
Skeletal Editing: Ring Insertion for Direct Access to Heterocycles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
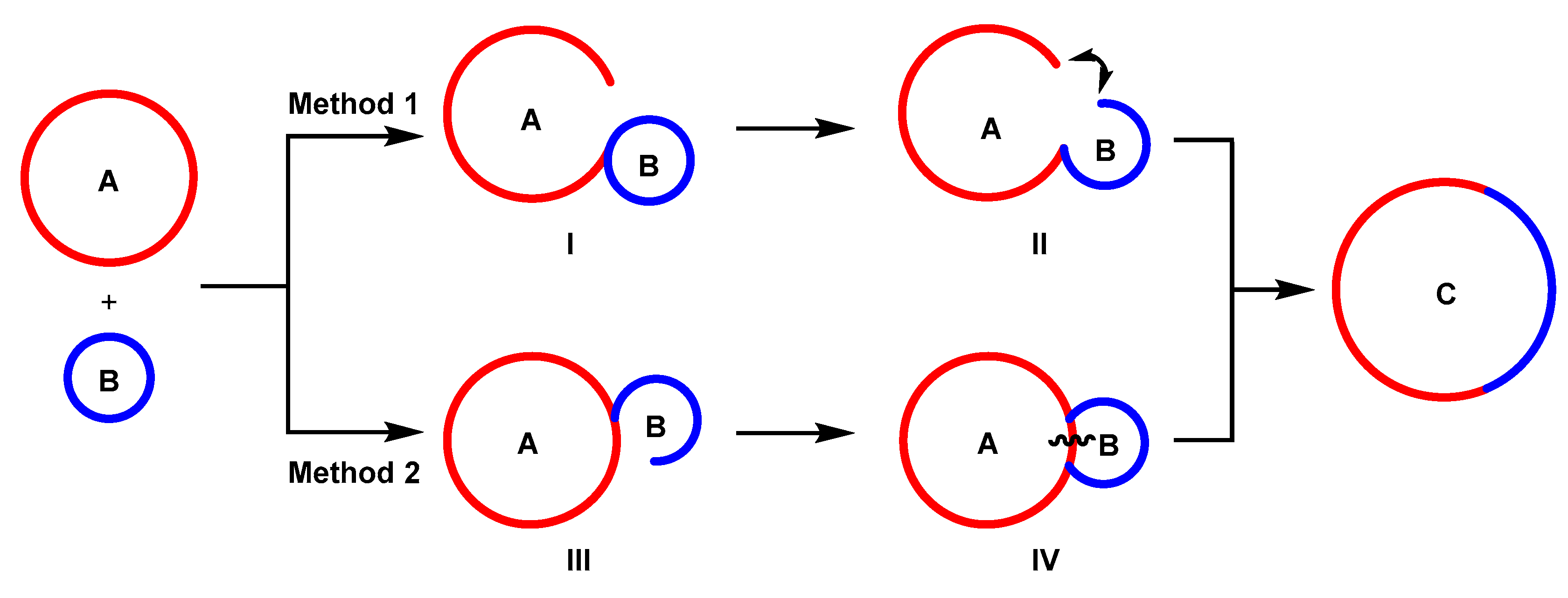
2. The Formation of a New Ring through Ring Opening and Reconnection
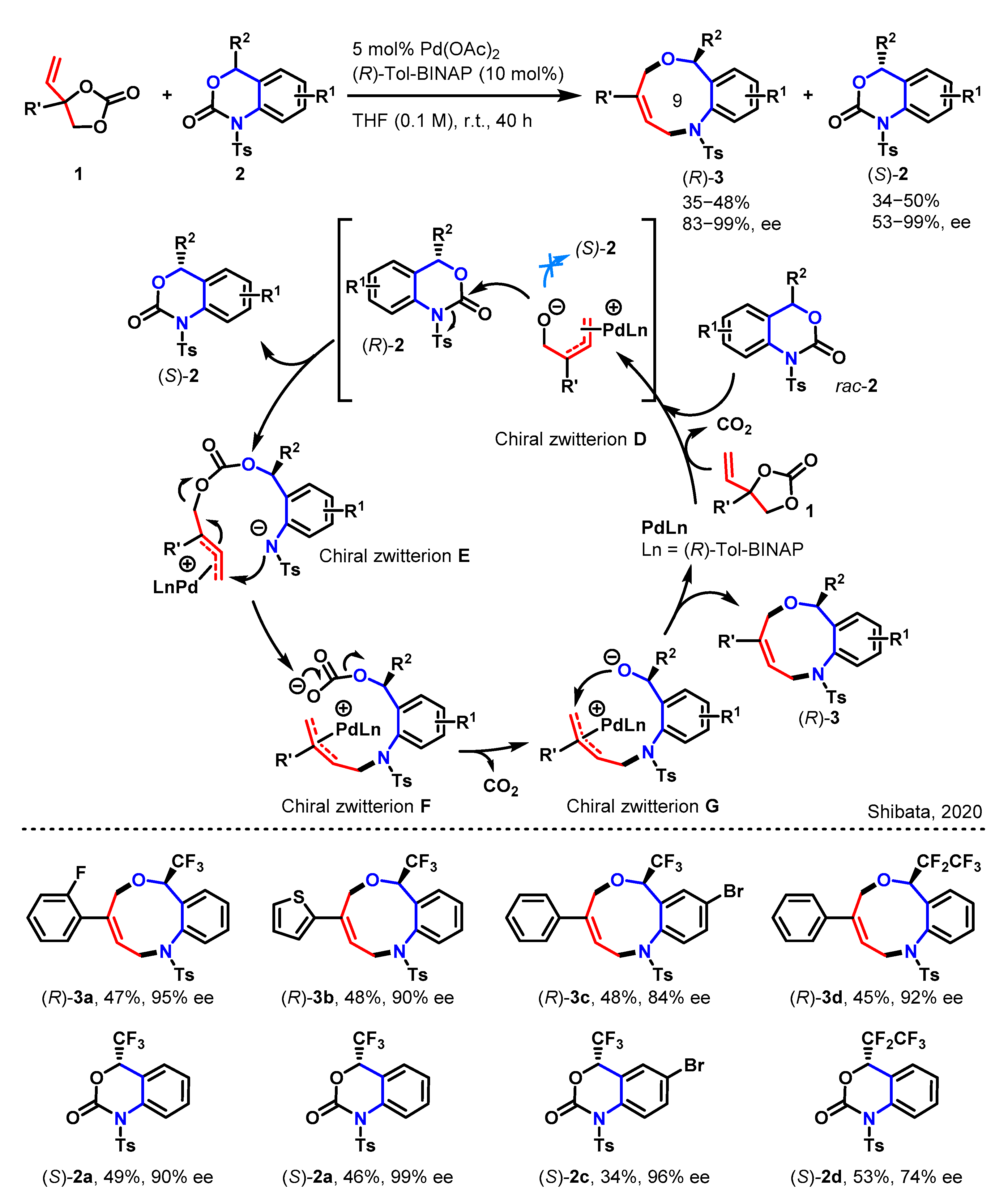
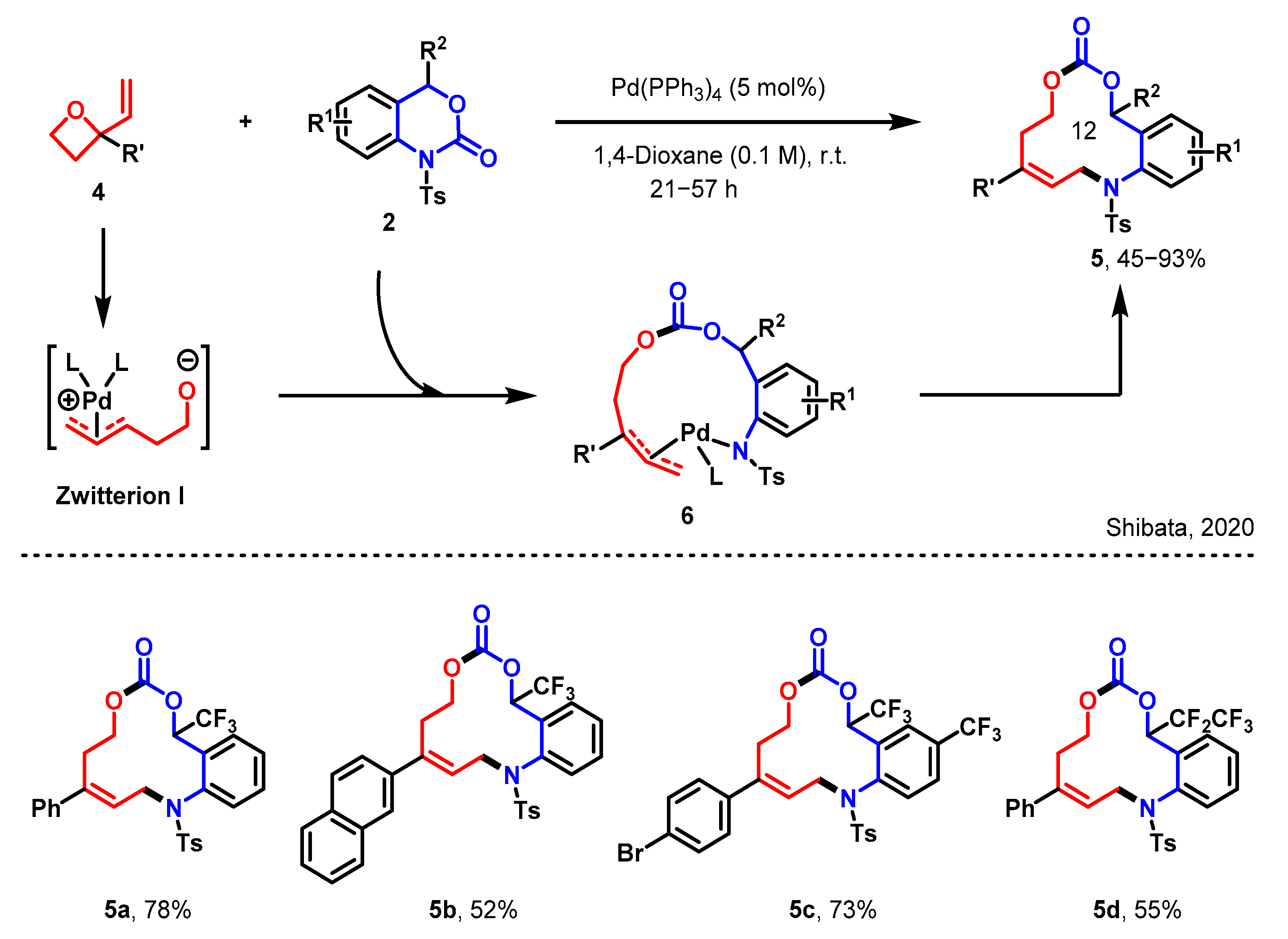
2.1. [5 + 4] Decarboxylative Ring Reconstruction Reaction
2.2. [6 + 6] Decarboxylative Ring Reconstruction Reaction
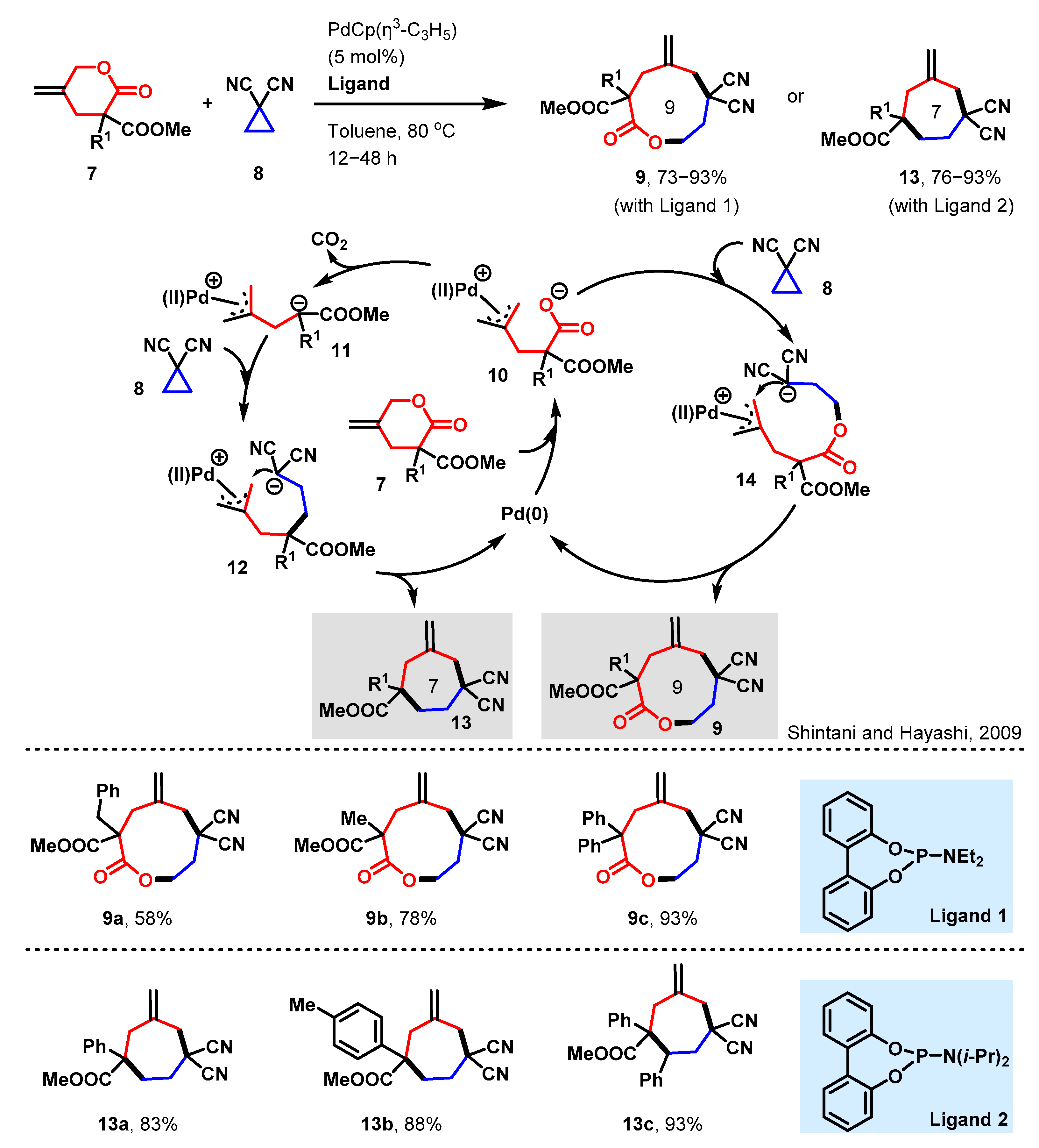
2.3. Nondecarboxylative [6 + 3] and Decarboxylative [4 + 3] Ring Reconstruction Reactions
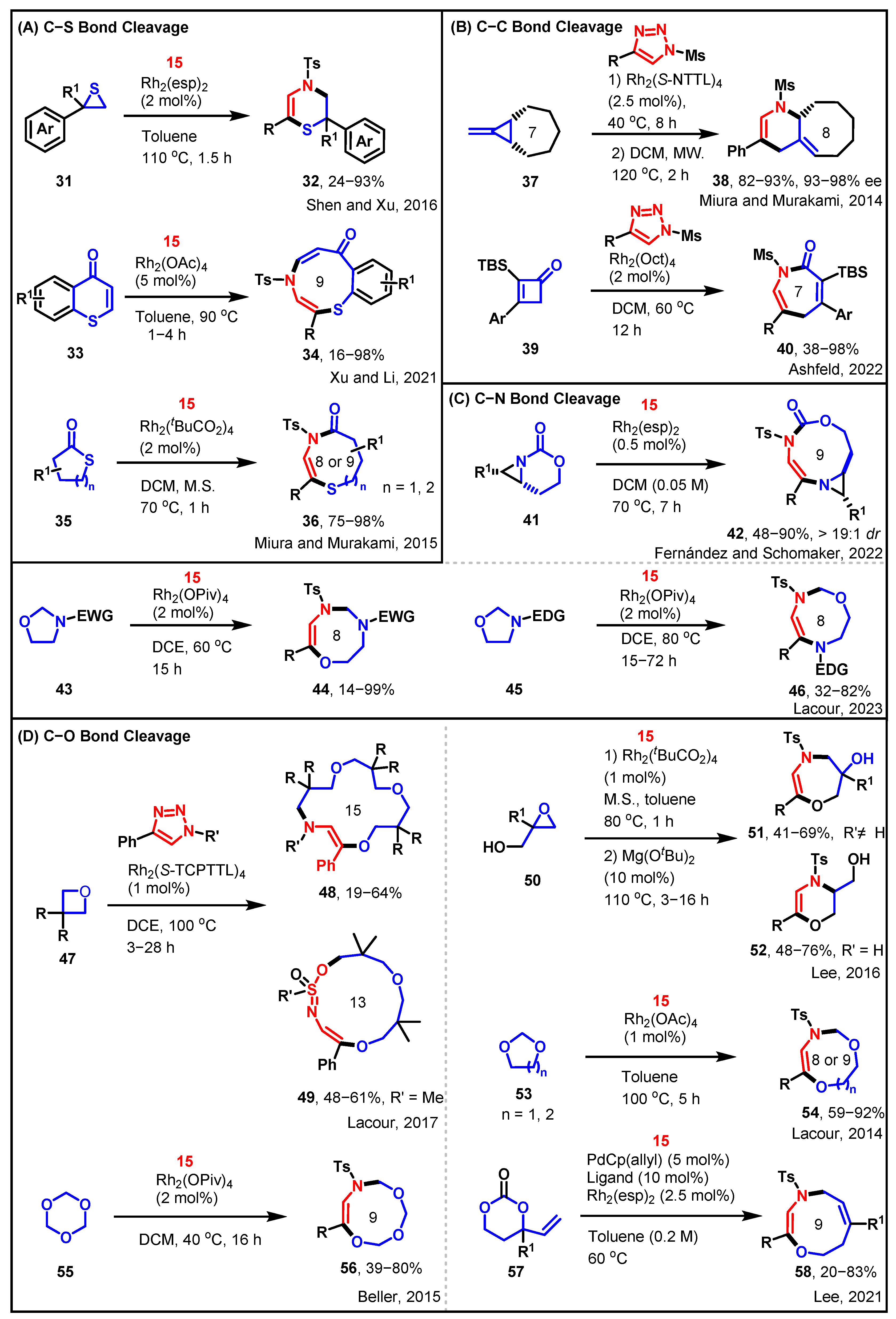
2.4. [3 + n] Cycloadditions of Sulfonyltriazole with Heterocycles
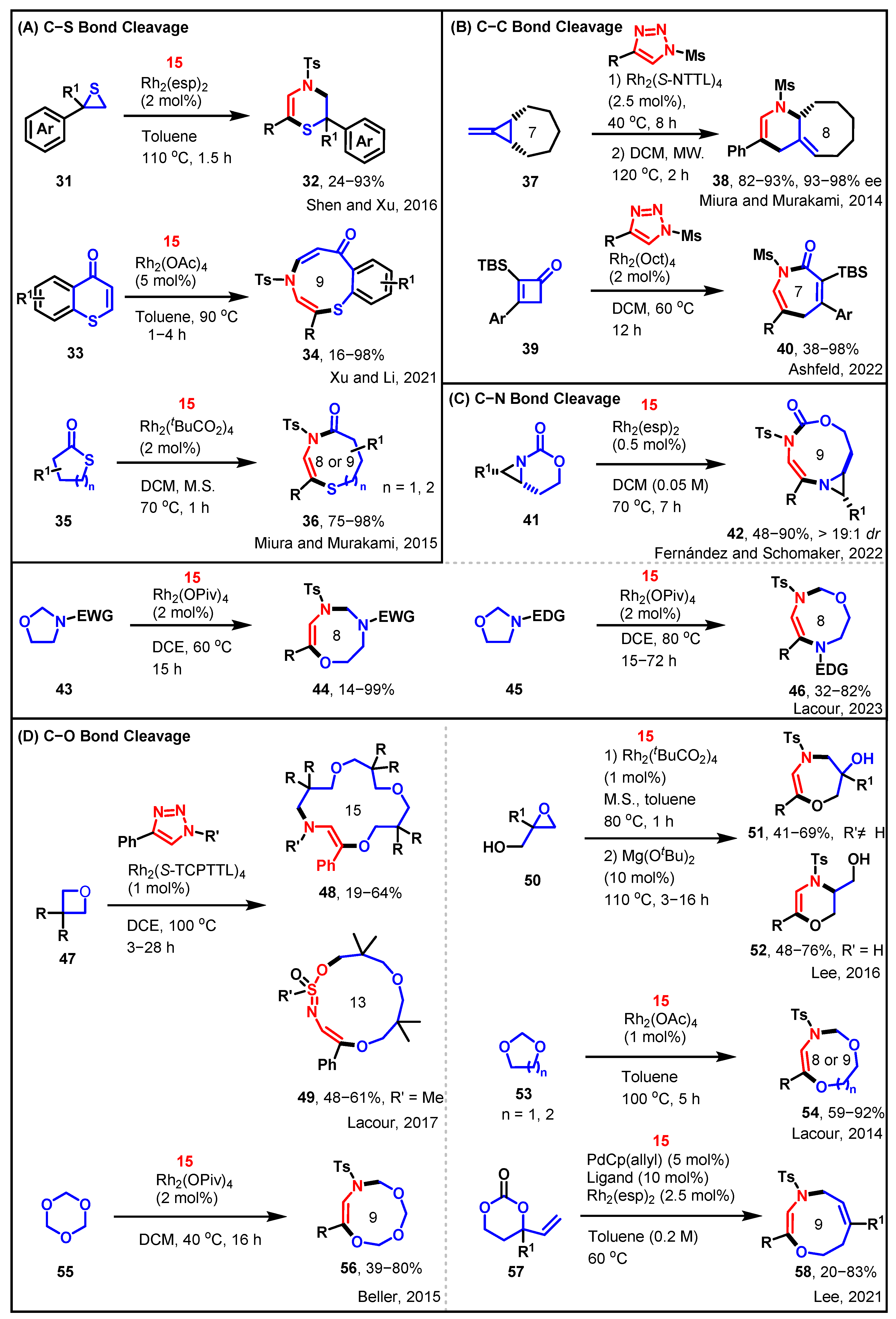
3. The Formation of a New Ring through Ring Closure and then Ring Opening
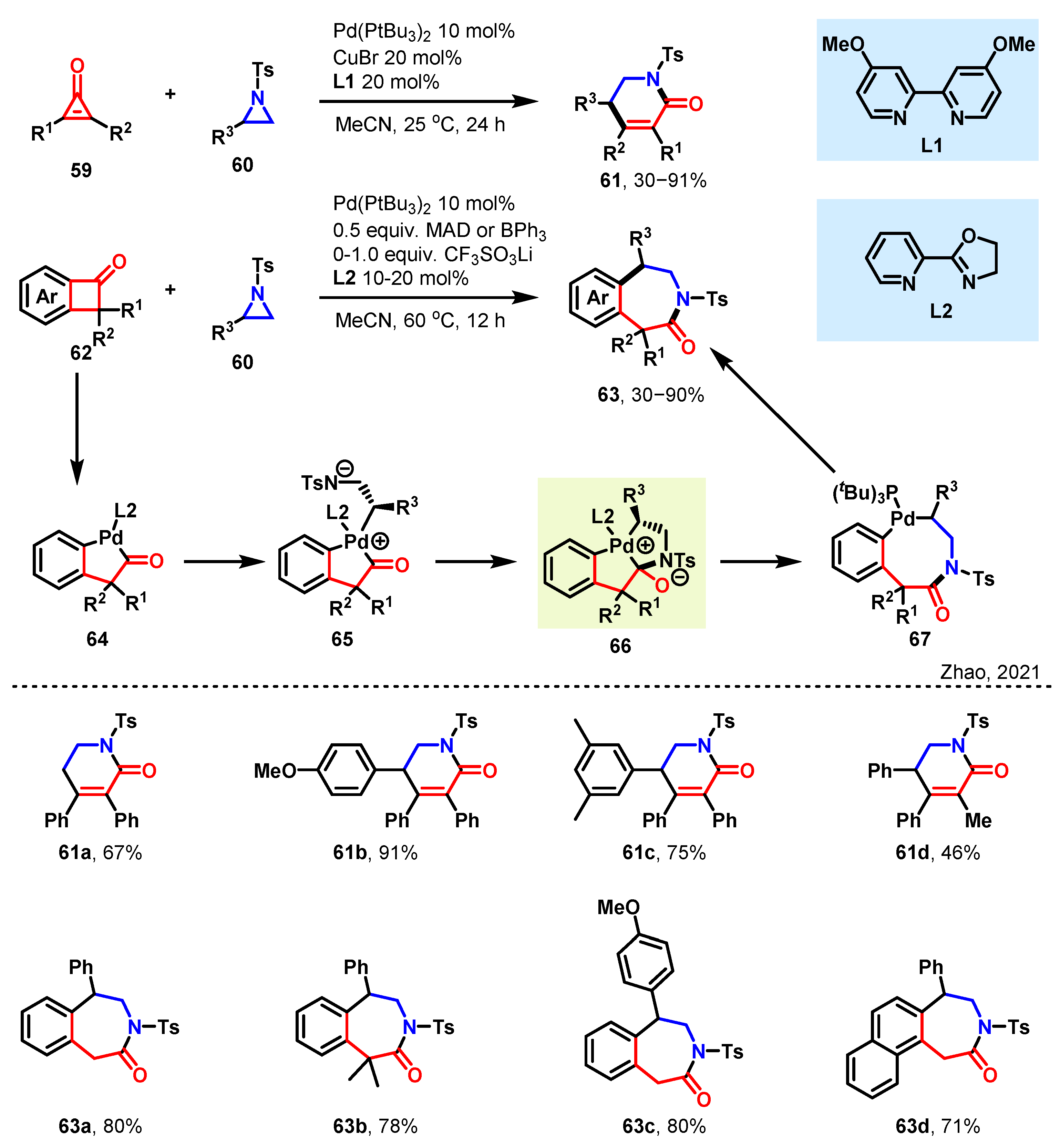
3.1. [3 + 3] and [3 + 4] towards Diverse Azaheterocycles
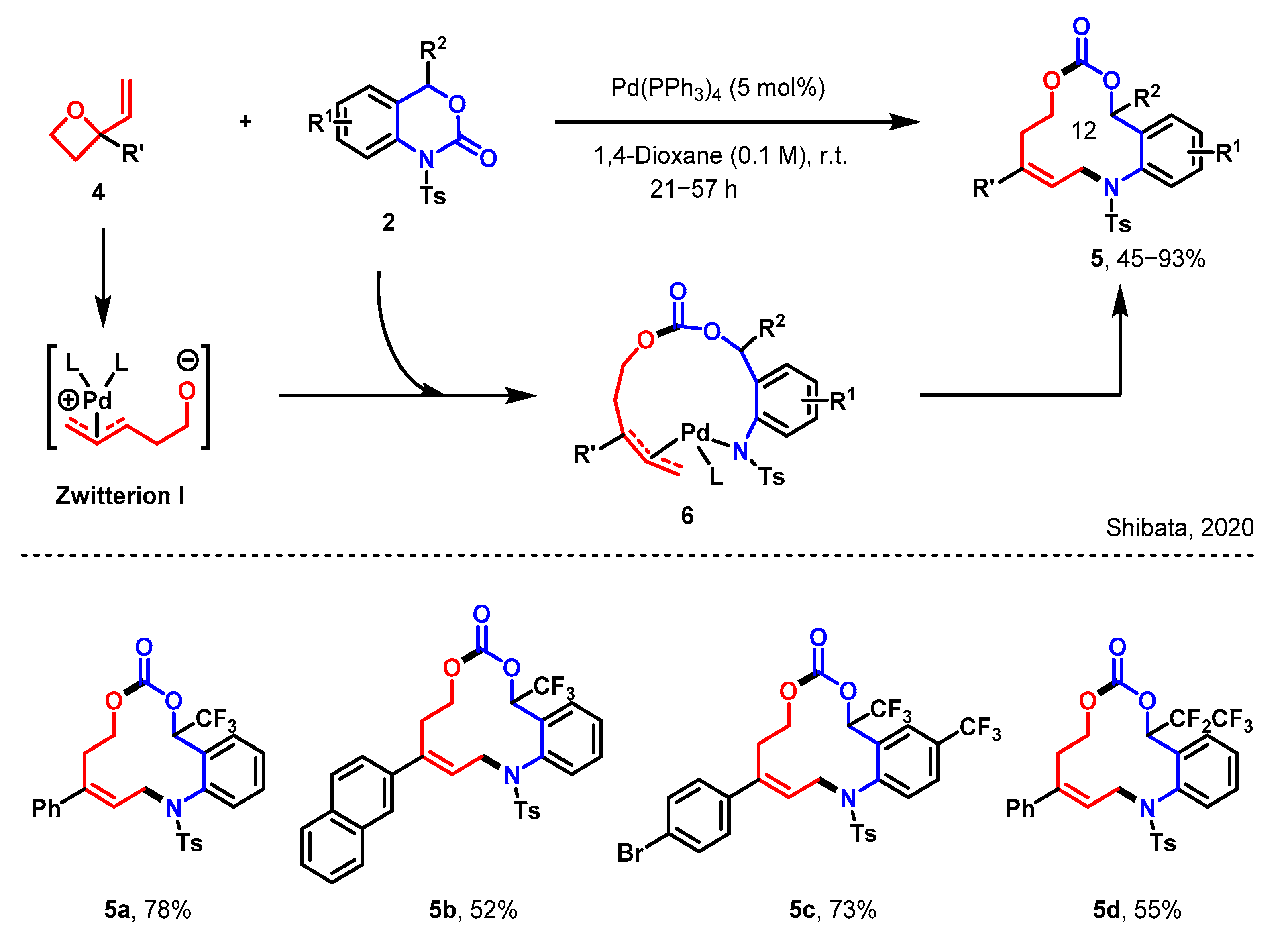
3.2. Expansion of Cyclic Ketones to Macrolactones
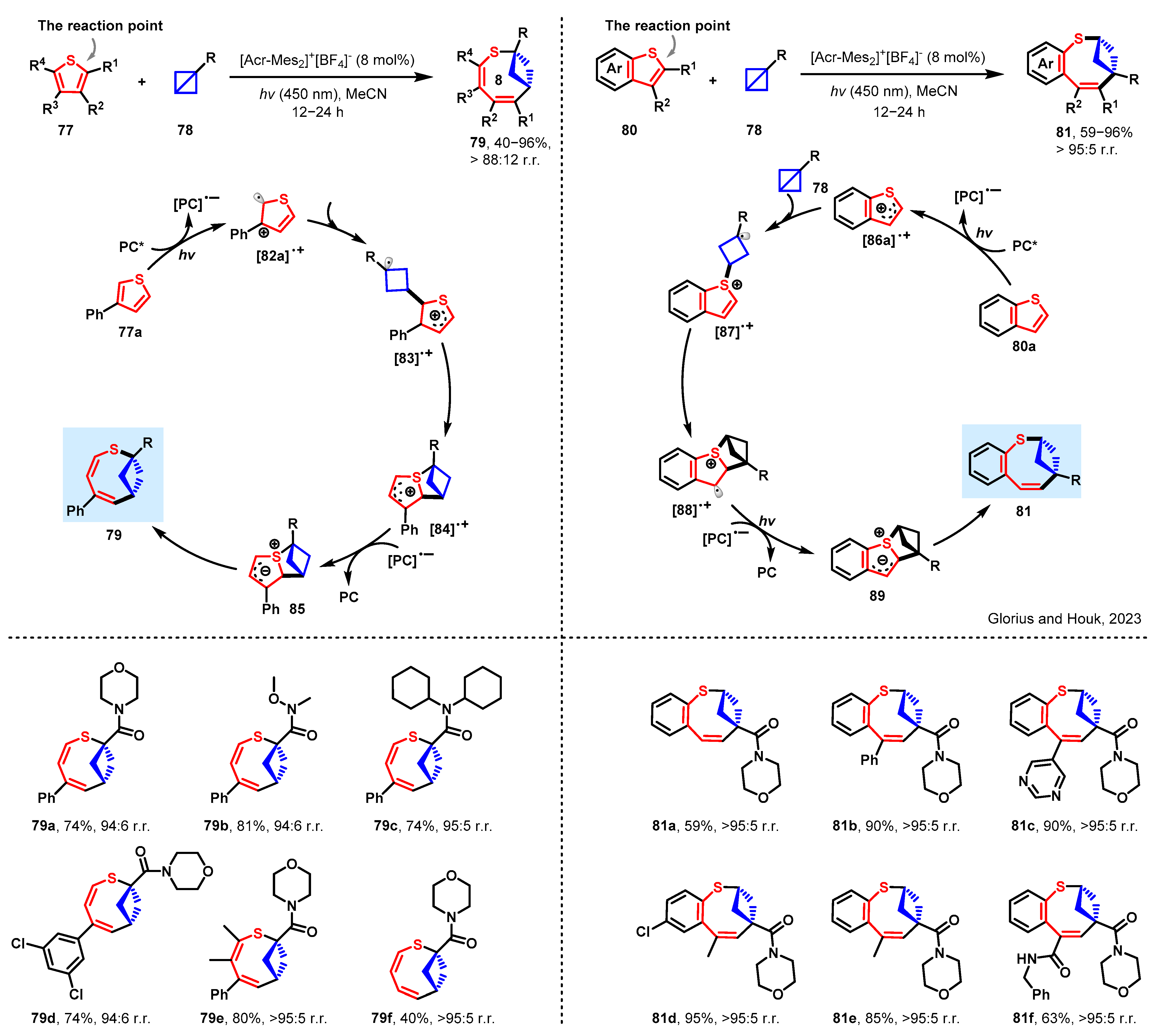
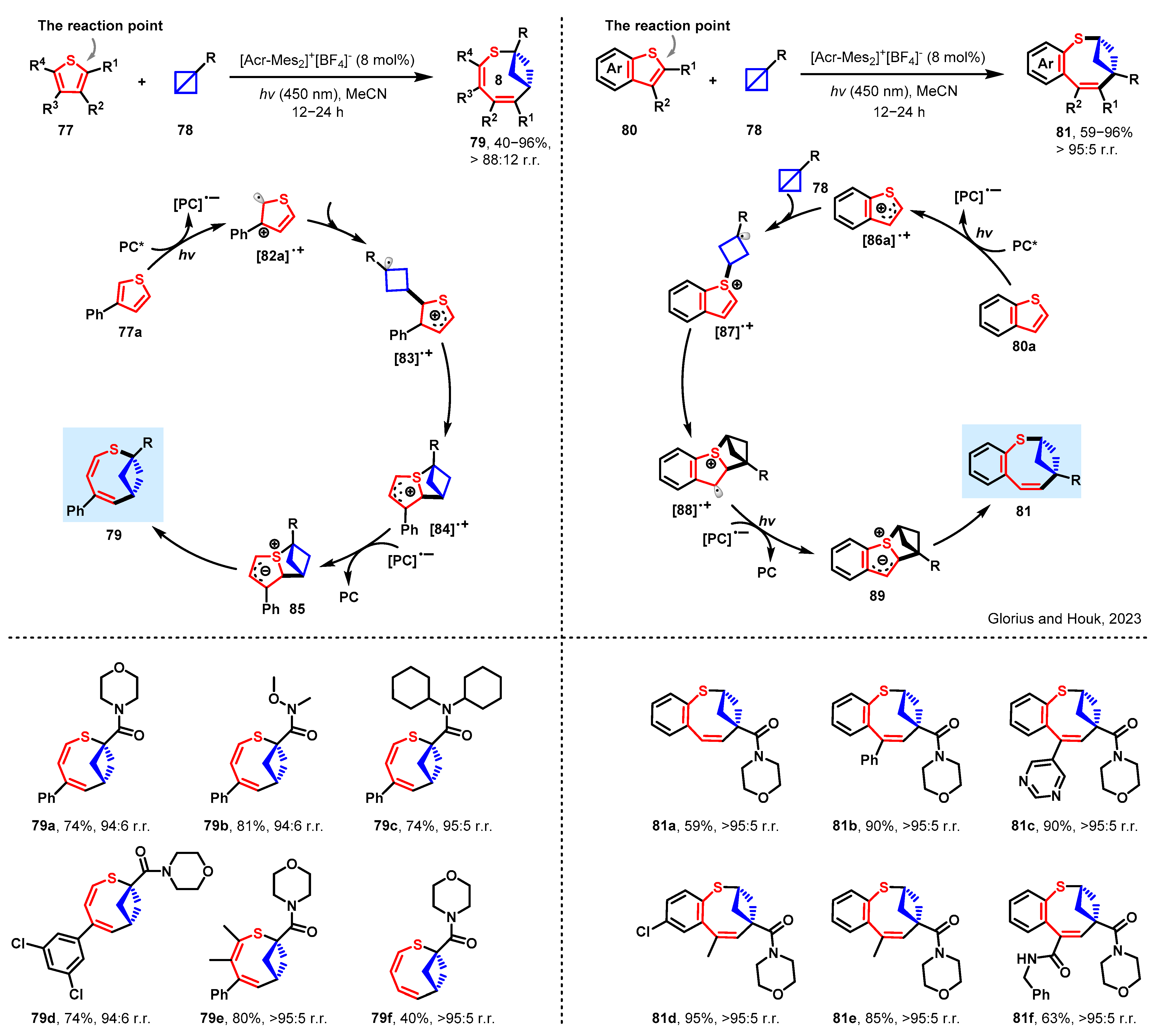
3.3. Expansion of Thiophenes by Bicyclobutane Insertion
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Yang, N.-N.; Ma, Q.-Y.; Yang, L.; Xie, Q.-Y.; Dai, H.-F.; Yu, Z.-F.; Zhao, Y.-X. A New Macrolide from the Strain Cordyceps spp. from Cell Fusion between Cordyceps militaris and Cordyceps cicadae. Chem. Nat. Compd. 2024, 60, 61–64. [Google Scholar] [CrossRef]
- Escolano, M.; Gaviña, D.; Alzuet-Piña, G.; Díaz-Oltra, S.; Sánchez-Roselló, M.; del Pozo, C. Recent Strategies in the Nucleophilic Dearomatization of Pyridines, Quinolines, and Isoquinolines. Chem. Rev. 2024, 124, 1122–1246. [Google Scholar] [CrossRef] [PubMed]
- Buttard, F.; Vigier, J.; Lebel, H.; Besset, T. C-Centered radical intermediates for C(sp3)-H bonds functionalization: An emerging approach towards alkyl thiocyanates. Eur. J. Org. Chem. 2024, 27, e2023012. [Google Scholar] [CrossRef]
