


Evaluation of Physicochemical Properties of Ipsapirone Derivatives Based on Chromatographic and Chemometric Approaches
 , , , , , ,
, , , , , , 
Abstract
1. Introduction
2. Results and Discussion
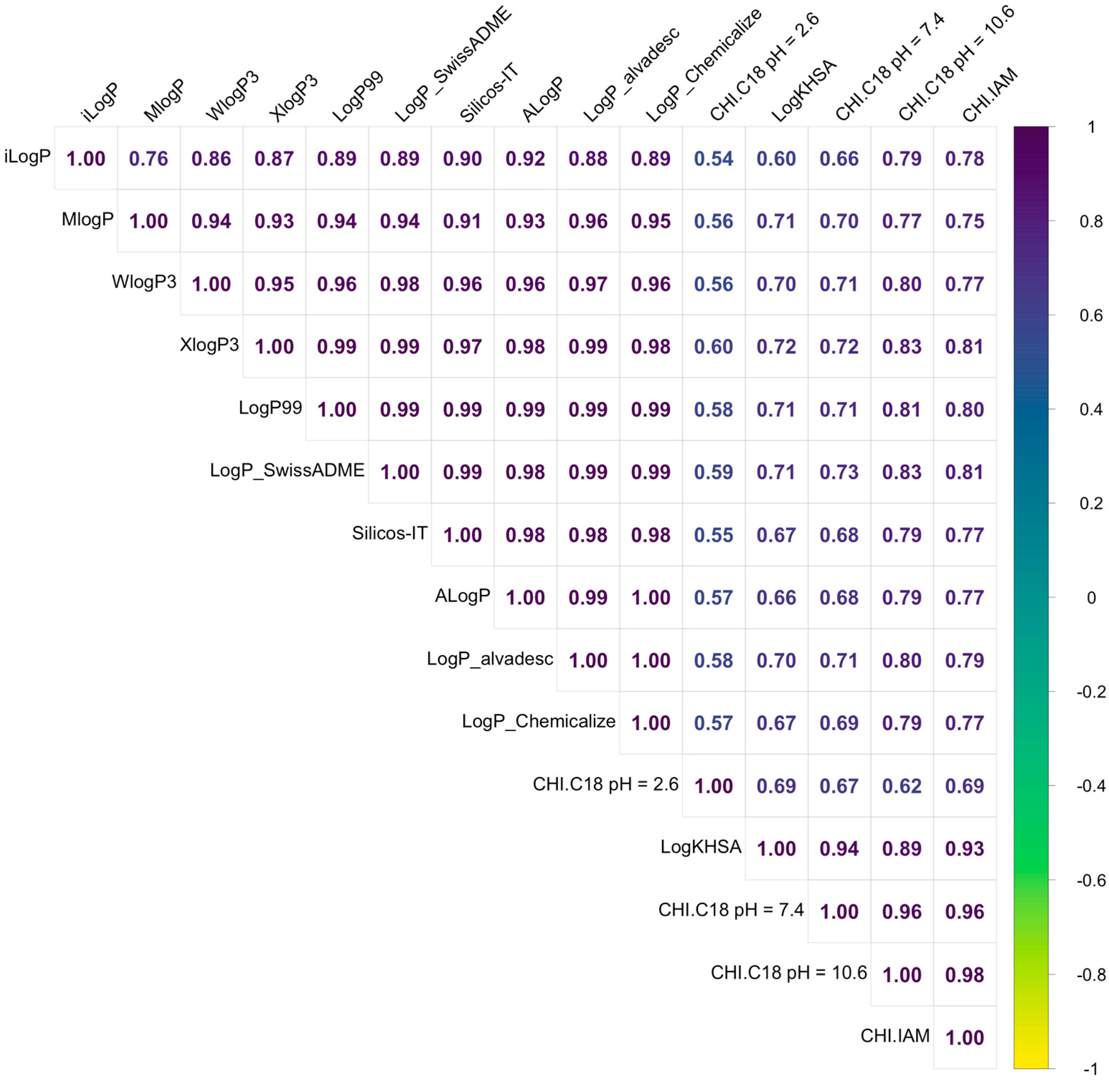
2.1. Lipophilicity Assessment
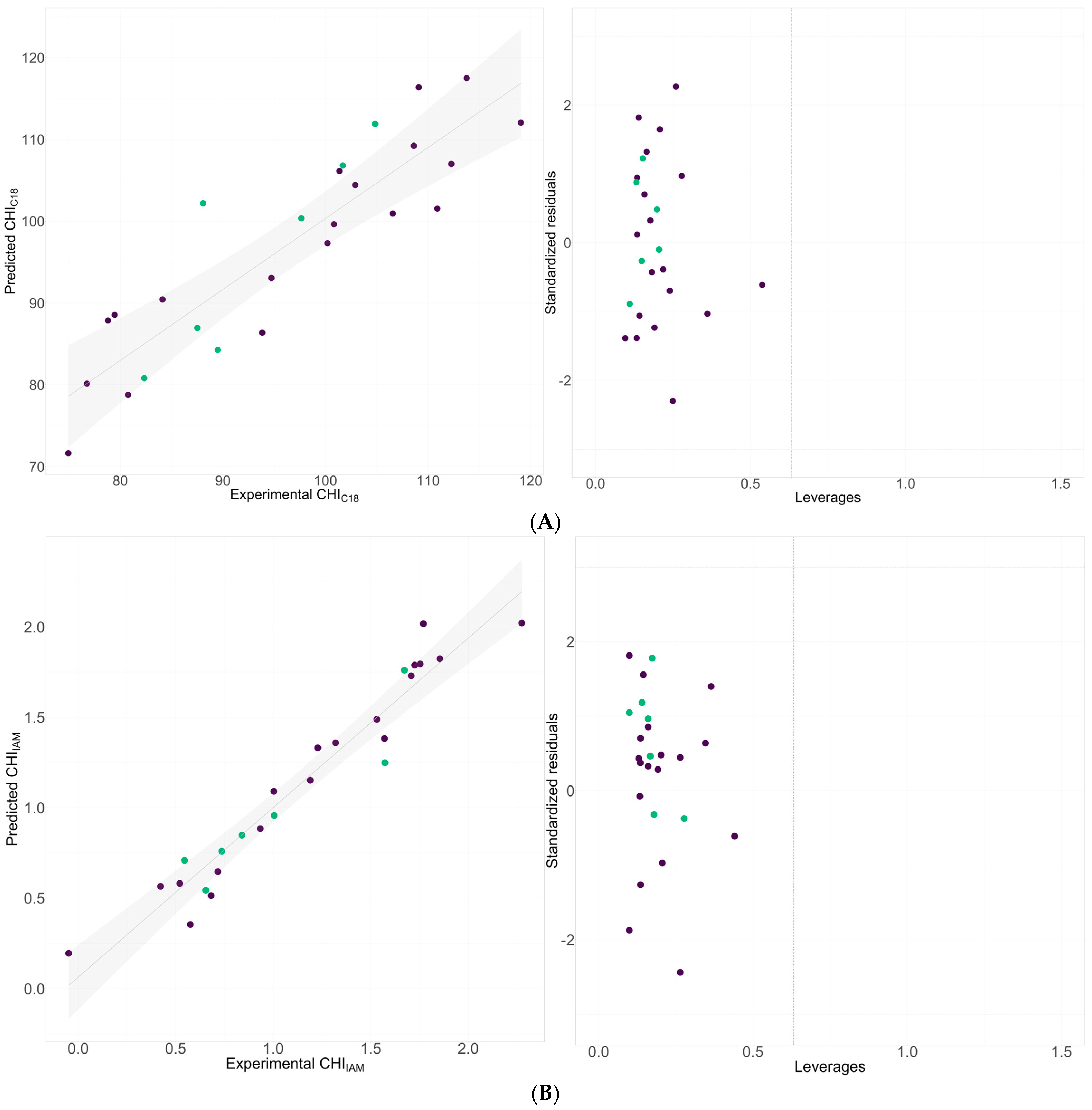
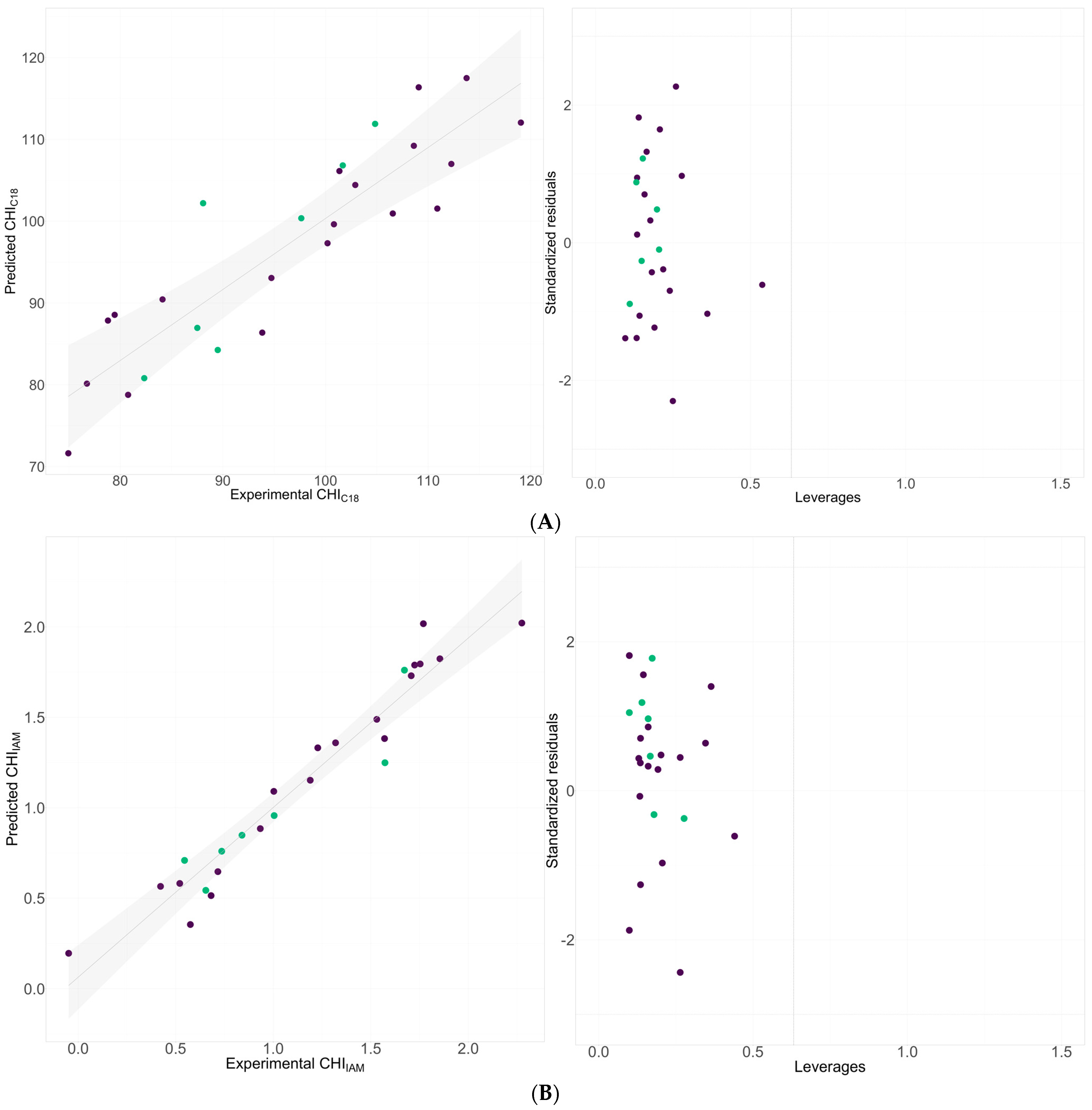
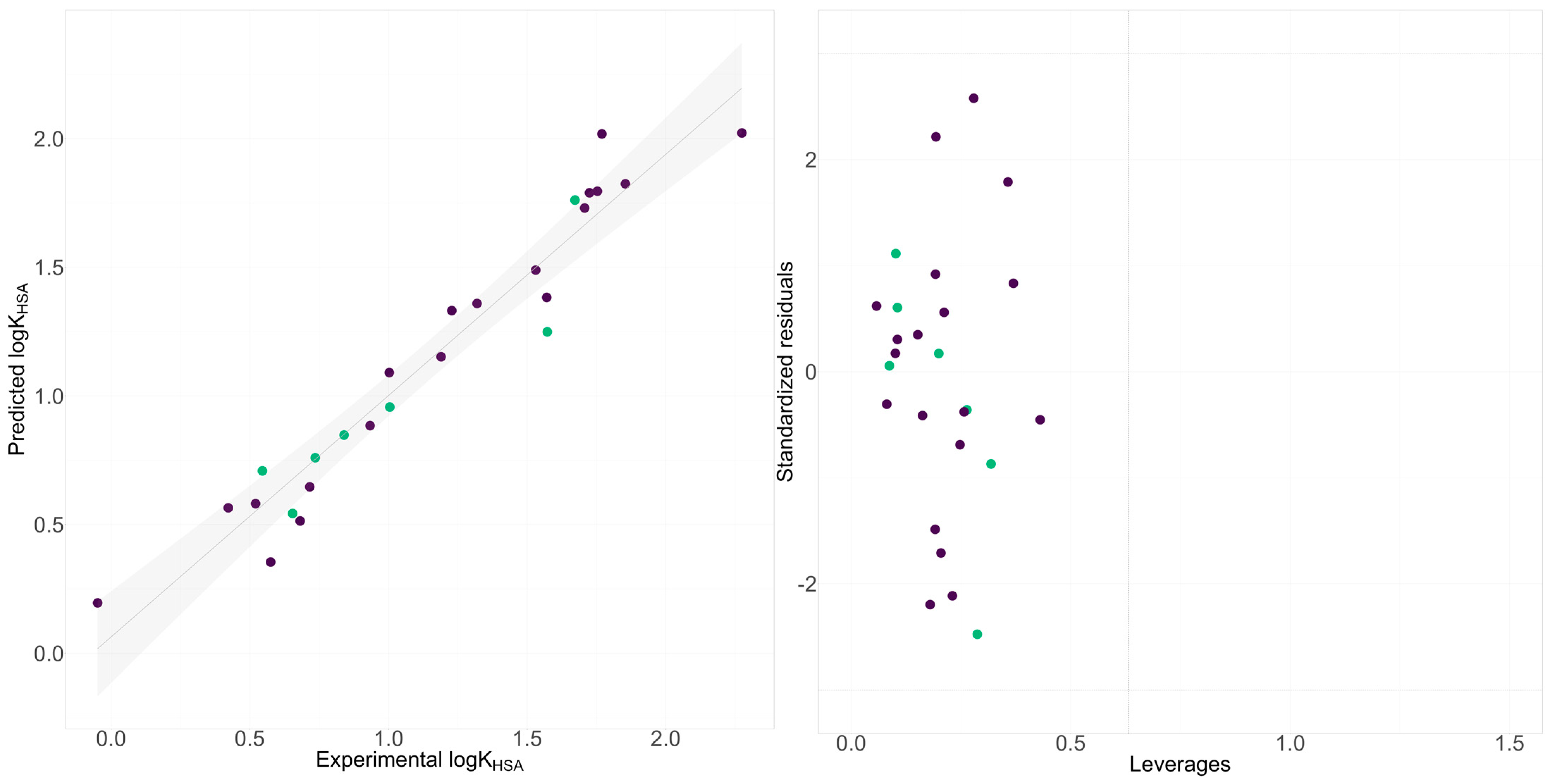
2.2. QSRR Modeling of Chromatography Determined Lipophilicity and Phospholipophilicity
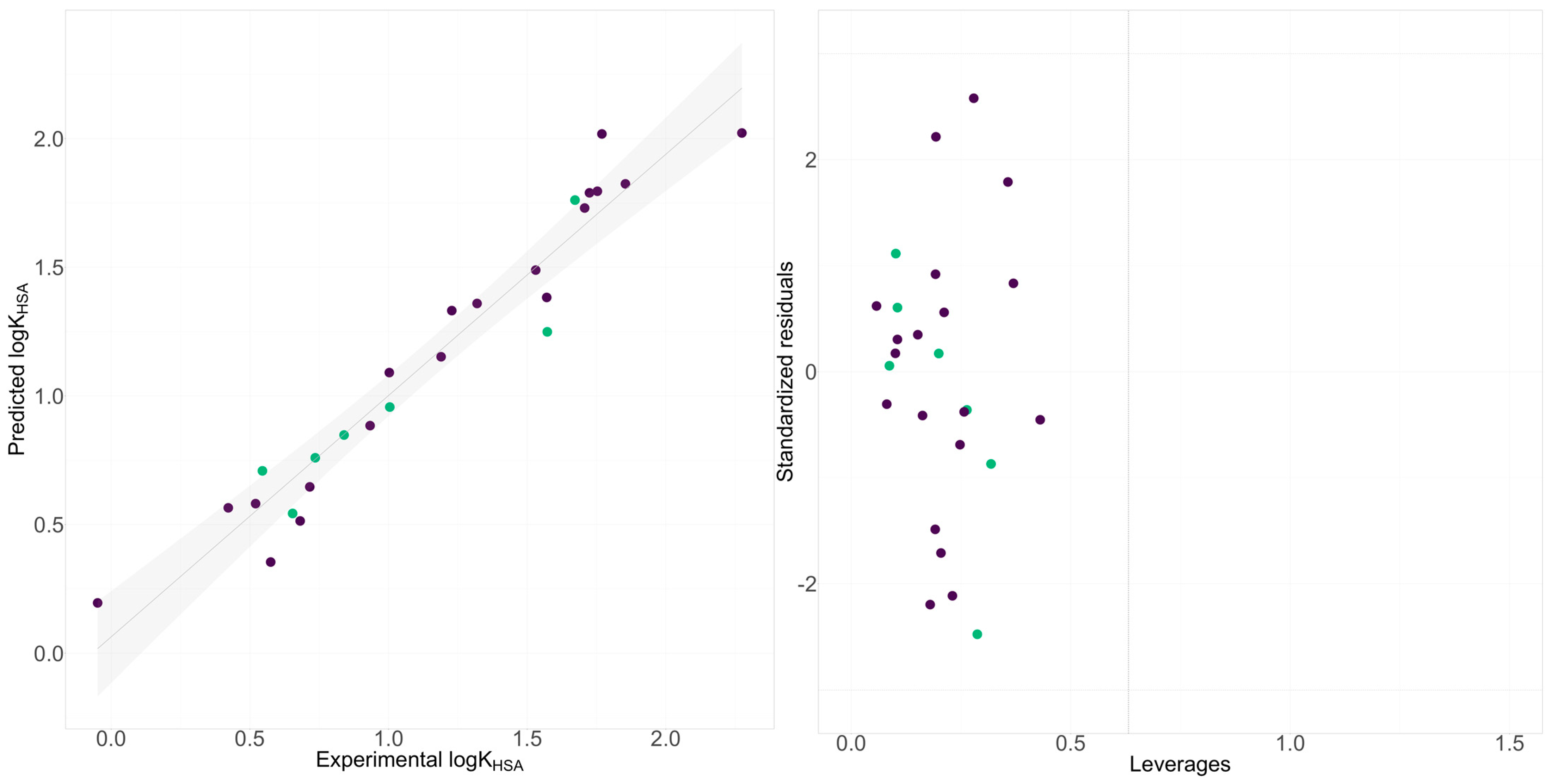
2.3. Interaction between Plasma Protein
3. Materials and Methods
3.1. Solvents
3.2. Analytes
Calibration Sets
3.3. Chromatographic Analysis
3.4. Theoretical Descriptors
3.5. CA Analysis
3.6. QSRR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valko, K.L. Biomimetic Chromatography—A Novel Application of the Chromatographic Principles. Anal. Sci. Adv. 2022, 3, 146–153. [Google Scholar] [CrossRef]
- Valkó, K.L. Lipophilicity and Biomimetic Properties Measured by HPLC to Support Drug Discovery. J. Pharm. Biomed. 2016, 130, 35–54. [Google Scholar] [CrossRef] [PubMed]
- van de Waterbeemd, H.; Gifford, E. ADMET in Silico Modelling: Towards Prediction Paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Valko, K.; Nunhuck, S.; Bevan, C.; Abraham, M.H.; Reynolds, D.P. Fast Gradient HPLC Method to Determine Compounds Binding to Human Serum Albumin. Relationships with Octanol/Water and Immobilized Artificial Membrane Lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. [Google Scholar] [CrossRef] [PubMed]
- Woziński, M.; Greber, K.E.; Pastewska, M.; Kolasiński, P.; Hewelt-Belka, W.; Żołnowska, B.; Sławiński, J.; Szulczyk, D.; Sawicki, W.; Ciura, K. Modification of Gradient HPLC Method for Determination of Small Molecules’ Affinity to Human Serum Albumin under Column Safety Conditions: Robustness and Chemometrics Study. J. Pharm. Biomed. Anal. 2023, 239, 115916. [Google Scholar] [CrossRef] [PubMed]
