
Asymmetric α-Fluoroalkyl-α-Amino Acids: Recent Advances in Their Synthesis and Applications

 , , , , and
, , , , and
Abstract
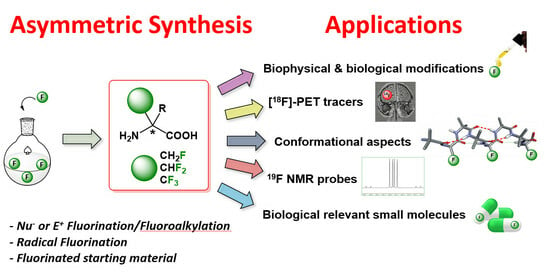
1. Introduction
2. Synthesis of α-Fluoroalkyl-α-Amino Acids
2.1. α-Monofluoroalkyl-α-Amino Acids
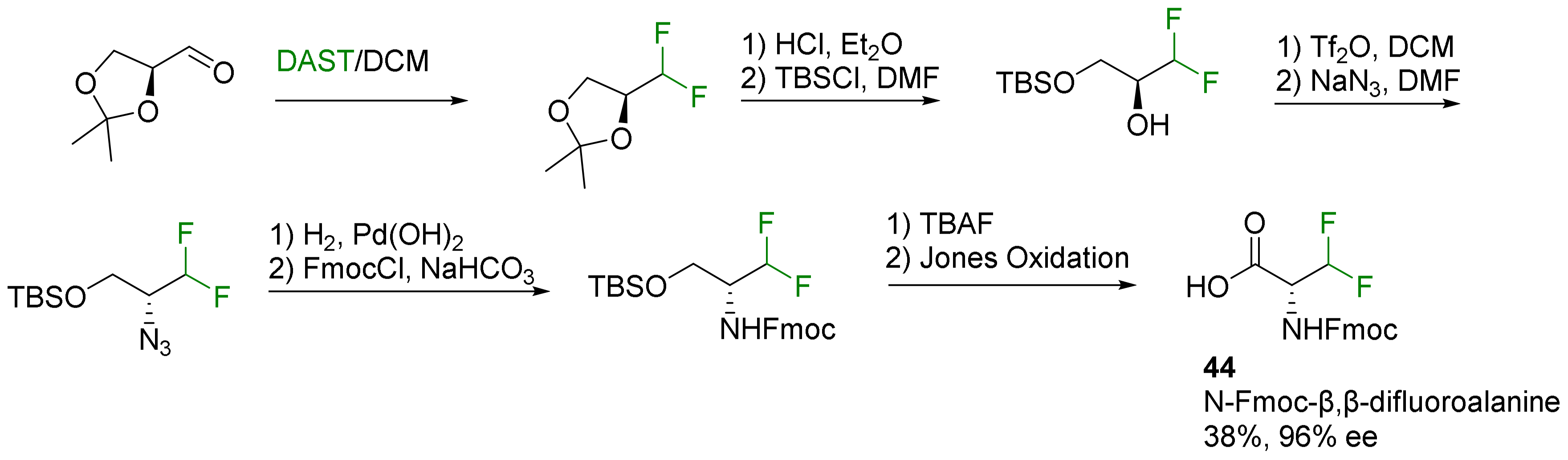
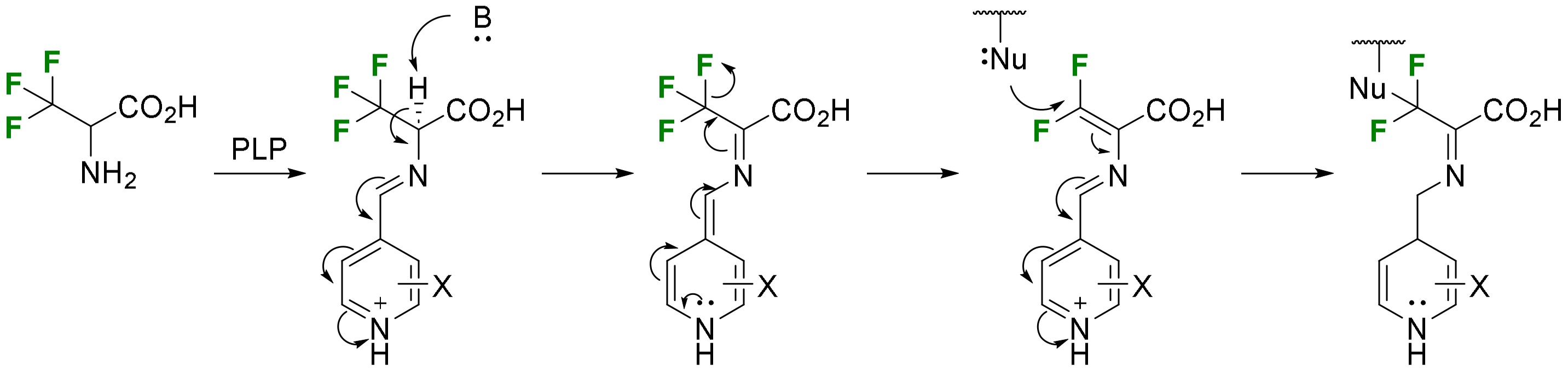
2.1.1. 3-Fluoroalanine Syntheses
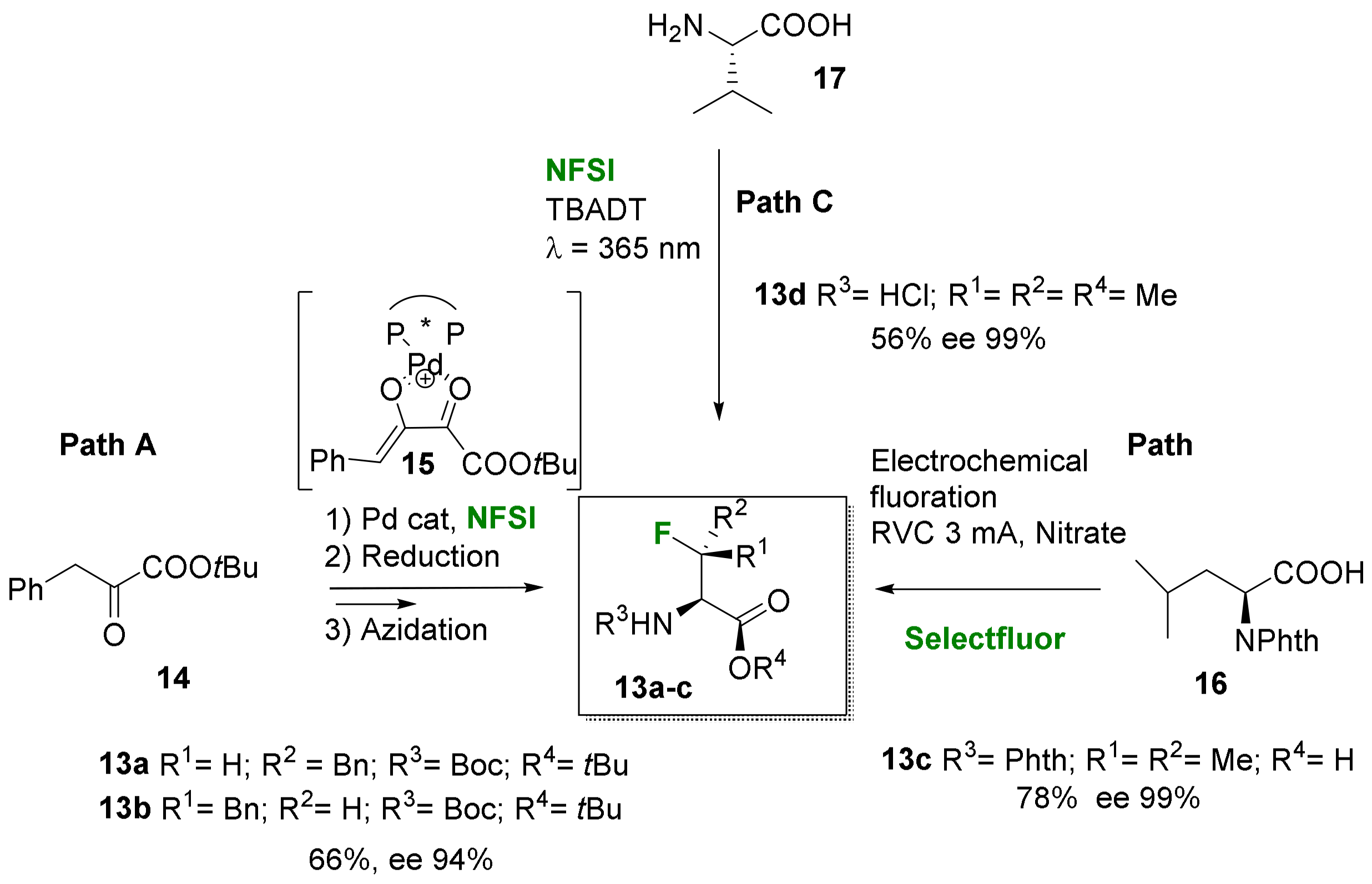
