
A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates


Abstract
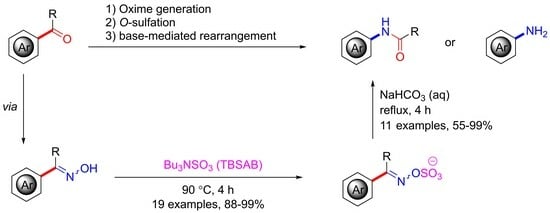
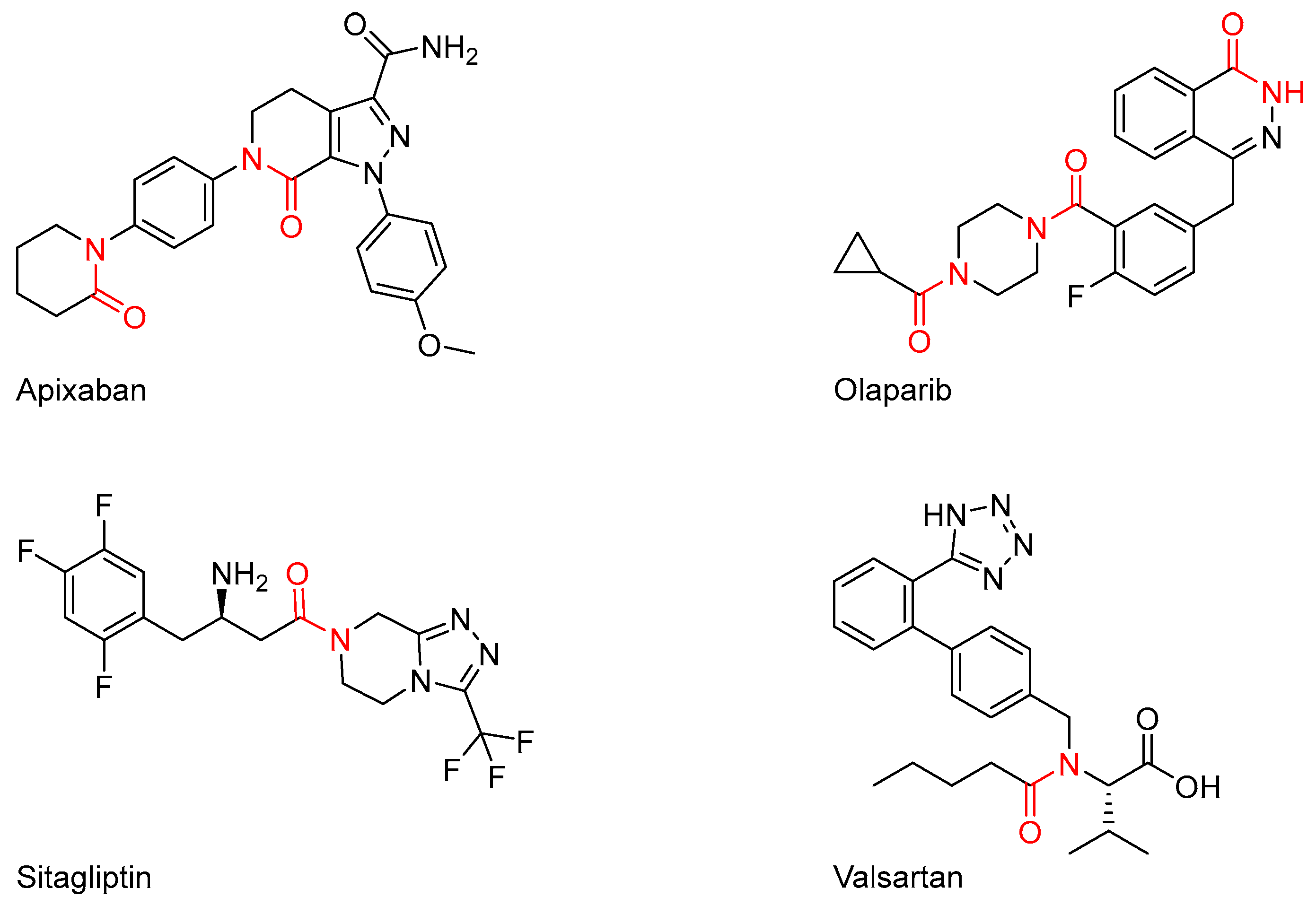
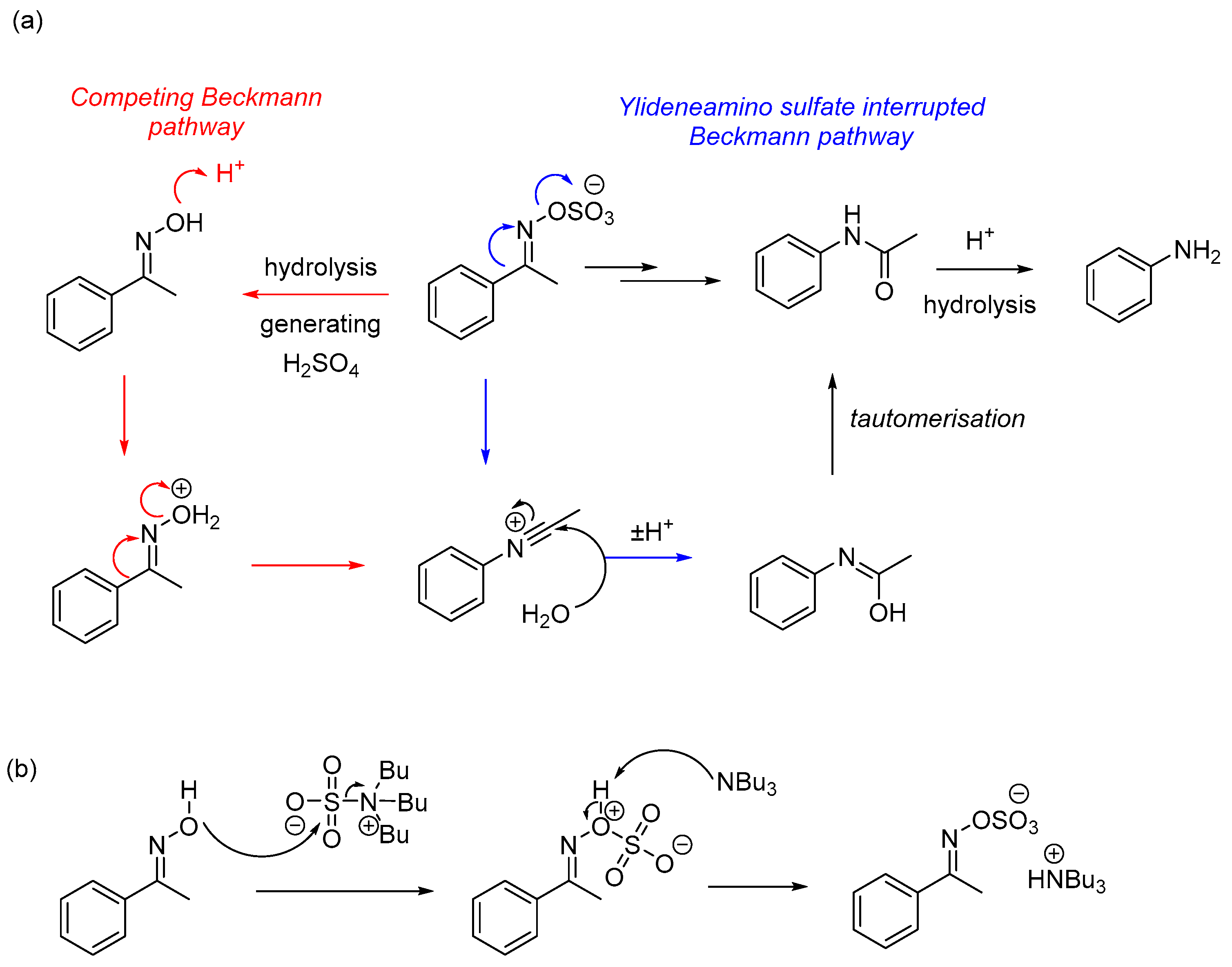
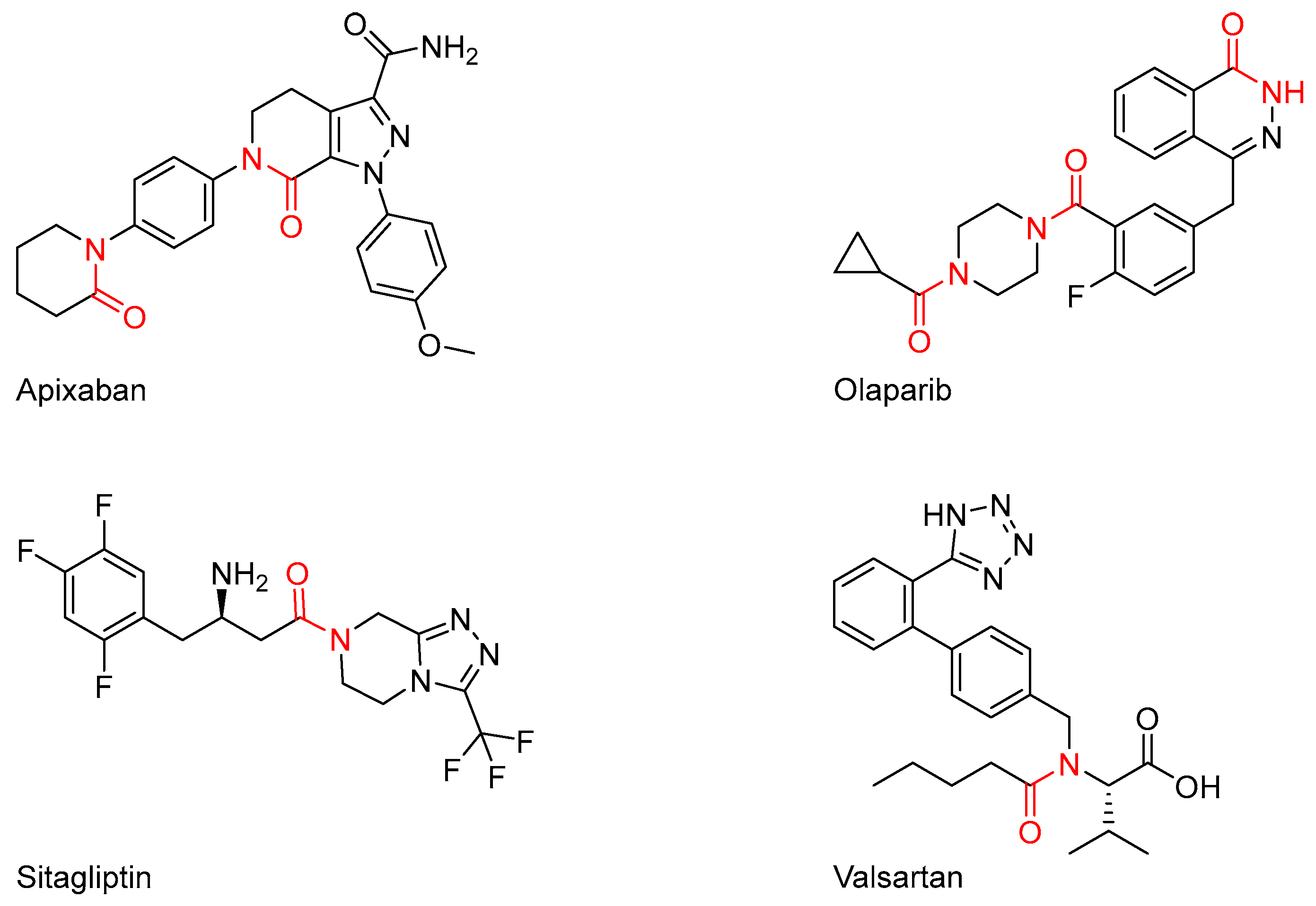
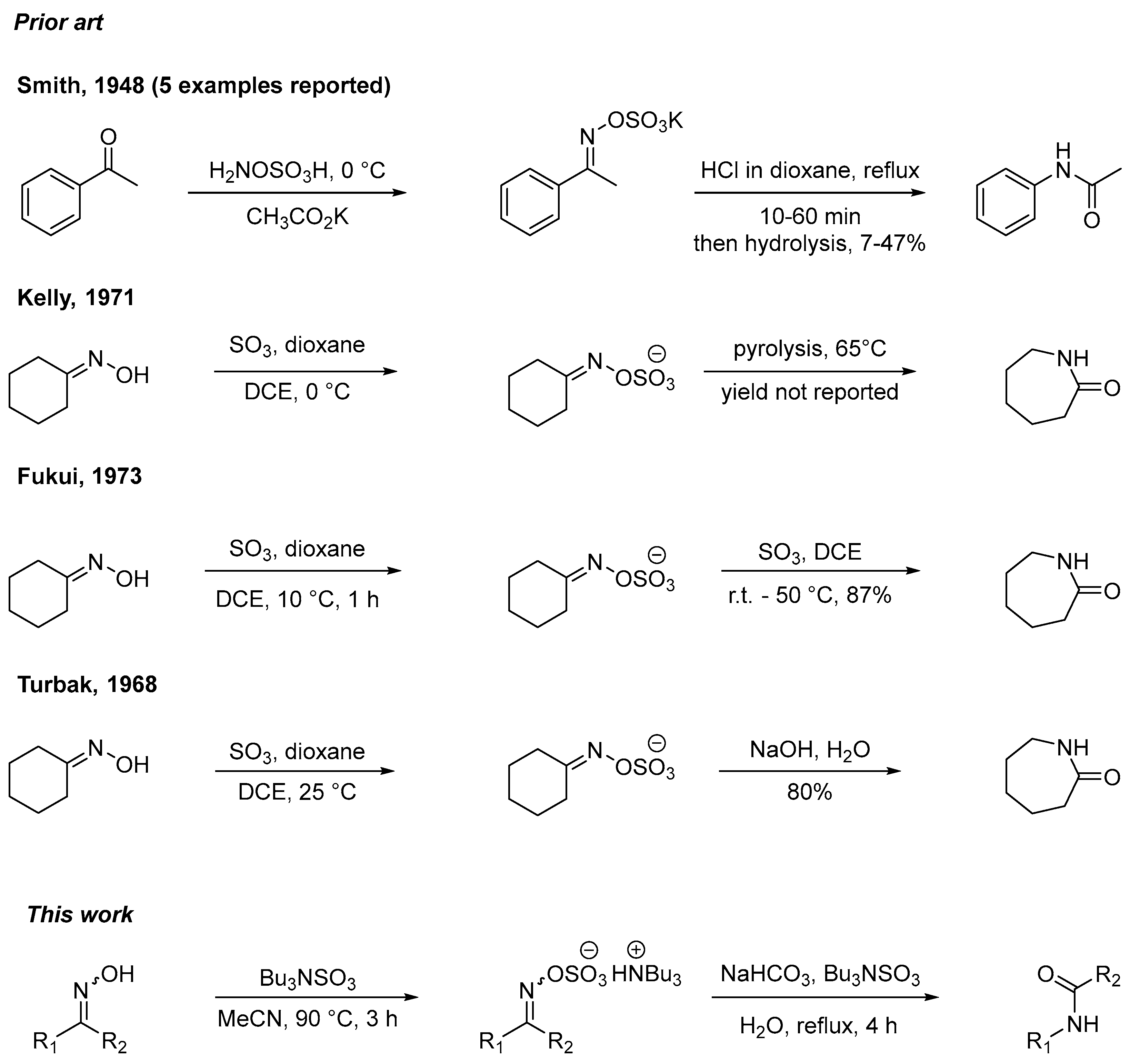
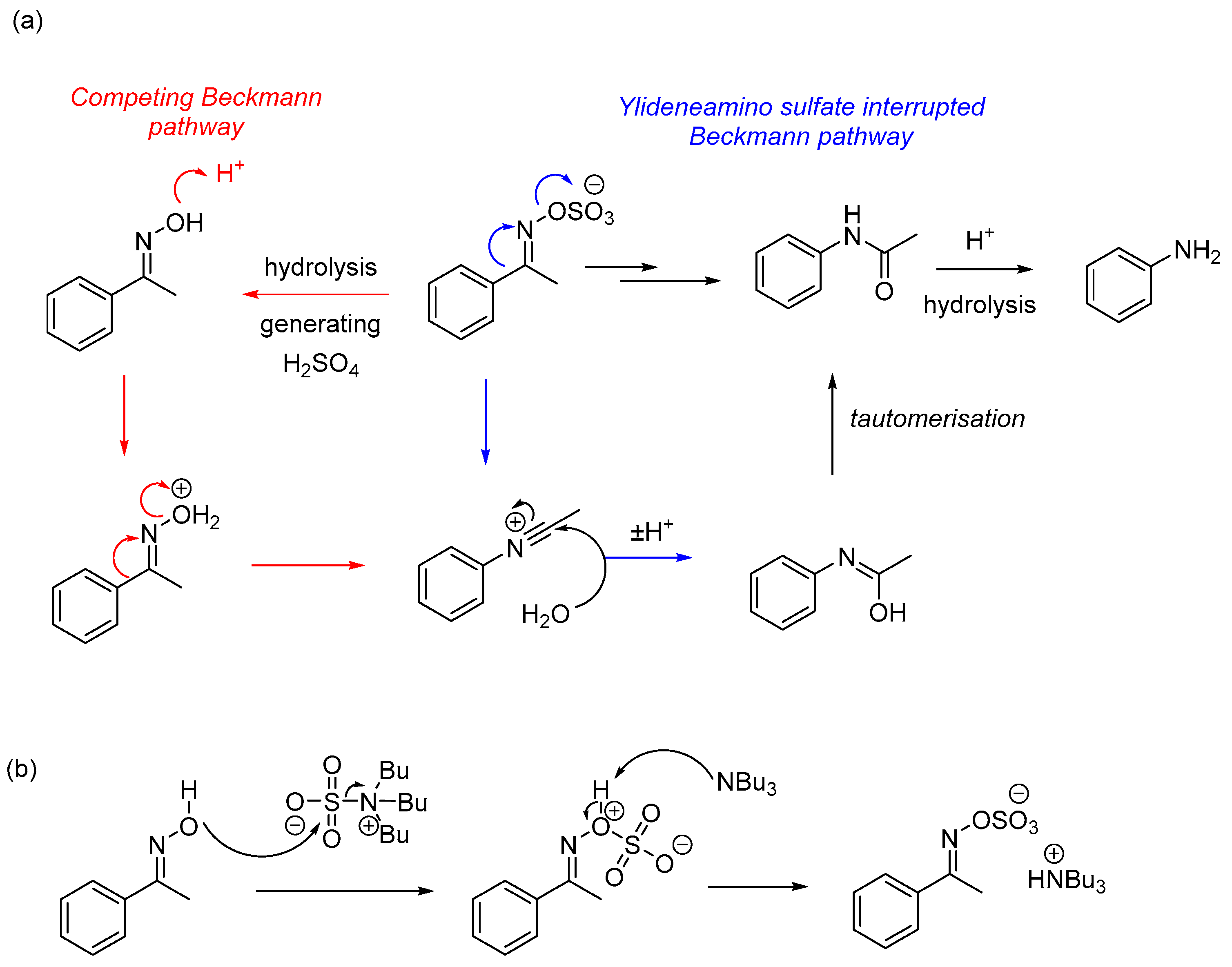
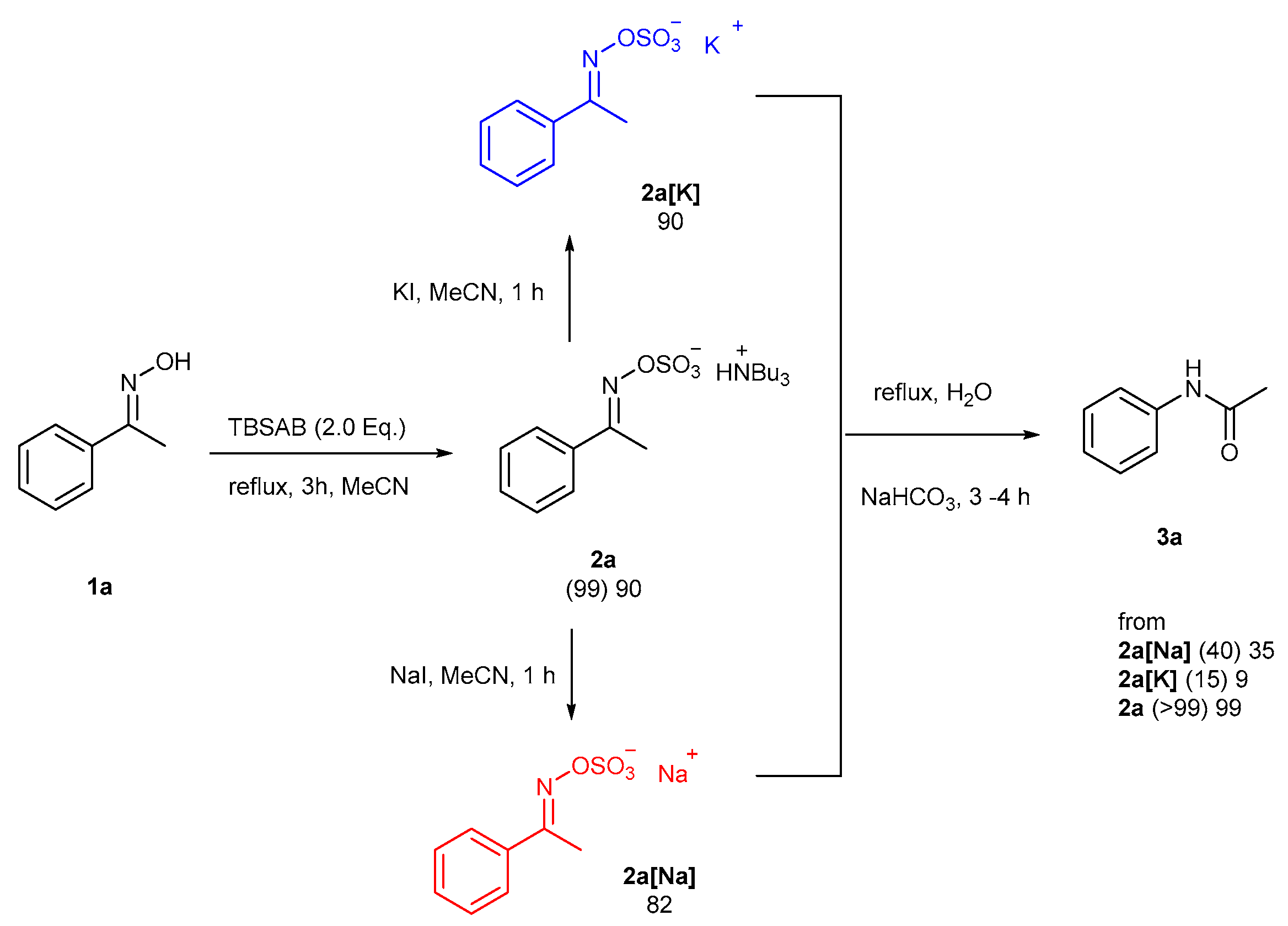
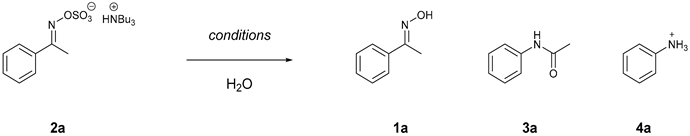
1. Introduction
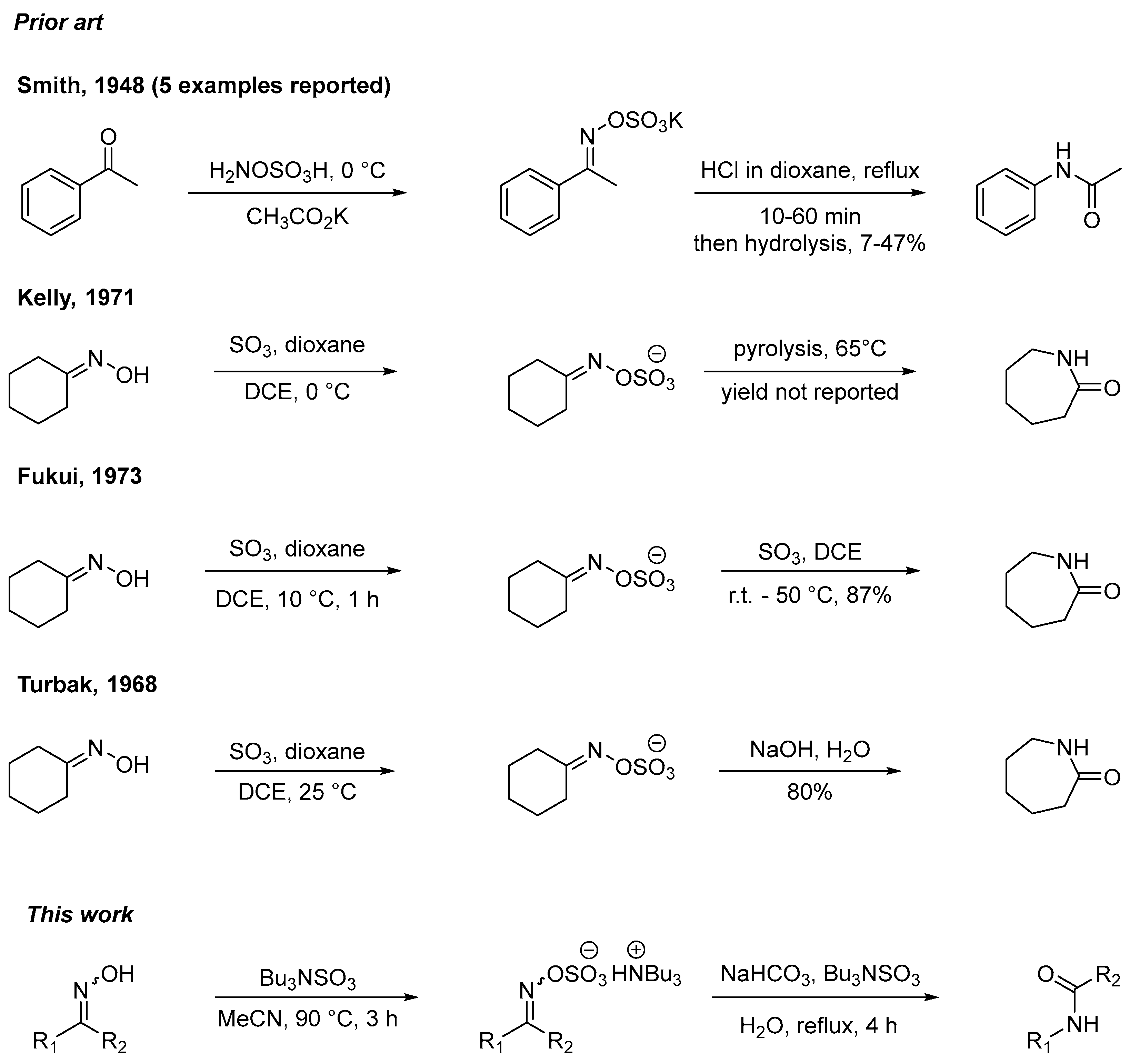
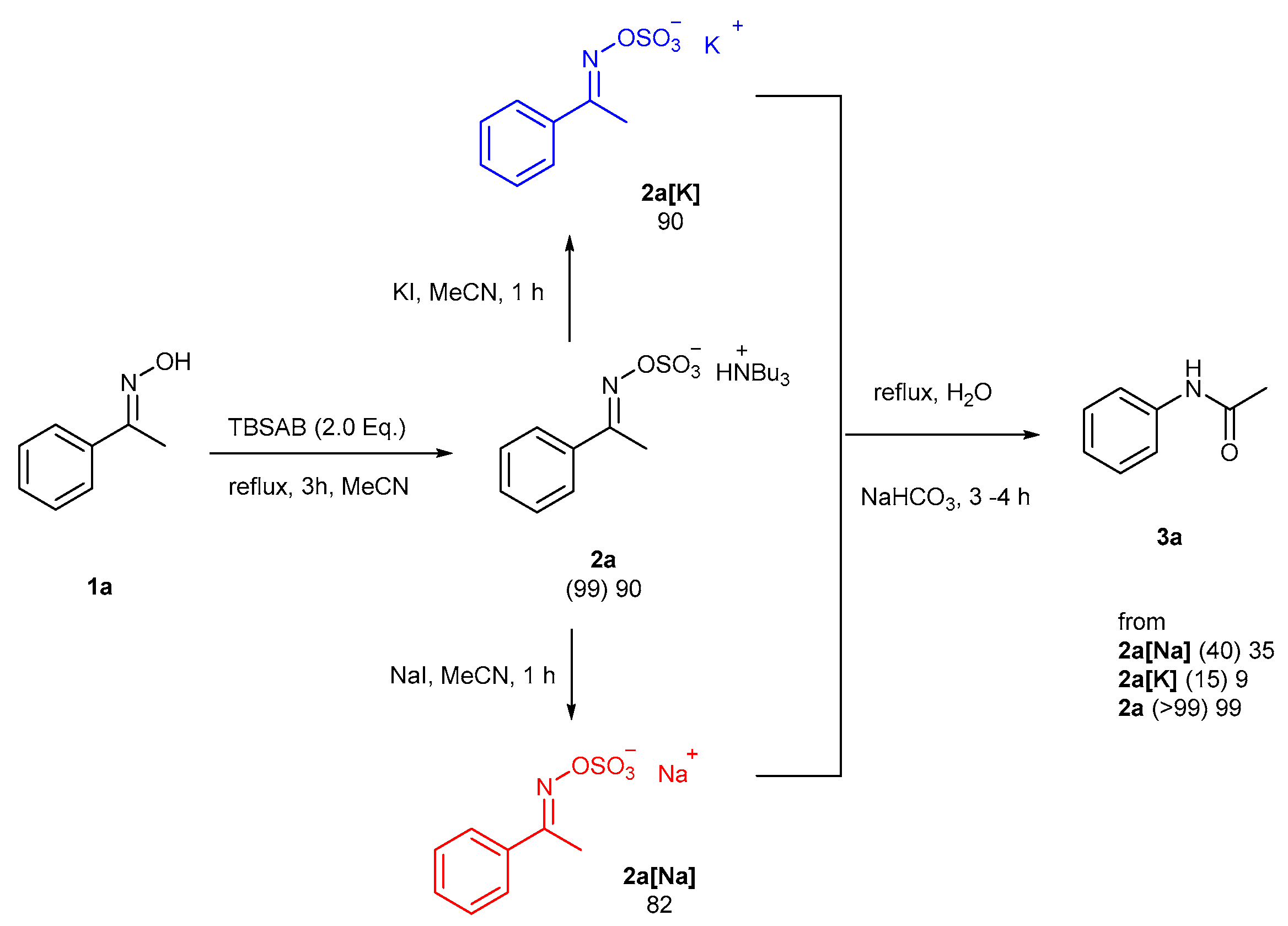
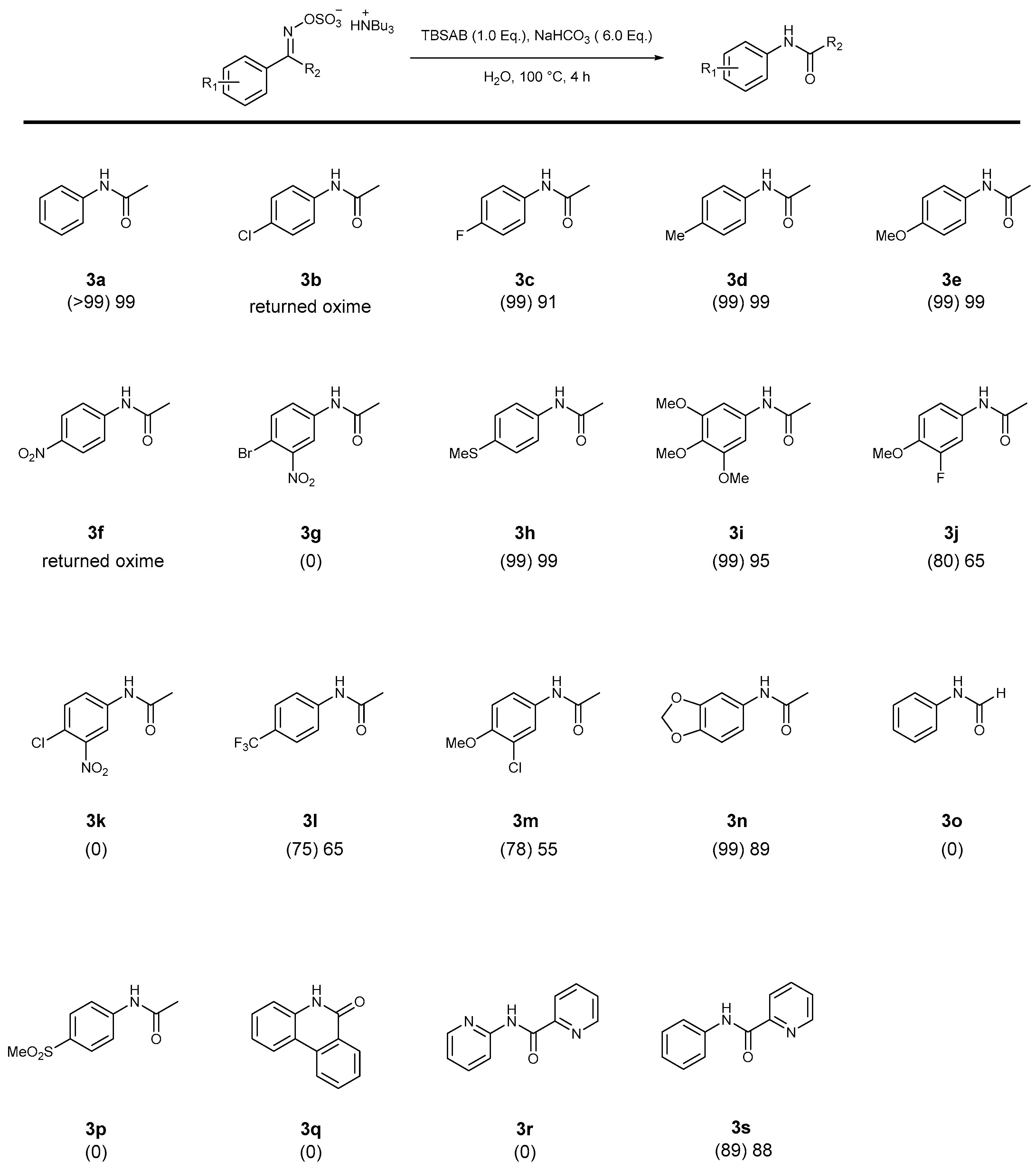
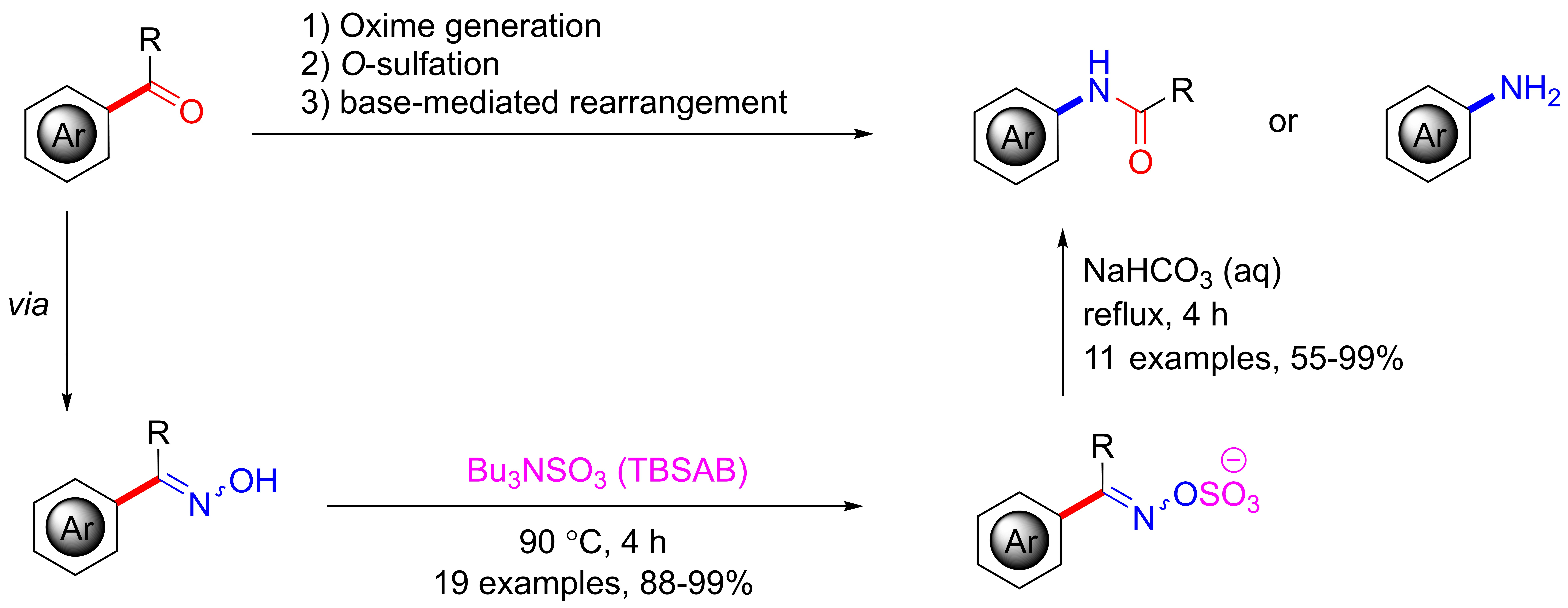
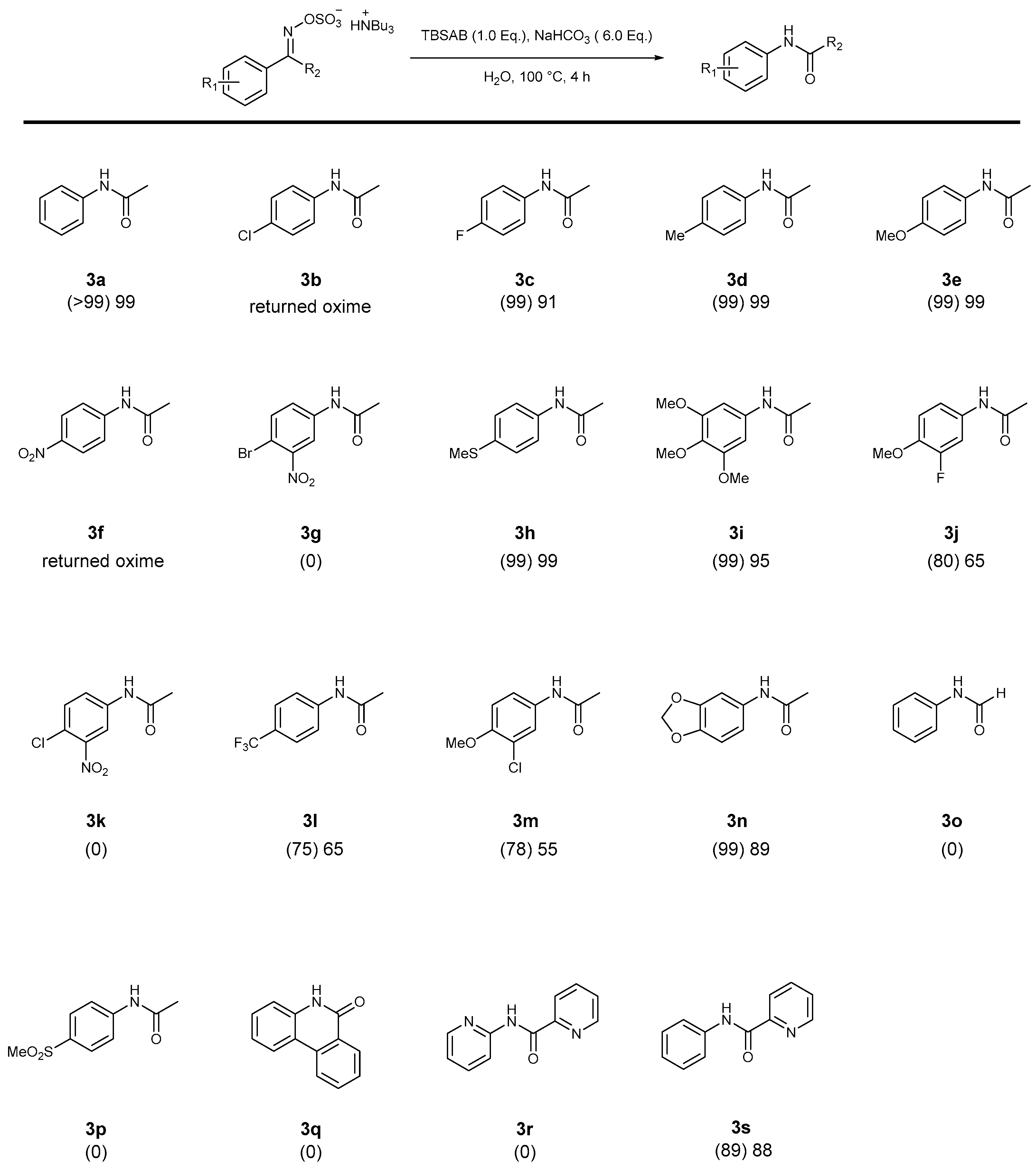
2. Results and Discussion
3. Conclusions
4. Experimental
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salim, H.; Jones, A.M. Angiotensin II receptor blockers (ARBs) and manufacturing contamination: A retrospective National Register Study into suspected associated adverse drug reactions. Br. J. Clin. Pharmacol. 2022, 88, 4812–4827. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Jones, A.M. Suspected adverse drug reactions of the type 2 antidiabetic drug class dipeptidyl-peptidase IV inhibitors (DPP4i): Can polypharmacology help explain? Pharmacol. Res. Perspect. 