- Kułaga, D.; Jaśkowska, J.; Satała, G. Radioligand and Computational Insight in Structure—Activity Relationship of Saccharin Derivatives Being Ipsapirone and Revospirone Analogues. Bioorganic Med. Chem. Lett. 2021, 42, 128028. [Google Scholar] [CrossRef] [PubMed]
- Kułaga, D.; Jaśkowska, J.; Satała, G. Design, Synthesis and Biological Evaluation of Novel Serotonin and Dopamine Receptor Ligands Being 6-Bromohexyl Saccharine Derivatives. Bioorganic Med. Chem. Lett. 2019, 29, 126667. [Google Scholar] [CrossRef]
- Vallianatou, T.; Tsopelas, F.; Tsantili-Kakoulidou, A. Cheminformatics, QSAR and Machine Learning Applications for Novel Drug Development; Elsevier: Amsterdam, The Netherlands, 2023; pp. 573–607. [Google Scholar] [CrossRef]
- Dąbrowska, M.; Starek, M.; Komsta, Ł.; Szafrański, P.; Stasiewicz-Urban, A.; Opoka, W. Assessment of the Chromatographic Lipophilicity of Eight Cephalosporins on Different Stationary Phases. Eur. J. Pharm. Sci. 2017, 101, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hubicka, U.; Żuromska-Witek, B.; Komsta, Ł.; Krzek, J. Lipophilicity Study of Fifteen Fluoroquinolones by Reversed-Phase Thin-Layer Chromatography. Anal. Methods 2015, 7, 3841–3848. [Google Scholar] [CrossRef]
- Starek, M.; Komsta, Ł.; Krzek, J. Reversed-Phase Thin-Layer Chromatography Technique for the Comparison of the Lipophilicity of Selected Non-Steroidal Anti-Inflammatory Drugs. J. Pharm. Biomed. Anal. 2013, 85, 132–137. [Google Scholar] [CrossRef]
- Valko, K.L.; Zhang, T. Biomimetic Properties and Estimated in Vivo Distribution of Chloroquine and Hydroxy-Chloroquine Enantiomers. ADMET DMPK 2021, 9, 151–165. [Google Scholar] [CrossRef]
- Valkó, K.L.; Nunhuck, S.B.; Hill, A.P. Estimating Unbound Volume of Distribution and Tissue Binding by in Vitro HPLC-based Human Serum Albumin and Immobilised Artificial Membrane-binding Measurements. J. Pharm. Sci. 2011, 100, 849–862. [Google Scholar] [CrossRef]