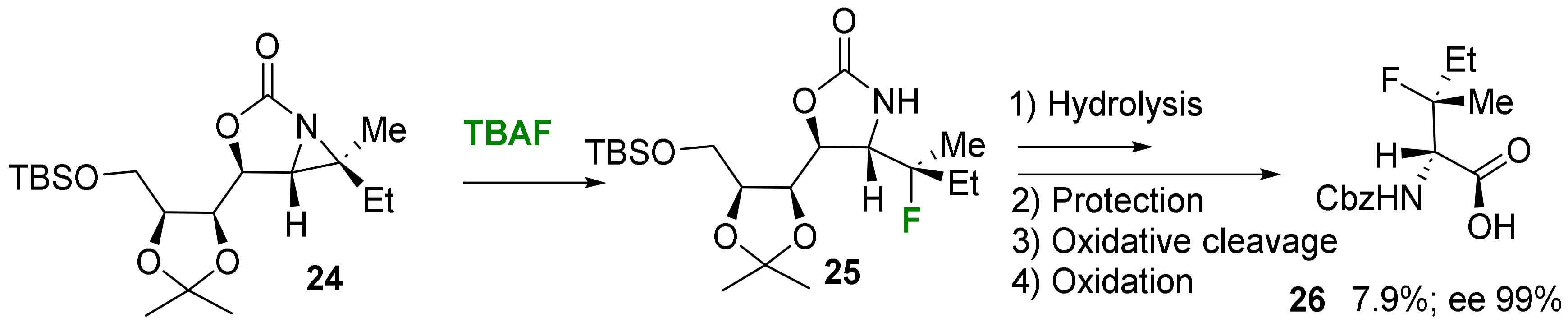
2.1.2. Acyclic 3-Substituted 3-Fluoroalanine Syntheses
- Electrophilic fluorination
- Nucleophilic fluorination
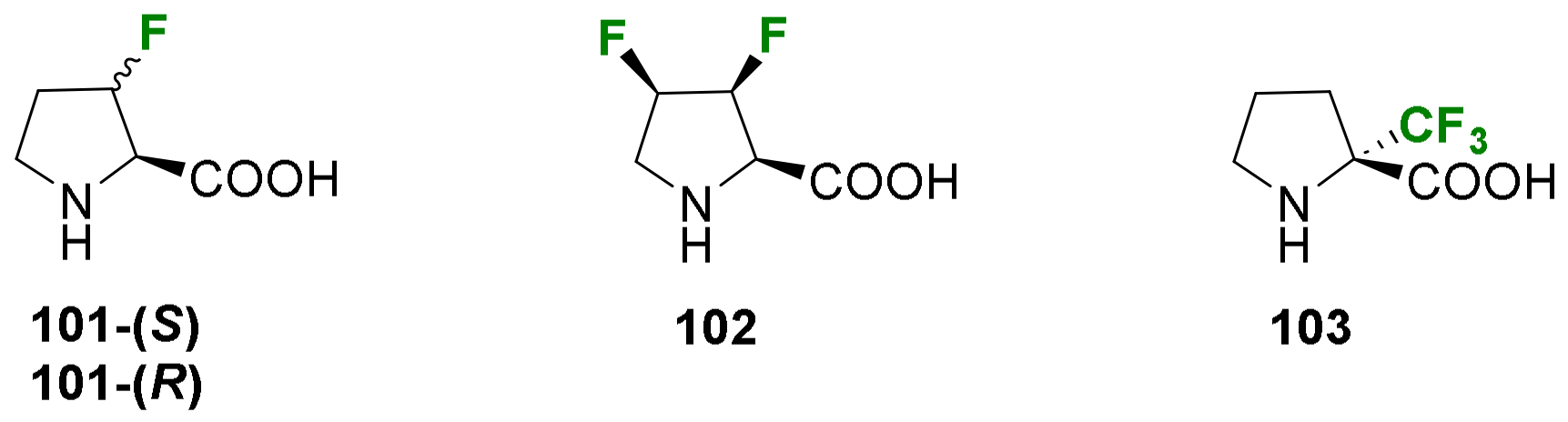
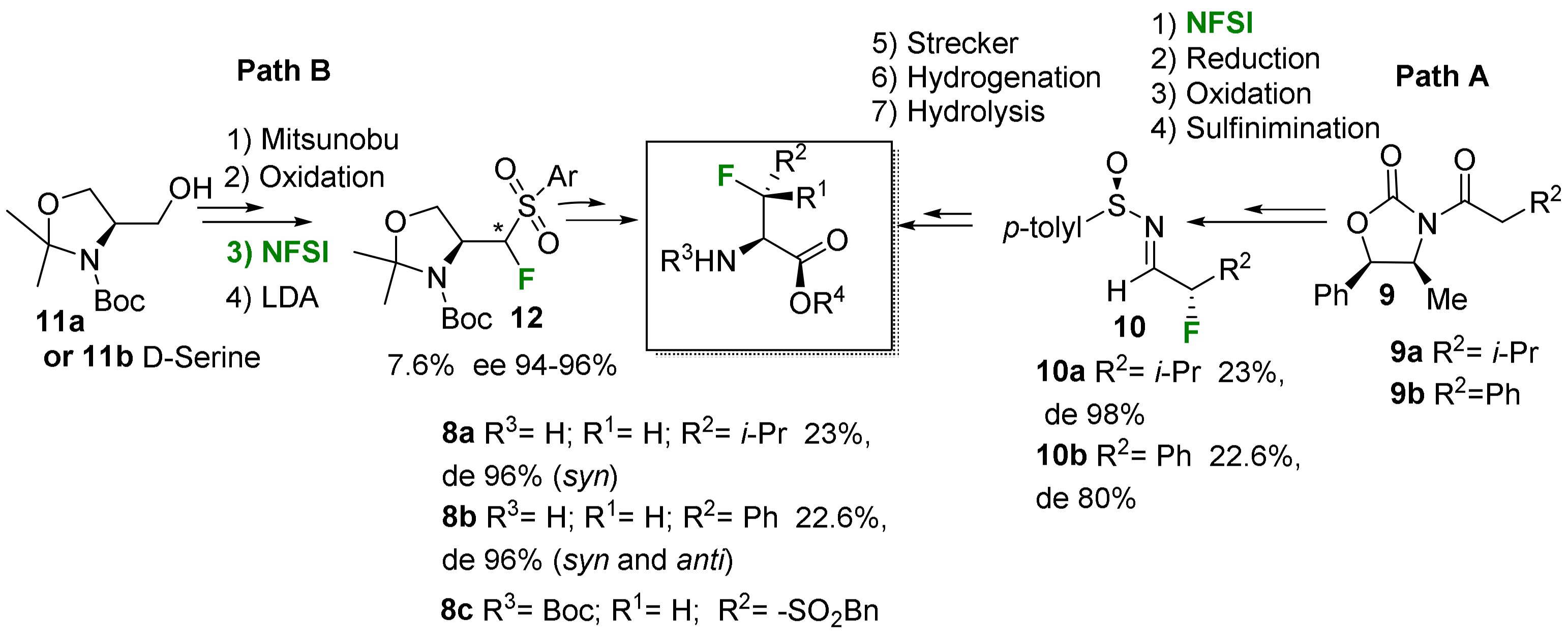