2022, 10, e01029. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, D.; Antolin, A.A.; Cox, A.R.; Jones, A.M. Identification of different side effects between PARP inhibitors and their polypharmacological multi-target rationale. Br. J. Clin. Pharmacol. 2022, 88, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Ferro, C.J.; Solkhon, F.; Jalal, Z.; Al-Hamid, A.M.; Jones, A.M. Relevance of physicochemical properties and functional pharmacology data to predict the clinical safety profile of direct oral anticoagulants. Pharmacol. Res. Perspect. 2020, 8, e00603. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, E. Zur Kenntniss der Isonitrosoverbindungen. Ber. Dtsch. Chem. Ges. 1886, 19, 988–993. [Google Scholar] [CrossRef]
- Kaur, K.; Srivastava, S. Beckmann rearrangement catalysis: A review of recent advances. New J. Chem. 2020, 44, 18530–18572. [Google Scholar] [CrossRef]
- Hashimoto, M.; Obora, Y.; Sakaguchi, S.; Ishii, Y. Beckmann Rearrangement of Ketoximes to Lactams by Triphosphazene Catalyst. J. Org. Chem. 2008, 73, 2894–2897. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Morgan, T.D.R.; Ang, H.T.; Hall, D.G. Scope and Mechanism of a True Organocatalytic Beckmann Rearrangement with a Boronic Acid/Perfluoropinacol System under Ambient Conditions. J. Am. Chem. Soc. 2018, 140, 5264–5271. [Google Scholar] [CrossRef] [PubMed]
- Brzeczek-Szafran, A.; Erfurt, K.; Swadzba-Kwasny, M.; Piotrowski, T.; Chrobok, A. Beckmann Rearrangement with Improved Atom Economy, Catalyzed by Inexpensive, Reusable, Bronsted Acidic Ionic Liquid. ACS Sustain. Chem. Eng. 2022, 10, 13568–13575. [Google Scholar] [CrossRef]
- Hu, H.; Cai, X.; Xu, Z.; Yan, Y.; Zhao, S. Beckmann Rearrangement of Ketoxime Catalyzed by N-methyl-imidazolium Hydrosulfate. Molecules 2018, 23, 1764. [Google Scholar] [CrossRef]
- Munnuri, S.; Verma, S.; Chandra, D.; Anugu, R.R.; Falck, J.R.; Jat, J.L. Cu(OTf)2-Catalyzed Beckmann Rearrangement of Ketones Using Hydroxylamine-O-sulfonic Acid (HOSA). Synthesis 2019, 51, 3709–3714. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.S.; Humne, V.T.; Tanpure, S.D.; Mhaske, S.B. Radical Beckmann Rearrangement and Its Application in the Formal Total Synthesis of Antimalarial Natural Product Isocryptolepine via