- Li, J.; Amatuni, A.; Renata, H. Recent advances in the chemoenzymatic synthesis of bioactive natural products. Curr. Opin. Chem. Biol. 2020, 55, 111–118. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, C. The future directions of synthetic chemistry. Pure Appl. Chem. 2021, 93, 1463–1472. [Google Scholar] [CrossRef]
- Yang, Z.; Zalessky, I.; Epton, R.G.; Whitwood, A.C.; Lynam, J.M.; Unsworth, W.P. Ring Expansion Strategies for the Synthesis of Medium Sized Ring and Macrocyclic Sulfonamides. Angew. Chem. Int. Ed. 2023, 62, e2022171. [Google Scholar]
- Donald, J.R.; Unsworth, W.P. Ring-Expansion Reactions in the Synthesis of Macrocycles and Medium-Sized Rings. Chem. Eur. J. 2017, 23, 8780–8799. [Google Scholar] [CrossRef]
- Cheng, Q.; Bhattacharya, D.; Haring, M.; Cao, H.; Mück-Lichtenfeld, C.; Studer, A. Skeletal editing of pyridines through atom-pair swap from CN to CC. Nat. Chem. 2024. [Google Scholar] [CrossRef]
- Jurczyk, J.; Woo, J.; Kim, S.F.; Dherange, B.D.; Sarpong, R.; Levin, M.D. Single-atom logic for heterocycle editing. Nat. Synth. 2022, 1, 352–364. [Google Scholar] [CrossRef]
- Liu, S.; Yang, Y.; Song, Q.; Liu, Z.; Lu, Y.; Wang, Z.; Sivaguru, P.; Bi, X. Tunable molecular editing of indoles with fluoroalkyl carbenes. Nat. Chem. 2024. [Google Scholar] [CrossRef]
- Kennedy, S.H.; Dherange, B.D.; Berger, K.J.; Levin, M.D. Skeletal editing through direct nitrogen deletion of secondary amines. Nature 2021, 593, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sivaguru, P.; Ning, Y.; Wu, Y.; Bi, X. Skeletal Editing of (Hetero)Arenes Using Carbenes. Chem. Eur. J. 2023, 29, e202301227. [Google Scholar] [CrossRef] [PubMed]