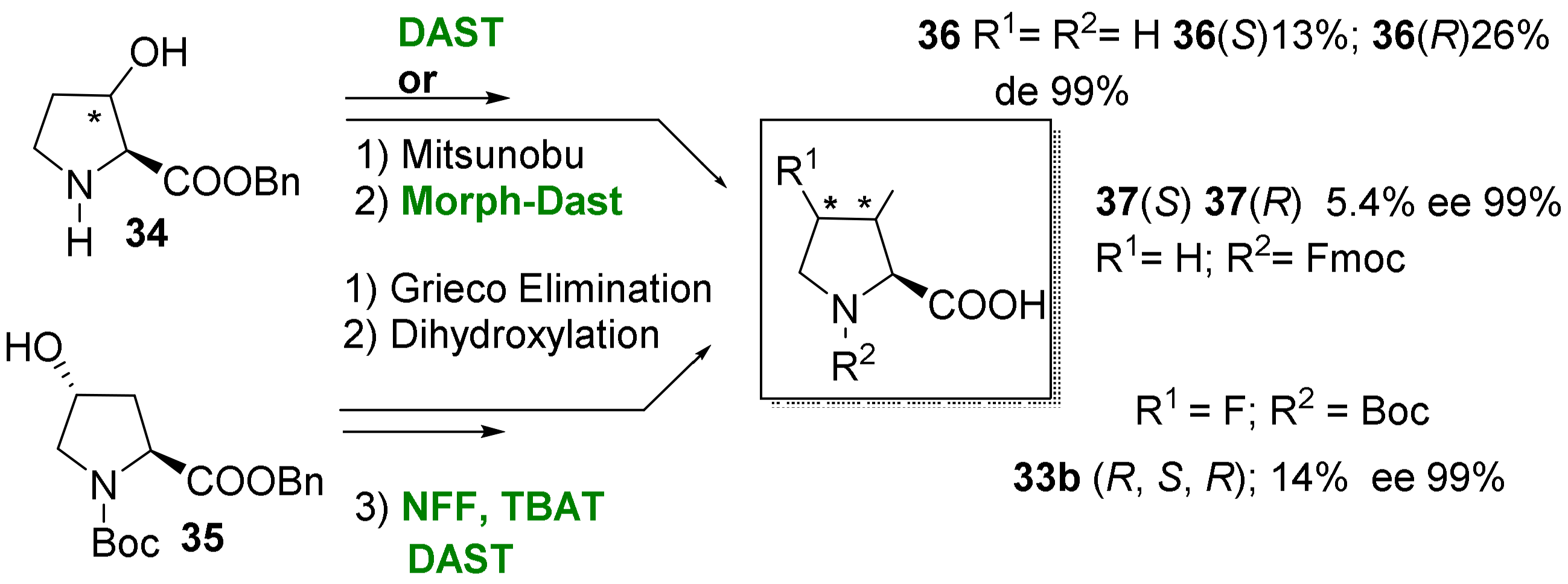
2.1.3. Syntheses of Cyclic 3-Fluoro-α-Amino Acids
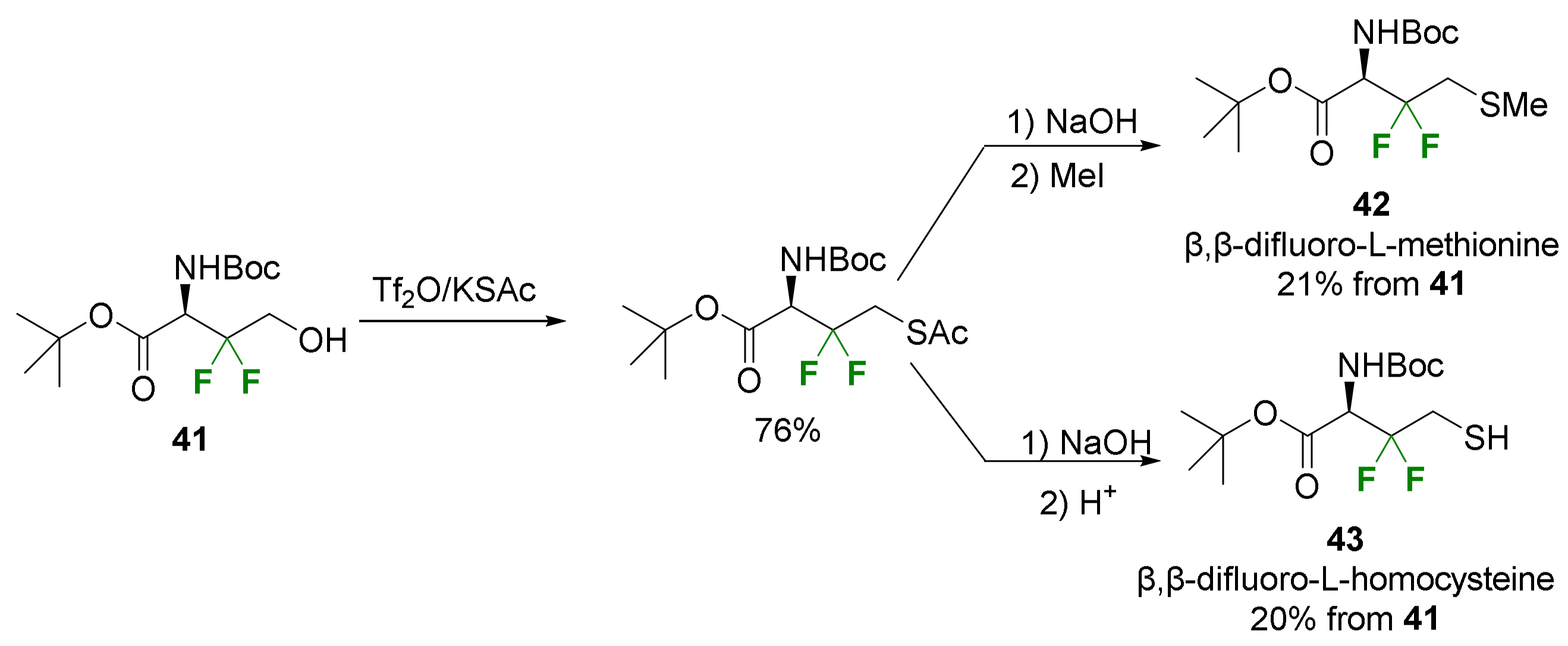
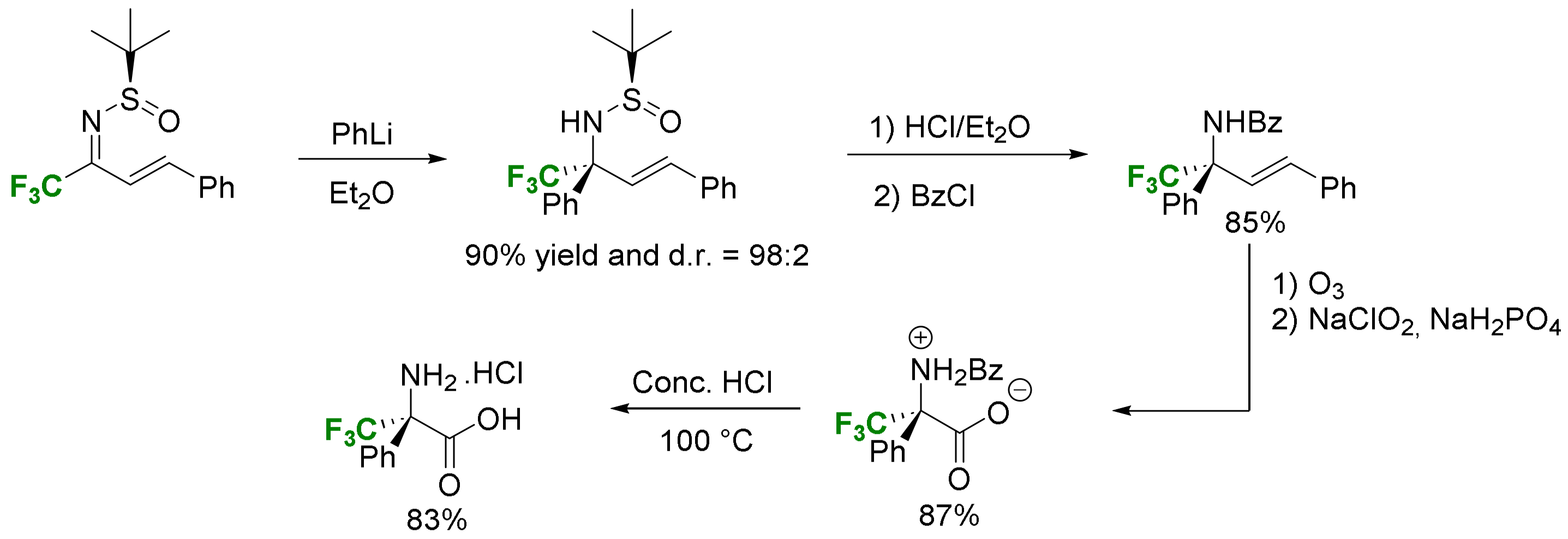
2.2. α-Difluoroalkyl-α-Amino Acids