C–H Activation. Org. Lett. 2016, 18, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cantillo, D.; Kappe, C.O. Visible Light-Promoted Beckmann Rearrangements: Separating Sequential Photochemical and Thermal Phenomena in a Continuous Flow Reactor. Eur. J. Org. Chem. 2019, 2019, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Giacomelli, G.; Porcheddu, A. Beckmann Rearrangement of Oximes under Very Mild Conditions. J. Org. Chem. 2002, 67, 6272–6274. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Ishihara, K.; Yamamoto, H. Cyanuric Chloride as a Mild and Active Beckmann Rearrangement Catalyst. J. Am. Chem. Soc. 2005, 127, 11240–11241. [Google Scholar] [CrossRef]
- Tochette, S.J.; Dunkley, E.M.; Lowder, L.L.; Wu, J. Nucleophile-intercepted Beckmann fragmentation reactions. Chem. Sci. 2019, 10, 7812–7815. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Súarez, P.; Kolliopoulos, A.V.; Smith, J.P.; Banks, C.E.; Jones, A.M. An experimentalist’s guide to electrosynthesis: The Shono oxidation. Tetrahedron Lett. 2015, 56, 6863–6867. [Google Scholar] [CrossRef]
- Bal, M.K.; Banks, C.E.; Jones, A.M. Metabolism Mimicry: An Electrosynthetic Method for the Selective Deethylation of Tertiary Benzamides. ChemElectroChem 2019, 6, 4284–4291. [Google Scholar] [CrossRef]
- Jones, A.M. Dialling-In New Reactivity into the Shono-Type Anodic Oxidation Reaction. Chem. Rec. 2021, 21, 2120–2129. [Google Scholar] [CrossRef]
- Jones, A.M.; Banks, C.E. The Shono-type electroorganic oxidation of unfunctionalised amides. Carbon–carbon bond formation via electrogenerated N-acyliminium ions. Beilstein J. Org. Chem. 2014, 10, 3056–3072. [Google Scholar] [CrossRef]
- Smith, P.A.S. Ketoxime-O-sulfonic Acids. J. Am. Chem. Soc. 1948, 70, 323–326. [Google Scholar] [CrossRef]
- Fukui, K.; Uchida, M.; Masaki, M. Formation and Reaction of Cyclohexanone Oxime Hydrogen Sulfate M. Bull. Jpn Chem. Soc. 1973, 46, 3168–3173. [Google Scholar] [CrossRef]
- Kelly, K.K.; Matthews, J.S. Use of Lewis base-sulfur trioxide complexes as reagents for the Beckmann rearrangement of ketoximes. J. Org. Chem. 1971, 36, 2159–2161. [Google Scholar] [CrossRef]
- Turbak, A.F. A Low Temperature Beckmann Rearrangement with SO3 Including Separation of Caprimidyl Sulfate as a New Composition. Ind. Eng. Chem. Prod. Res. Dev. 1968, 7, 189–191. [Google Scholar] [CrossRef]
- Zhou, Y.; Jones, A.M. Rearrangement of Arylsulfamates and Sulfates to Para-Sulfonyl Anilines and Phenols. Molecules 2024, 29, 1445. [Google Scholar] [CrossRef]
- Alshehri, J.A.; Gill, D.M.; Jones, A.M. A Sulfuryl Group Transfer Strategy to Selectively Prepare Sulfated Steroids and Isotopically Labelled Derivatives. Front. Mol. Biosci. 2021, 8, 776900. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M.; Povinelli, A.P.R.; Zazeri, G.; Mahmoud, A.M.; Shamir, S.A.; Wilkinson, F.L.; Alexander, M.Y.; Cornelio, M.L.; Jones, A.M. The modulatory role of sulfated and non-sulfated small molecule heparan sulfate-glycomimetics in endothelial dysfunction: Absolute structural clarification, molecular docking and simulated dynamics, SAR analyses and ADMET studies. RSC Med. Chem. 2021, 12, 779–790. [Google Scholar] [CrossRef]
- Jones, A.M. Tributylsulfoammonium betaine. In The Encyclopaedia of Reagents for Organic Synthesis (e-EROS); John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2021; Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/047084289X.RN02393 (accessed on 27 March 2024).