- Joynson, B.W.; Ball, L.T. Skeletal Editing: Interconversion of Arenes and Heteroarenes. Helv. Chim. Acta 2023, 106, e2022001. [Google Scholar] [CrossRef]
- Zippel, C.; Seibert, J.; Bräse, S. Skeletal Editing-Nitrogen Deletion of Secondary Amines by Anomeric Amide Reagents. Angew. Chem. Int. Ed. 2021, 60, 19522–19524. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Anand, L.; Szostak, M. Diversification of Indoles and Pyrroles by Molecular Editing: New Frontiers in Heterocycle-to-Heterocycle Transmutation. Chem. Eur. J. 2023, 29, e202300096. [Google Scholar] [CrossRef] [PubMed]
- Reisenbauer, J.C.; Green, O.; Franchino, A.; Finkelstein, P.; Morandi, B. Late-stage diversification of indole skeletons throughnitrogen atom insertion. Science 2022, 377, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, G.L.; Carpaneto, F.; Sarpong, R. Skeletal Editing of Pyrimidines to Pyrazoles by Formal Carbon Deletion. J. Am. Chem. Soc. 2022, 144, 22309–22315. [Google Scholar] [CrossRef]
- Hui, C.; Wang, Z.; Wang, S.; Xu, C. Molecular editing in natural product synthesis. Org. Chem. Front. 2022, 9, 1451–1457. [Google Scholar] [CrossRef]
- Bartholomew, G.L.; Kraus, S.L.; Karas, L.J.; Carpaneto, F.; Bennett, R.; Sigman, M.S.; Yeung, C.S.; Sarpong, R. 14N to 15N Isotopic Exchange of Nitrogen Heteroaromatics through Skeletal Editing. J. Am. Chem. Soc. 2024, 146, 2950–2958. [Google Scholar] [CrossRef]
- Unsworth, W.P.; Avestro, A.-J. Unprecedented reactionsfor molecular editing. Nature 2021, 593, 203. [Google Scholar] [CrossRef]
- He, Y.; Wang, J.; Zhu, T.; Zheng, Z.; Wei, H. Nitrogen atom insertion into arenols to access benzazepines. Chem. Sci. 2024, 15, 2612–2617. [Google Scholar] [CrossRef]
- Qiu, X.; Sang, Y.; Wu, H.; Xue, X.-S.; Yan, Z.-Z.; Wang, Y.; Cheng, Z.; Wang, X.; Tan, H.; Song, S.; et al. Cleaving arene rings for acyclic alkenylnitrile synthesis. Nature 2021, 597, 64–69. [Google Scholar] [CrossRef]
