 9, 487–516. [Google Scholar] [CrossRef] [PubMed]
- Almahmoud, S.; Jin, W.; Geng, L.; Wang, J.; Wang, X.; Vennerstrom, J.L.; Zhong, H.A. Ligand-based design of GLUT inhibitors as potential antitumor agents. Bioorganic Med. Chem. 2020, 28, 115395. [Google Scholar] [CrossRef]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor-ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, A.; Fujiyoshi, Y.; Stowell, M.; Unwin, N. Nicotinic acetylcholine receptor at 4.6 A resolution: Transverse tunnels in the channel wall. J. Mol. Biol. 1999, 288, 765–786. [Google Scholar] [CrossRef] [PubMed]
- Benoit, E.; Couesnon, A.; Lindovsky, J.; Iorga, B.I.; Aráoz, R.; Servent, D.; Zakarian, A.; Molgó, J. Synthetic Pinnatoxins A and G Reversibly Block Mouse Skeletal Neuromuscular Transmission In Vivo and In Vitro. Mar. Drugs 2019, 17, 306. [Google Scholar] [CrossRef] [PubMed]
- Majumder, R.; Mandal, M. Screening of plant-based natural compounds as a potential COVID-19 main protease inhibitor: An in silico docking and molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2022, 40, 696–711. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Fan, Y.; Chen, X.; Yang, Y.; Zhu, L.; Zhao, J.; Chen, Y.; Zhang, Y. In Silico Prediction of Human Intravenous Pharmacokinetic Parameters with Improved Accuracy. J. Chem. Inf. Model. 2019, 59, 3968–3980. [Google Scholar] [CrossRef]
- Ali, M.I.; Thirukovela, N.S.; Kumar, G.B.; Dasari, G.; Badithapuram, V.; Manchal, R.; Bandari, S. Design, synthesis, in silico molecular docking, and ADMET studies of quinoxaline-isoxazole-piperazine conjugates as EGFR-targeting agents. Chem. Biol. Drug Des. 2024, 103, e14499. [Google Scholar] [CrossRef] [PubMed]
- Viana Nunes, A.M.; das Chagas Pereira de Andrade, F.; Filgueiras, L.A.; de Carvalho Maia, O.A.; Cunha, R.; Rodezno, S.V.A.; Maia Filho, A.L.M.; de Amorim Carvalho, F.A.; Braz, D.C.; Mendes, A.N. preADMET analysis and clinical aspects of dogs treated with the Organotellurium compound RF07: A possible control for canine visceral leishmaniasis? Environ. Toxicol. Pharmacol. 2020, 80, 103470. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Yin, K.; Zhao, G.; Xu, C.; Qiu, X.; Wen, B.; Sun, H.; Liu, G.; Liu, Y.; Zhao, Q.; Wei, Q.; et al. Prediction of Toxoplasma gondii virulence factor ROP18 competitive inhibitors by virtual screening. Parasites Vectors 2019, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Mooers, B.H.M.; Brown, M.E. Templates for writing PyMOL scripts. Protein Sci. 2021, 30, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., 3rd. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Simmonett, A.C.; Brooks, B.R. A compression strategy for particle mesh Ewald theory. J. Chem. Phys. 2021, 154, 054112. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | ΔGbind | ΔEvdw | ΔEele | ΔGps | ΔGnps |
|---|---|---|---|---|---|
| ZINC257357801 | −40.52 ± 5.49 | −42.87 ± 3.28 | −726.05 ± 4.22 | 735.10 ± 1.23 | −6.70 ± 0.05 |
| ZINC257459695 | −50.40 ± 3.61 | −33.82 ± 0.84 | −1453.27 ± 0.09 | 1442.28 ± 3.51 | −5.59 ± 0.00 |
| ZINC8926303 | −31.01 ± 6.22 | −37.21 ± 2.20 | −586.86 ± 3.01 | 598.35 ± 4.97 | −5.28 ± 0.02 |
| d-tubocurarine | −39.57 ± 3.11 | −55.84 ± 0.11 | −657.70 ± 2.99 | 680.80 ± 0.83 | −6.82 ± 0.16 |
| Cisatracurium | −66.86 ± 3.07 | −92.50 ± 2.01 | −669.38 ± 0.33 | 706.26 ± 2.30 | −11.24 ± 0.08 |
| Rocuronium | −57.65 ± 2.50 | −62.59 ± 1.72 | −340.73 ± 1.67 | 353.45 ± 0.70 | −7.78 ± 0.01 |
| Compound | Buffer Solubility 1 | BBB 2 | PPB 3 | CYP2D6 Inhibition | Ames Test | hERG Inhibition |
|---|---|---|---|---|---|---|
| ZINC257357801 | 1132 | 0.04 | 3.83 | Inhibitor | Non-mutagen | Ambiguous |
| ZINC257459695 | 0.30 | 0.11 | 13.97 | Non | Non-mutagen | High |
| ZINC8926303 | 88,380 | 0.05 | 19.16 | Inhibitor | Mutagen | Ambiguous |
| d-tubocurarine | 1.18 | 1.22 | 67.68 | Non | Non-mutagen | High risk |
| Cisatracurium | 0.0081 | 0.80 | 72.49 | Non | Non-mutagen | Medium risk |
| Rocuronium | 121.69 | 0.26 | 18.97 | Inhibitor | Mutagen | Low risk |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Ge, G.; Xu, X.; Wu, J. Ensemble-Based Virtual Screening Led to the Discovery of Novel Lead Molecules as Potential NMBAs. Molecules 2024, 29, 1955. https://doi.org/10.3390/molecules29091955
Zhang Y, Ge G, Xu X, Wu J. Ensemble-Based Virtual Screening Led to the Discovery of Novel Lead Molecules as Potential NMBAs. Molecules. 2024; 29(9):1955. https://doi.org/10.3390/molecules29091955
Chicago/Turabian StyleZhang, Yi, Gonghui Ge, Xiangyang Xu, and Jinhui Wu. 2024. "Ensemble-Based Virtual Screening Led to the Discovery of Novel Lead Molecules as Potential NMBAs" Molecules 29, no. 9: 1955. https://doi.org/10.3390/molecules29091955
APA StyleZhang, Y., Ge, G., Xu, X., & Wu, J. (2024). Ensemble-Based Virtual Screening Led to the Discovery of Novel Lead Molecules as Potential NMBAs. Molecules, 29(9), 1955. https://doi.org/10.3390/molecules29091955