- Benedetti, A.M.; Gill, D.M.; Tsang, C.W.; Jones, A.M. Chemical Methods for N- and O-Sulfation of Small Molecules, Amino Acids and Peptides. ChemBioChem 2020, 21, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M.; Male, L.; Jones, A.M. Sulfation made simple: A strategy for synthesising sulfated molecules. Chem. Commun. 2019, 55, 4319–4322. [Google Scholar] [CrossRef]
- Nishijima, K.; Nishida, H.; Yamashita, Y.; Ito, M.; Onuki, Y.; Mizota, M.; Miyano, S. Synthesis and diuretic activity of bicyclic fused heterocycles containing oxime-O-sulfonic acid moiety. Eur. J. Med. Chem. 2000, 35, 227–240. [Google Scholar] [CrossRef]
- Chen, Y.; Gardiner, M.G.; Lan, P.; Banwell, M.G. α-Iodo-α,β-Unsaturated Ketones as Vicinal Dielectrophiles: Their Reactions with Dinucleophiles Provide New Annulation Protocols for the Formation of Carbo- and Heterocyclic Ring Systems. J. Org. Chem. 2022, 87, 6146–6160. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.B.; Boronat, M.; Blasco, T.; Corma, A. Establishing a Molecular Mechanism for the Beckmann Rearrangement of Oximes over Microporous Molecular Sieves. Angew. Chem. Int. Ed. 2005, 44, 2370–2373. [Google Scholar] [CrossRef] [PubMed]
- Sharghi, H.; Sarvari, M.H. One-step Beckmann rearrangement from carbonyl compounds and hydroxylamine hydrochloride in Al2O3/CH3SO3H (AMA) as a new reagent. J. Chem. Res. 2001, 2001, 446–449. [Google Scholar] [CrossRef]
- Han, Z.; Lv, J.; Zhang, J. One-pot synthesis of 2-amino-3,4-dicyanopyridines from ketoximes and tetracyanoethylene via Cu(I)-catalyzed cyclization. Tetrahedron 2019, 75, 2162–2168. [Google Scholar] [CrossRef]
- Sonn, A. Über einige synthetische Versuche mit O-Trimethyl-gallusaldehyd. (Mitbearbeitet von Ernst Müller, Wolfgang Bülow und Walter Meyer). Ber. Chem. Ges. A/B 1925, 58, 1103–1110. [Google Scholar] [CrossRef]
- Mahajan, P.S.; Mahajan, J.P.; Mhaske, S.B. Malonic ester amide synthesis. An efficient methodology for synthesis of amides. Synth. Commun. 2013, 43, 2508–2516. [Google Scholar] [CrossRef]
- Tang, L.; Wang, Z.-L.; Wan, H.-L.; He, Y.-H.; Guan, Z. Visible-Light-Induced Beckmann Rearrangement by Organic Photoredox Catalysis. Org. Lett. 2020, 22, 6182–6186. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Ghosh, P.; Basu, B. Graphene oxide (GO) catalyzed transamidation of aliphatic amides: An efficient metal-free procedure. Tetrahedron Lett. 2018, 59, 899–903. [Google Scholar] [CrossRef]
- Jat, J.L.; Kumar, P.; Verma, S.; Chandra, D.; Singh, V.; Tiwari, B. Metal-free synthesis of secondary amides using N-Boc-O-tosylhydroxylamine as nitrogen source via Beckmann rearrangement. New J. Chem. 2022, 46, 14782–14785. [Google Scholar] [CrossRef]