- Chaudhary, A. Recent development in the synthesis of heterocycles by 2-naphthol-based multicomponent reactions. Mol. Div. 2021, 25, 1211–1245. [Google Scholar] [CrossRef]
- Biletskyi, B.; Colonna, P.; Masson, K.; Parrain, J.-L.; Commeiras, L.; Chouraqui, G. Small rings in the bigger picture: Ring expansion of three- and four-membered rings to access larger all-carbon cyclic systems. Chem. Soc. Rev. 2021, 50, 7513–7538. [Google Scholar] [CrossRef]
- Huber, T.; Wildermuth, R.E.; Magauer, T. 9-Membered Carbocycles: Strategies and Tactics for their Synthesis. Chem. Eur. J. 2018, 24, 12107–12120. [Google Scholar] [CrossRef]
- Sindlinger, M.; Ströbele, M.; Grunenberg, J.; Bettinger, H.F. Accessing unusual heterocycles: Ring expansion of benzoborirenes by formal cycloaddition reactions. Chem. Sci. 2023, 14, 10478. [Google Scholar] [CrossRef]
- Baidya, R.; Das, P.; Pratihar, P.; Maiti, D.K. Ring expansion and fused cyclization catalysis to construct indoloquinazolinones with functionalization. Chem. Commun. 2023, 59, 7978. [Google Scholar] [CrossRef]
- Babar, K.; Zahoor, A.F.; Ahmad, S.; Akhtar, R. Recent synthetic strategies toward the synthesis of spirocyclic compounds comprising six-membered carbocyclic/heterocyclic ring systems. Mol. Div. 2021, 25, 2487–2532. [Google Scholar] [CrossRef]
- Stephens, T.C.; Unsworth, W.P. Consecutive Ring-Expansion Reactions for the Iterative Assembly of Medium-Sized Rings and Macrocycles. Synlett 2020, 31, 133–146. [Google Scholar] [CrossRef]
- Uno, H.; Punna, N.; Tokunaga, E.; Shiro, M.; Shibata, N. Synthesis of Both Enantiomers of Nine-Membered CF3-Substituted Heterocycles Using a Single Chiral Ligand: Palladium-Catalyzed Decarboxylative Ring Expansion with Kinetic Resolution. Angew. Chem. Int. Ed. 2020, 59, 8187–8194. [Google Scholar] [CrossRef]