- Valko, K.L.; Rava, S.; Bunally, S.; Anderson, S. Revisiting the Application of Immobilized Artificial Membrane (IAM) Chromatography to Estimate in Vivo Distribution Properties of Drug Discovery Compounds Based on the Model of Marketed Drugs. ADMET DMPK 2020, 8, 78–97. [Google Scholar] [CrossRef]
- Kaliszan, R. QSRR: Quantitative Structure-(Chromatographic) Retention Relationships. Chem. Rev. 2007, 107, 3212–3246. [Google Scholar] [CrossRef]
- Kumari, P.; Laethem, T.V.; Duroux, D.; Fillet, M.; Hubert, P.; Sacré, P.-Y.; Hubert, C. A Multi-Target QSRR Approach to Model Retention Times of Small Molecules in RPLC. J. Pharm. Biomed. Anal. 2023, 236, 115690. [Google Scholar] [CrossRef]
- Naylor, B.C.; Catrow, J.L.; Maschek, J.A.; Cox, J.E. QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics. Metabolites 2020, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Wiczling, P.; Kamedulska, A. Comparison of Chromatographic Stationary Phases Using a Bayesian-Based Multilevel Model. Anal. Chem. 2024, 96, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, S.; Banjac, M.K.; Milošević, N.; Ćurčić, J.; Marjanović, D.; Todorović, N.; Krmar, J.; Podunavac-Kuzmanović, S.; Banjac, N.; Ušćumlić, G. Comparative Chemometric and Quantitative Structure-Retention Relationship Analysis of Anisotropic Lipophilicity of 1-Arylsuccinimide Derivatives Determined in High-Performance Thin-Layer Chromatography System with Aprotic Solvents. J. Chromatogr. A 2020, 1628, 461439. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, S.; Banjac, M.K.; Anojčić, J.; Podunavac-Kuzmanović, S.; Jevrić, L.; Nikolić, A.; Savić, M.; Kuzminac, I. Chemometrics of Anisotropic Lipophilicity of Anticancer Androstane Derivatives Determined by Reversed-Phase Ultra High Performance Liquid Chromatography with Polar Aprotic and Protic Modifiers. J. Chromatogr. A 2022, 1673, 463197. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘Rule of Three’ for fragment-based lead discovery. Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Reutlinger, M.; Koch, C.P.; Reker, D.; Todoroff, N.; Schneider, P.; Rodrigues, T.; Schneider, G. Chemically Advanced Template Search (CATS) for Scaffold-Hopping and Prospective Target Prediction for “Orphan” Molecules. Mol. Inf. 2013, 32, 133–138. [Google Scholar] [CrossRef]
- Kratz, F.; Elsadek, B. Clinical Impact of Serum Proteins on Drug Delivery. J. Control. Release 2012, 161, 429–445. [Google Scholar] [CrossRef]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin–Drug Interaction and Its Clinical Implication. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Patt, M.; Becker, G.A.; Grossmann, U.; Habermann, B.; Schildan, A.; Wilke, S.; Deuther-Conrad, W.; Graef, S.; Fischer, S.; Smits, R.; et al. Evaluation of Metabolism, Plasma Protein Binding and Other Biological Parameters after Administration of (−)-[18F]Flubatine in Humans. Nucl. Med. Biol. 2014, 41, 489–494. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A New Software for the Development, Analysis, and Validation of QSAR MLR Models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria Datasets and New QSAR/QSPR Models for Environmental Pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | MLogP | ALogP | LogP99 | LogAlvaDesc | LogChemicalize | iLogP | XLogP3 | WLogP | Silicos-IT | LogPSwissADME | CHI LogP * |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.15 | 2.40 | 2.04 | 2.20 | 2.49 | 2.92 | 2.57 | 1.98 | 1.59 | 2.19 | 3.48 |
| 2 | 1.89 | 2.38 | 2.05 | 2.11 | 2.33 | 3.40 | 2.55 | 1.99 | 1.65 | 2.23 | 4.44 |
| 3 | 2.86 | 3.72 | 3.35 | 3.31 | 3.70 | 3.32 | 3.83 | 3.29 | 2.88 | 3.18 | 3.27 |