- Ito, A.; Asami, Y.; Asato, M.; Fukuda, K.; Yamasaki, R.; Okamoto, I. Synthesis and conformational analysis of N-aryl-N-(3-thienyl)acetamides. Tetrahedron Lett. 2018, 59, 2454–2458. [Google Scholar] [CrossRef]
- Ran, M.; He, J.; Yan, B.; Liu, W.; Li, Y.; Fu, Y.; Li, S.-J.; Yao, Q. Catalyst-free generation of acyl radicals induced by visible light in water to construct C–N bonds. Org. Biomol. Chem. 2021, 19, 1970–1975. [Google Scholar] [CrossRef] [PubMed]
- Chip, G.K.; Grossert, J.S. Aromatic Halogenation with Titanium (IV) Chloride in Presence of Peroxytrifluoroacetic Acid. Can. J. Chem. 1972, 50, 1233–1240. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Sokolova, O.O.; Bower, J.F. Branch-Selective Alkene Hydroarylation by Cooperative Destabilization: Iridium-Catalyzed ortho-Alkylation of Acetanilides. Angew. Chem. Int. Ed. 2015, 54, 14866–14870. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, X.; Tao, S.; Bu, Q.; Wei, D.; Liu, N. Manganese Catalyzed Direct Amidation of Esters with Amines. J. Org. Chem. 2021, 86, 2339–2358. [Google Scholar] [CrossRef] [PubMed]

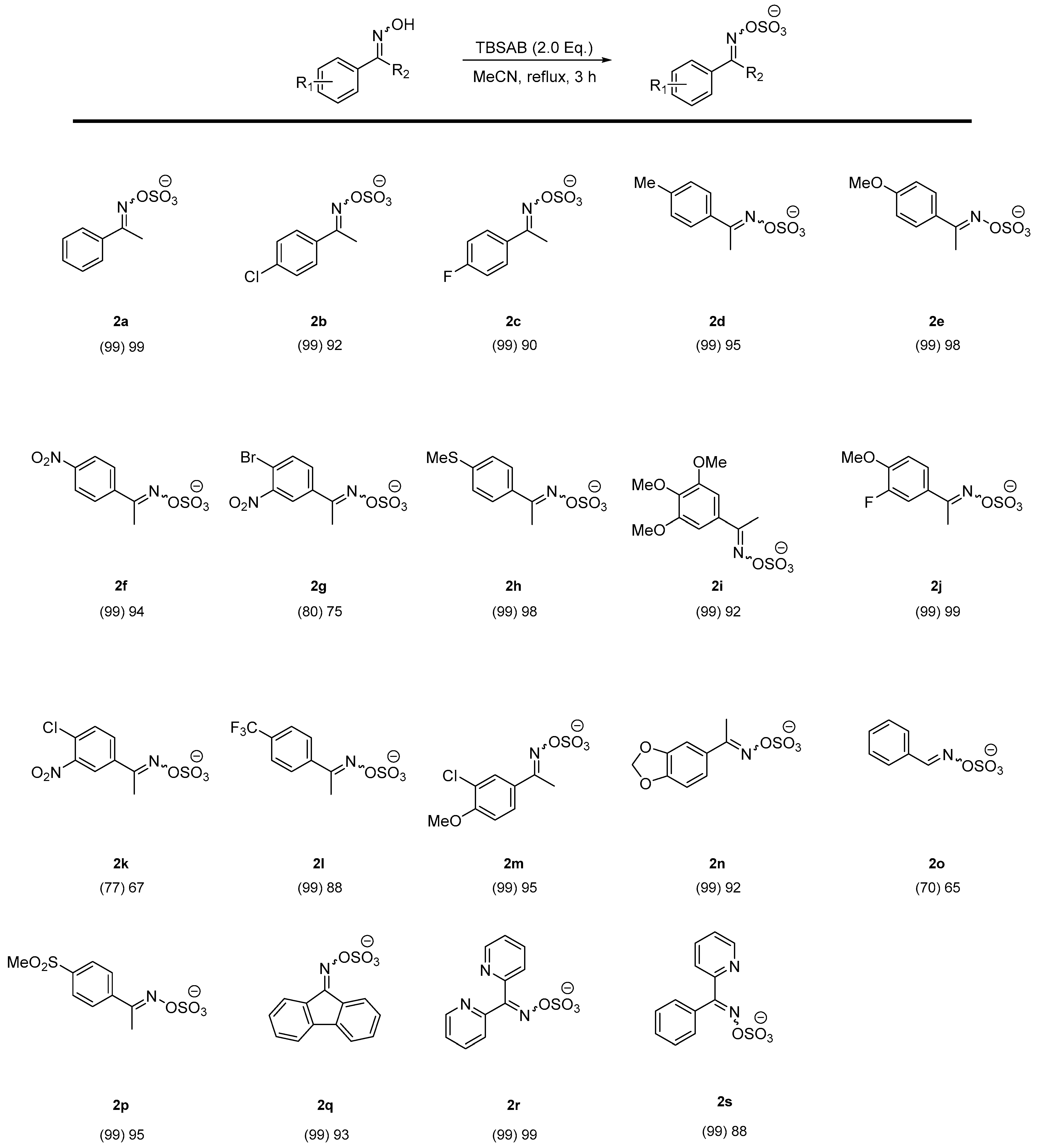

{kind=link}
{kind=link}
{kind=link}
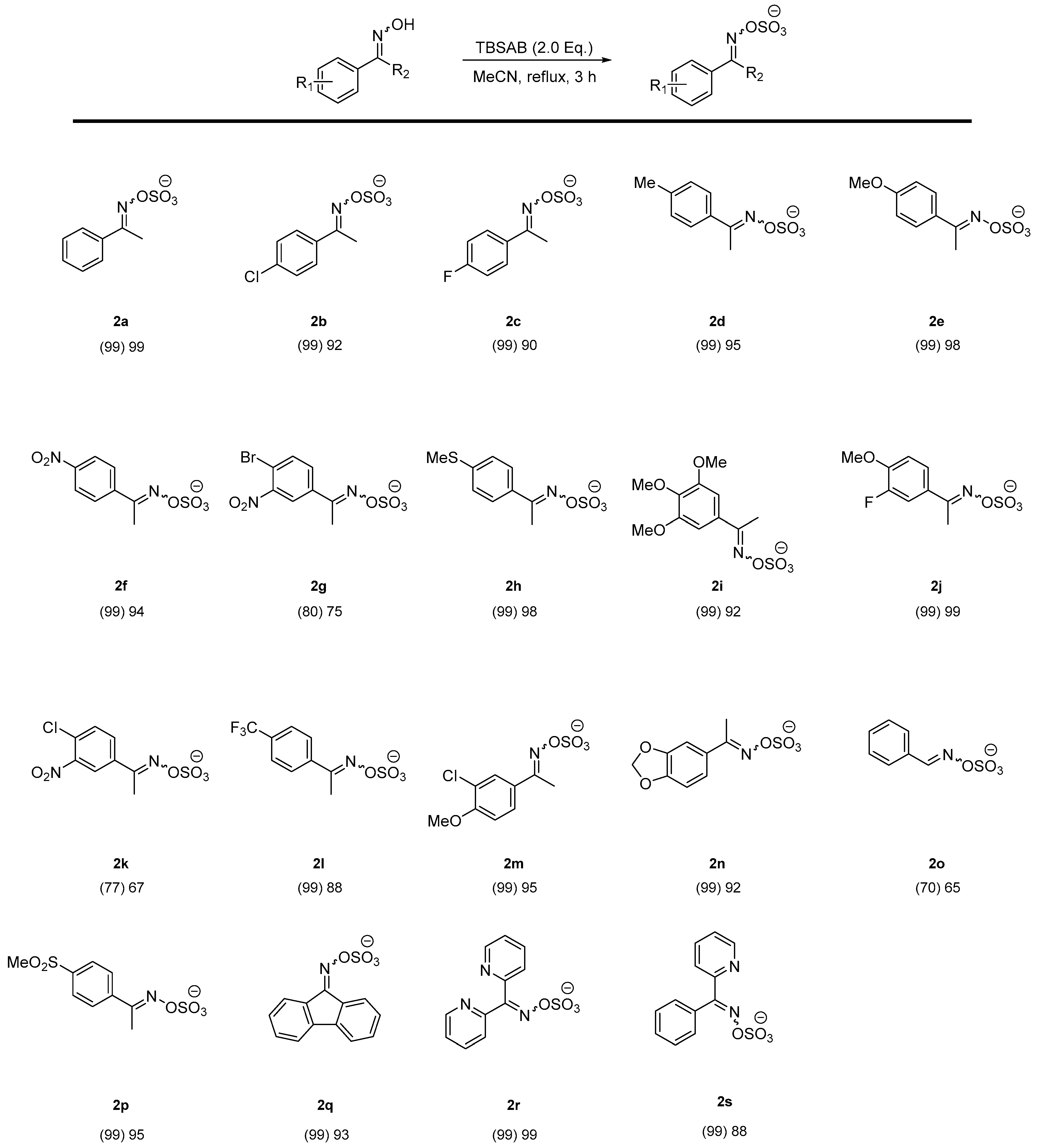{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Entry | Solvent | Temperature [°C] | Time [h] | Conv. [%] |
| 1 | MeCN | 90 | 3 | >99 |
| 2 | MeCN | 60 | 3 | 90 |
| 3 | MeCN | 30 | >24 | <10 |
 | |||||||
| Entry | T (°C) | Time (h) | TBSAB (1.0 Equiv.) | Base [6.0 Equiv.] | Conv. [%] a 1a | Conv. [%] a 3a | Conv. [%] a 4a |
| 1 | 80 | 8 | - | - | 9 | 26 | trace |
| 2 | 100 | 8 | - | - | 6 | 54 | 14 |
| 3 | 120 | 8 | - | - | 7 | 37 | 20 |
| 4 | 80 | 8 | Yes | - | 5 | 40 | trace |
| 5 | 100 | 8 | Yes | - | 10 | 57 | 10 |
| 6 | 120 | 8 | Yes | - | 9 | 37 | 25 |
| 7 b | 100 | 4 | - | NaHCO3 | trace | >99 | 0 |
| 8 b | 100 | 4 | Yes | NaHCO3 | 0 | >99 | 0 |
| 9 b | 100 | 4 | Yes | KHCO3 | 0 | 90 | 0 |
| 10 b | 100 | 4 | Yes | pyridine | 0 | 60 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Jones, A.M. A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates. Molecules 2024, 29, 1667. https://doi.org/10.3390/molecules29071667
Zhou Y, Jones AM. A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates. Molecules. 2024; 29(7):1667. https://doi.org/10.3390/molecules29071667
Chicago/Turabian StyleZhou, Yifei, and Alan M. Jones. 2024. "A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates" Molecules 29, no. 7: 1667. https://doi.org/10.3390/molecules29071667
APA StyleZhou, Y., & Jones, A. M. (2024). A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates. Molecules, 29(7), 1667. https://doi.org/10.3390/molecules29071667