- Das, P.; Gondo, S.; Nagender, P.; Uno, H.; Tokunaga, E.; Shibata, N. Access to benzo-fused nine-membered heterocyclic alkenes with a trifluoromethyl carbinol moiety via a double decarboxylative formal ring-expansion process under palladium catalysis. Chem. Sci. 2018, 9, 3276–3281. [Google Scholar] [CrossRef]
- Uno, H.; Imai, T.; Harada, K.; Shibata, N. Synthesis of Highly Functionalized 12-Membered Trifluoromethyl Heterocycles via a Nondecarboxylative Pd-Catalyzed [6 + 6] Annulation. ACS Catal. 2020, 10, 1454–1459. [Google Scholar] [CrossRef]
- Shintani, R.; Murakami, M.; Tsuji, T.; Tanno, H.; Hayashi, T. Palladium-Catalyzed Decarboxylative [4 + 3] Cyclization of γ-Methylidene-δ-valerolactones with 1,1-Dicyanocyclopropanes. Org. Lett. 2009, 11, 5642–5645. [Google Scholar] [CrossRef]
- Yuan, W.; Xu, J. Ring Expansions of Oxetanes. Chin. J. Org. Chem. 2021, 41, 947–958. [Google Scholar] [CrossRef]
- Zhao, Y.-Z.; Yang, H.-B.; Tang, X.-Y.; Shi, M. RhII-Catalyzed [3 + 2] Cycloaddition of 2H-Azirines with N-Sulfonyl1,2,3-Triazoles. Chem. Eur. J. 2015, 21, 3562–3566. [Google Scholar] [CrossRef]
- Ma, X.; Pan, S.; Wang, H.; Chen, W. Rhodium-Catalyzed Transannulation of N-Sulfonyl-1,2,3-triazoles and Epoxides: Regioselective Synthesis of Substituted 3,4-Dihydro-2H-1,4-oxazines. Org. Lett. 2014, 16, 4554–4557. [Google Scholar] [CrossRef]
- Mishra, D.R.; Chakroborty, S.; Lalitha, P.; Panda, B.S.; Mishra, N.P.; Barik, A.; Panda, A.R.; Malviya, J.; Khatarkar, K.; Mukopadhyay, M. Advances in Metal-Catalyzed Denitrogenative Pathways for N-Heterocycle Synthesis. Top Catal. 2024, 67, 246–262. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, X.; Tang, Y. Rh(II)-catalyzed cycloadditions of 1-tosyl 1,2,3-triazoles with 2H-azirines: Switchable reactivity of Rh-azavinylcarbene as [2C]- or aza-[3C]-synthon. Chem. Commun. 2015, 51, 4507–4510. [Google Scholar] [CrossRef]