| 4 | 2.64 | 3.06 | 2.70 | 2.80 | 3.09 | 3.27 | 3.2 | 2.64 | 2.23 | 2.74 | 3.97 |
| 5 | 2.60 | 3.43 | 2.82 | 2.95 | 3.45 | 3.36 | 3.29 | 2.76 | 2.37 | 2.82 | 3.70 |
| 6 | 2.32 | 3.41 | 2.83 | 2.86 | 3.29 | 3.58 | 3.26 | 2.77 | 2.44 | 2.81 | 3.49 |
| 7 | 3.29 | 4.76 | 4.13 | 4.06 | 4.66 | 4.03 | 4.54 | 4.07 | 3.66 | 3.86 | 4.63 |
| 8 | 3.08 | 4.10 | 3.48 | 3.55 | 4.06 | 3.71 | 3.92 | 3.42 | 3.01 | 3.37 | 3.98 |
| 9 | 1.86 | 2.17 | 1.61 | 1.88 | 2.21 | 3.12 | 1.90 | 1.55 | 1.25 | 1.78 | 2.91 |
| 10 | 2.43 | 2.82 | 2.22 | 2.49 | 2.83 | 3.18 | 2.55 | 2.16 | 1.81 | 2.28 | 3.10 |
| 11 | 3.29 | 4.34 | 3.98 | 3.87 | 4.44 | 3.77 | 4.54 | 3.92 | 3.40 | 3.72 | 4.55 |
| 12 | 2.81 | 3.89 | 3.21 | 3.30 | 3.90 | 3.68 | 3.65 | 3.15 | 2.77 | 3.16 | 3.98 |
| 13 | 2.53 | 3.87 | 3.22 | 3.21 | 3.74 | 3.86 | 3.62 | 3.16 | 2.84 | 3.14 | 3.76 |
| 14 | 2.53 | 3.87 | 3.22 | 3.21 | 3.74 | 3.92 | 3.62 | 3.16 | 2.84 | 3.15 | 3.92 |
| 15 | 2.53 | 3.87 | 3.22 | 3.21 | 3.74 | 3.96 | 3.62 | 3.16 | 2.84 | 3.16 | 3.76 |
| 16 | 3.29 | 4.55 | 3.87 | 3.90 | 4.50 | 3.94 | 4.27 | 3.81 | 3.41 | 3.69 | 4.58 |
| 17 | 3.29 | 4.55 | 3.87 | 3.90 | 4.50 | 3.92 | 4.27 | 3.81 | 3.41 | 3.69 | 4.49 |
| 18 | 3.50 | 5.22 | 4.52 | 4.41 | 5.10 | 4.20 | 4.90 | 4.46 | 4.05 | 4.17 | 4.93 |
| 19 | 3.60 | 4.83 | 4.23 | 4.22 | 4.77 | 4.01 | 4.53 | 5.32 | 3.86 | 4.21 | 4.69 |
| 20 | 3.18 | 4.09 | 3.35 | 3.54 | 4.04 | 3.70 | 3.75 | 3.71 | 3.19 | 3.45 | 4.19 |
| 21 | 3.49 | 4.80 | 4.37 | 4.22 | 4.89 | 4.11 | 4.90 | 4.31 | 3.79 | 4.06 | 4.91 |
| 22 | 2.65 | 3.28 | 2.61 | 2.84 | 3.27 | 3.47 | 2.91 | 2.55 | 2.20 | 2.61 | 3.44 |
| 23 | 2.09 | 2.63 | 2.00 | 2.24 | 2.65 | 3.33 | 2.26 | 1.94 | 1.64 | 2.10 | 3.37 |
| 24 | 3.00 | 4.64 | 3.82 | 3.82 | 4.56 | 4.03 | 4.38 | 3.76 | 3.85 | 3.70 | 4.33 |
| 25 | 3.16 | 4.75 | 4.43 | 4.11 | 4.77 | 4.09 | 4.66 | 4.37 | 4.42 | 4.12 | 4.40 |
| 26 | 2.72 | 3.84 | 3.36 | 3.31 | 3.92 | 3.54 | 3.48 | 3.00 | 2.81 | 3.05 | 3.93 |