2.2.1. Fluorination of a Chiral Precursor as the Key Step
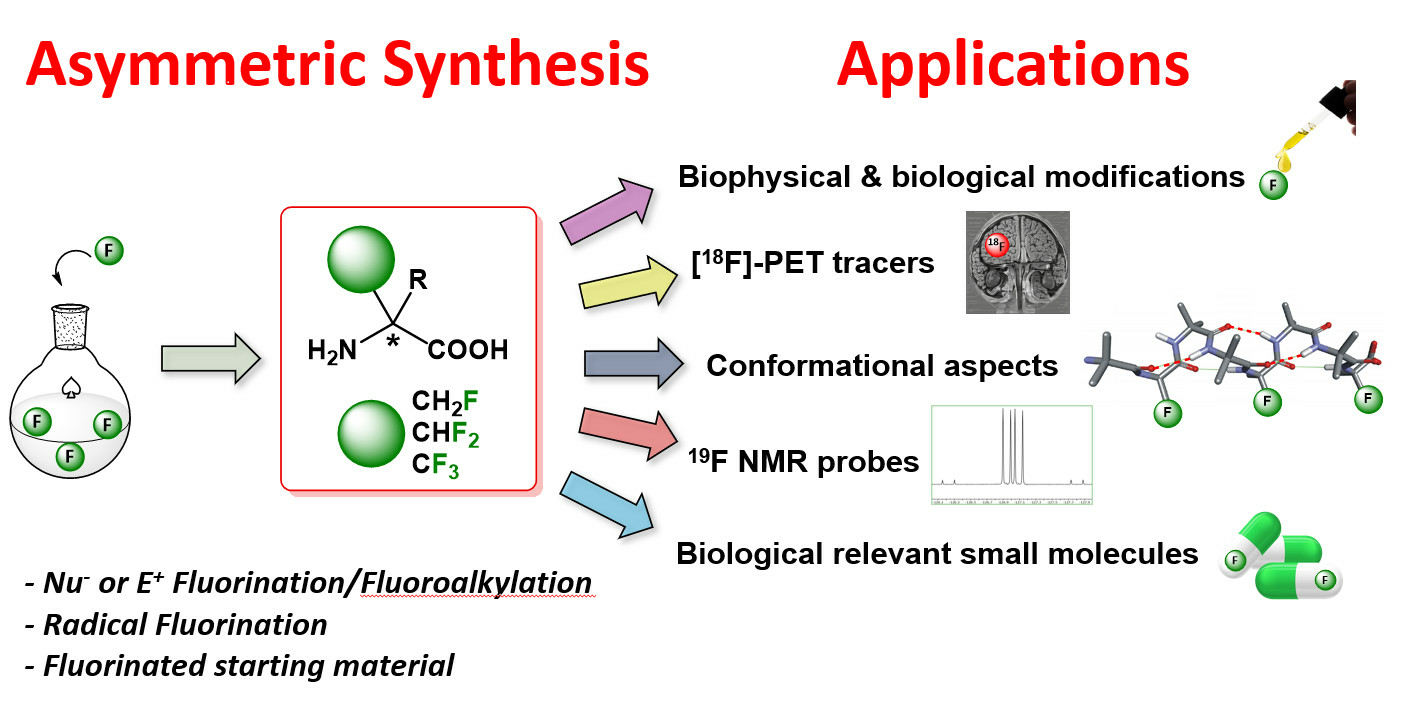
2.2.2. Syntheses from Fluorinated Starting Materials
- Catalytic methods
- Chiral auxiliary-based methods