- Ding, H.; Hong, S.; Zhang, N. Rhodium(II)-catalyzed transannulation of 1-sulfonyl-1,2,3-triazoles with 2H-azirines: A new method to dihydropyrazines. Tetrahedron Lett. 2015, 56, 507–510. [Google Scholar] [CrossRef]
- Liu, Z.-K.; Gao, Y.; Hu, X.-Q. Recent advances in catalytic synthesis of mediumring lactones and their derivatives. Catal. Sci. Technol. 2021, 11, 6931–6946. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Wang, J. Recent developments in copper-catalyzed reactions of diazo compounds. Chem. Commun. 2012, 48, 10162–10173. [Google Scholar] [CrossRef]
- Yang, D.; Guan, Z.; Peng, Y.; Zhu, S.; Wang, P.; Huang, Z.; Alhumade, H.; Gu, D.; Yi, H.; Lei, A. Electrochemical oxidative difunctionalization of diazo compounds with two different nucleophiles. Nat. Commun. 2023, 14, 1476. [Google Scholar] [CrossRef]
- Lu, X.-L.; Liu, Y.-T.; Wang, Q.-X.; Shen, M.-H.; Xu, H.-D. Straightforward regioselective construction of 3,4-dihydro-2H-1,4-thiazine by rhodium catalysed [3 + 3] cycloaddition of thiirane with 1-sulfonyl-1,2,3-triazole: A pronounced acid additive effect. Org. Chem. Front. 2016, 3, 725–729. [Google Scholar] [CrossRef]
- Jablasone, S.T., Jr.; Ye, Z.; Duan, S.; Xu, Z.-F.; Li, C.-Y. Synthesis of benzothiazonine by rhodiumcatalyzed denitrogenative transannulation of 1-sulfonyl-1,2,3-triazole and thiochromone. Org. Biomol. Chem. 2021, 19, 5758–5761. [Google Scholar] [CrossRef]
- Miura, T.; Fujimoto, Y.; Funakoshi, Y.; Murakami, M. A Reaction of Triazoles with Thioesters to Produce β-Sulfanyl Enamides by Insertion of an Enamine Moiety into the Sulfur-Carbonyl Bond. Angew. Chem. Int. Ed. 2015, 54, 9967–9970. [Google Scholar] [CrossRef]