| Name | Description | Software |
|---|---|---|
| MLogP | Based on quantitative structure–logP relationships, using topological indexes. | AlvaDesc |
| ALogP | Ghose–Crippen octanol–water partition coefficient | AlvaDesc |
| LogP99 | Wildmann–Crippen octanol–water partition coefficient—atom-based method | AlvaDesc |
| LogPconsAlvaDesc | Consensus model of LogP from AlvaDesc | AlvaDesc |
| LogPChemicalize | Atomic correction using the contribution of individual molecular fragments | Chemicalize |
| iLogP | In-house physics-based method relying on free energies of solvation in n-octanol and water calculated using Generalized Born and solvent-accessible surface area (GB/SA) model—atomic- and knowledge-based method | SwissADME |
| XLogP3 | Atomistic method including corrective factors and knowledge-based library—atomistic- and knowledge-based method | SwissADME |
| WLogP | Implementation of a purely atomistic method based on 27 fragments and 7 topological descriptors—hybrid fragmental/topological method | SwissADME |
| Silicos-IT | Hybrid method relying on 27 fragments and 7 topological descriptors—hybrid fragmental/topological method | SwissADME |
| Consensus LogPSwissADME | The arithmetic mean of the values predicted using the five propose methods—average of all predictions calculated using SwissADME | SwissADME |
| Model | Equation | |||||
|---|---|---|---|---|---|---|
| 1 | CHIC18 pH = 7.4 = 3.375(±1.761)RDF020i + 3.262(±0.456)CATS3D_12_LL − 141.299(±0.842) LLS_01 + 90.055(±20.430) | |||||
| 2 | CHIIAM = 0.872(±0.269)CATS3D_07_AL − 33.834(±0.565)LLS_01 − 1.615(±0.425) N% + 58.328(±6.162) | |||||
| 3 | LogKHSA = 0.114(±0.952) CHIIAM − 0.857(±0.141)GATS2e − 0.050(±0.274) RDF155u − 2.751(±0.994) | |||||
| R2 | RMSEtr | Q2LOO | R2EXT | RMSEP | CCCExt | |
| 1 | 0.838 | 5.561 | 0.748 | 0.751 | 6.687 | 0.770 |