- Miura, T.; Nakamuro, T.; Liang, C.-J.; Murakami, M. Synthesis of trans-Cycloalkenes via Enantioselective Cyclopropanation and Skeletal Rearrangement. J. Am. Chem. Soc. 2014, 136, 15905–15908. [Google Scholar] [CrossRef]
- Hill, H.M.; Tucker, Z.D.; Rodriguez, K.X.; Wendt, K.A.; Ashfeld, B.L. Generation of Functionalized Azepinone Derivatives via a (4 + 3)-Cycloaddition of Vinyl Ketenes and α-Imino Carbenes Derived from N-Sulfonyl-triazoles. J. Org. Chem. 2022, 87, 3825–3833. [Google Scholar] [CrossRef]
- Dequina, H.J.; Eshon, J.; Schmid, S.C.; Raskopf, W.T.; Sanders, K.M.; Fernández, I.; Schomaker, J.M. Re-Evaluation of Product Outcomes in the Rh-Catalyzed Ring Expansion of Aziridines with N-Sulfonyl-1,2,3-Triazoles. J. Org. Chem. 2022, 87, 10902–10907. [Google Scholar] [CrossRef]
- Viudes, O.; Guarnieri-Ibáñez, A.; Besnard, C.; Lacour, J. Regiodivergent Synthesis of Oxadiazocines via Dirhodium-Catalyzed Reactivity of Oxazolidines and α-Imino Carbenes. Synlett 2023, 34, 1472–1476. [Google Scholar]
- Guarnieri-Ibáñez, A.; Medina, F.; Besnard, C.; Kidd, S.L.; Spring, D.R.; Lacour, J. Diversity-oriented synthesis of heterocycles and macrocycles by controlled reactions of oxetanes with α-iminocarbenes. Chem. Sci. 2017, 8, 5713–5720. [Google Scholar] [CrossRef]
- Ko, Y.O.; Jeon, H.J.; Jung, D.J.; Kim, U.B.; Lee, S.-G. Rh(II)/Mg(OtBu)2-Catalyzed Tandem One-Pot Synthesis of 1,4-Oxazepines and 1,4-Oxazines from N-Sulfonyl-1,2,3-triazoles and Glycidols. Org. Lett. 2016, 18, 6432–6435. [Google Scholar] [CrossRef]
- Medina, F.; Besnard, C.; Lacour, J. One-Step Synthesis of Nitrogen-Containing Medium-Sized Rings via α-Imino Diazo Intermediates. Org. Lett. 2014, 16, 3232–3235. [Google Scholar] [CrossRef]
- Pospech, J.; Ferraccioli, R.; Neumann, H.; Beller, M. Rhodium(II)-Catalyzed Annulation of Azavinyl Carbenes Through Ring-Expansion of 1,3,5-Trioxane: Rapid Access to NineMembered 1,3,5,7-Trioxazonines. Chem. Asian J. 2015, 10, 2624–2630. [Google Scholar] [CrossRef]
- Lee, K.R.; Ahn, S.; Lee, S.-G. Synergistic Pd(0)/Rh(II) Dual Catalytic [6 + 3] Dipolar Cycloaddition for the Synthesis of Monocyclic Nine-Membered N,O-Heterocycles and Their Alder-ene Rearrangement to Fused Bicyclic Compounds. Org. Lett. 2021, 23, 3735–3740. [Google Scholar] [CrossRef]
- Li, R.; Li, B.; Zhang, H.; Ju, C.-W.; Qin, Y.; Xue, X.-S.; Zhao, D. A ring expansion strategy towards diverse azaheterocycles. Nat. Chem. 2021, 13, 1006–1016. [Google Scholar] [CrossRef]
- Du, J.; Yang, X.; Wang, X.; An, Q.; He, X.; Pan, H.; Zuo, Z. Photocatalytic Aerobic Oxidative Ring Expansion of Cyclic Ketones to Macrolactones by Cerium and Cyanoanthracene Catalysis. Angew. Chem. Int. Ed. 2021, 60, 5370–5376. [Google Scholar] [CrossRef]
- Ye, J. Macrolactones via Photoinduced Ring Expansion of Cyclic Ketones. Chin. J. Org. Chem. 2021, 41, 1755–1756. [Google Scholar] [CrossRef]
- Wang, H.; Shao, H.; Das, A.; Dutta, S.; Chan, H.T.; Daniliuc, C.; Houk, K.N.; Glorius, F. Dearomative ring expansion of thiophenes by bicyclobutane insertion. Science 2023, 381, 75–81. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Xu, Z. Skeletal Editing: Ring Insertion for Direct Access to Heterocycles. Molecules 2024, 29, 1920. https://doi.org/10.3390/molecules29091920
Li X, Xu Z. Skeletal Editing: Ring Insertion for Direct Access to Heterocycles. Molecules. 2024; 29(9):1920. https://doi.org/10.3390/molecules29091920
Chicago/Turabian StyleLi, Xue, and Zhigang Xu. 2024. "Skeletal Editing: Ring Insertion for Direct Access to Heterocycles" Molecules 29, no. 9: 1920. https://doi.org/10.3390/molecules29091920
APA StyleLi, X., & Xu, Z. (2024). Skeletal Editing: Ring Insertion for Direct Access to Heterocycles. Molecules, 29(9), 1920. https://doi.org/10.3390/molecules29091920