| 2 | 0.844 | 1.944 | 0.736 | 0.852 | 2.019 | 0.863 |
| 3 | 0.946 | 0.138 | 0.910 | 0.876 | 0.149 | 0.930 |
| Model | Name | Description | Block |
|---|---|---|---|
| 1 | RDF020i | Radial Distribution Function—020/weighted using ionization potential | RDF descriptors |
| CATS3D_12_LL | CATS3D Lipophilic-Lipophilic BIN 12 (12.000–13.000 Å) | CATS 3D | |
| LLS_01 | modified lead-like score | Drug-like indices | |
| 2 | CATS3D_07_AL | CATS3D Acceptor-Lipophilic BIN 07 (7.000–8.000 Å) | CATS 3D |
| LLS_01 | modified lead-like score | Drug-like indices | |
| N% | percentage of N atoms | Constitutional indices | |
| 3 | CHI IAM | CHIIAM values from the experiment | Experiment |
| GATS2e | Geary autocorrelation of lag 2 weighted using Sanderson electronegativity | 2D autocorrelations | |
| RDF155u | Radial Distribution Function—155/unweighted | RDF descriptors |
| No. | CHIC18 pH = 2.6 | CHIC18 pH = 7.4 | CHIC18 pH = 10.6 | CHIIAM | LogKHSA | %HSA |
|---|---|---|---|---|---|---|
| 1 | 48.96 | 88.07 | 91.64 | 36.93 | 0.74 | 85.32 |
| 2 | 60.08 | 110.90 | 109.41 | 46.90 | 1.72 | 99.13 |
| 3 | 49.19 | 78.80 | 87.69 | 35.71 | 0.52 | 77.60 |
| 4 | 56.31 | 100.82 | 100.70 | 42.61 | 1.32 | 96.38 |
| 5 | 52.20 | 89.50 | 95.60 | 39.09 | 0.84 | 88.23 |
| 6 | 52.94 | 79.45 | 91.77 | 38.03 | 0.68 | 83.59 |
| 7 | 72.79 | 112.26 | 112.92 | 48.28 | 1.77 | 99.31 |
| 8 | 97.99 | 101.36 | 100.81 | 44.30 | 1.53 | 98.11 |
| 9 | 48.71 | 74.93 | 81.12 | 34.40 | 0.58 | 79.78 |
| 10 | 38.96 | 76.74 | 84.51 | 35.73 | 0.72 | 84.71 |
| 11 | 63.51 | 104.83 | 111.43 | 47.26 | 1.67 | 98.90 |
| 12 | 55.26 | 93.84 | 100.78 | 41.59 | 1.00 | 91.87 |
| 13 | 54.89 | 84.12 | 96.78 | 40.54 | 0.42 | 73.28 |
| 14 | 55.41 | 94.73 | 99.75 | 41.42 | 0.93 | 90.46 |
| 15 | 55.04 | 87.50 | 96.78 | 40.04 | 0.65 | 82.67 |
| 16 | 61.89 | 108.61 | 111.92 | 46.73 | 1.71 | 99.06 |
| 17 | 61.34 | 106.56 | 110.38 | 46.32 | 1.57 | 98.36 |
| 18 | 65.39 | 119.04 | 118.39 | 50.19 | 2.27 | <99.80 |
| 19 | 63.86 | 113.75 | 114.05 | 46.84 | 1.85 | 99.61 |
| 20 | 57.21 | 97.64 | 104.68 | 42.69 | 1.00 | 91.90 |
| 21 | 65.07 | 109.08 | 118.03 | 48.85 | 1.75 | 99.25 |
| 22 | 43.25 | 82.33 | 90.87 | 38.48 | 0.55 | 78.60 |
| 23 | 41.03 | 80.76 | 89.54 | 34.77 | −0.05 | 47.66 |
| 24 | 60.18 | 102.90 | 107.33 | 45.78 | 1.23 | 95.36 |
| 25 | 60.65 | 101.68 | 108.64 | 45.96 | 1.57 | 98.37 |
| 26 | 55.76 | 100.20 | 100.01 | 41.48 | 1.19 | 94.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nisterenko, W.; Kułaga, D.; Woziński, M.; Singh, Y.R.; Judzińska, B.; Jagiello, K.; Greber, K.E.; Sawicki, W.; Ciura, K. Evaluation of Physicochemical Properties of Ipsapirone Derivatives Based on Chromatographic and Chemometric Approaches. Molecules 2024, 29, 1862. https://doi.org/10.3390/molecules29081862
Nisterenko W, Kułaga D, Woziński M, Singh YR, Judzińska B, Jagiello K, Greber KE, Sawicki W, Ciura K. Evaluation of Physicochemical Properties of Ipsapirone Derivatives Based on Chromatographic and Chemometric Approaches. Molecules. 2024; 29(8):1862. https://doi.org/10.3390/molecules29081862
Chicago/Turabian StyleNisterenko, Wiktor, Damian Kułaga, Mateusz Woziński, Yash Raj Singh, Beata Judzińska, Karolina Jagiello, Katarzyna Ewa Greber, Wiesław Sawicki, and Krzesimir Ciura. 2024. "Evaluation of Physicochemical Properties of Ipsapirone Derivatives Based on Chromatographic and Chemometric Approaches" Molecules 29, no. 8: 1862. https://doi.org/10.3390/molecules29081862
APA StyleNisterenko, W., Kułaga, D., Woziński, M., Singh, Y. R., Judzińska, B., Jagiello, K., Greber, K. E., Sawicki, W., & Ciura, K. (2024). Evaluation of Physicochemical Properties of Ipsapirone Derivatives Based on Chromatographic and Chemometric Approaches. Molecules, 29(8), 1862. https://doi.org/10.3390/molecules29081862