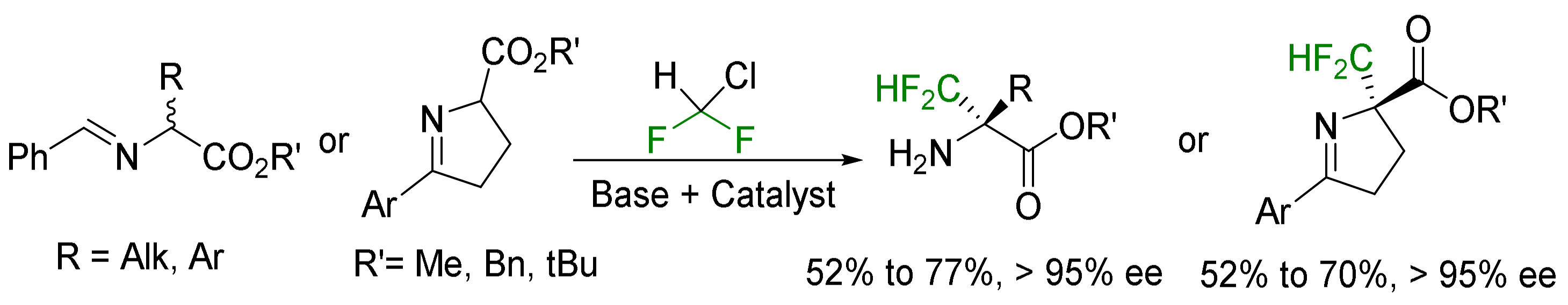
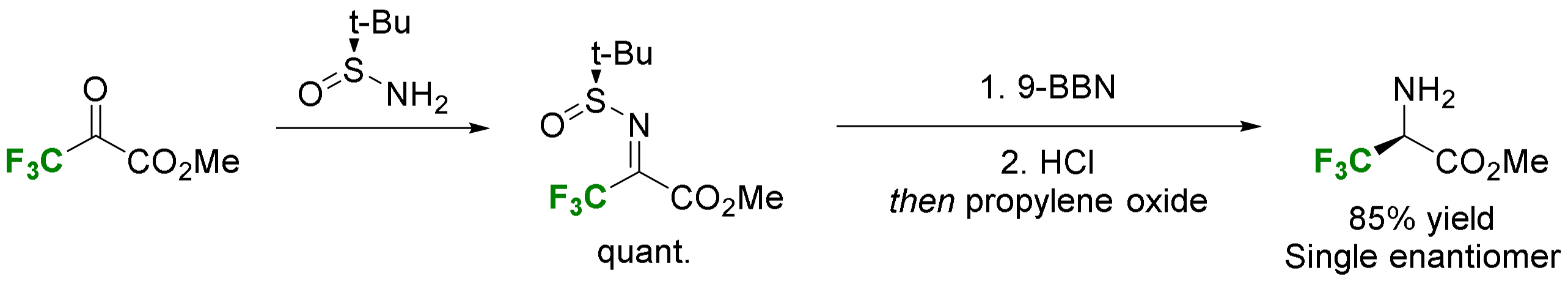
2.3. α-CF3-α-Amino Acids
2.3.1. Via the Introduction of the Side Chain
- Alkynylation and allylation
- α-Arylation: the Friedel–Crafts reaction
- Cycloadditions
- Others
2.3.2. Via Introduction of the Carboxyl Group
2.3.3. Via Introduction of the Amino Group
3. Applications
3.1. Biological Applications of α-Fluoroalkyl-α-Amino Acids and Analogues
3.1.1. Enzyme Inhibitors and Bioactive Small Molecules
3.1.2. Building Blocks for Heterocyclic Compounds
3.2. Incorporation in Peptides and Modulation of the Biophysical and Biological Properties of Peptides
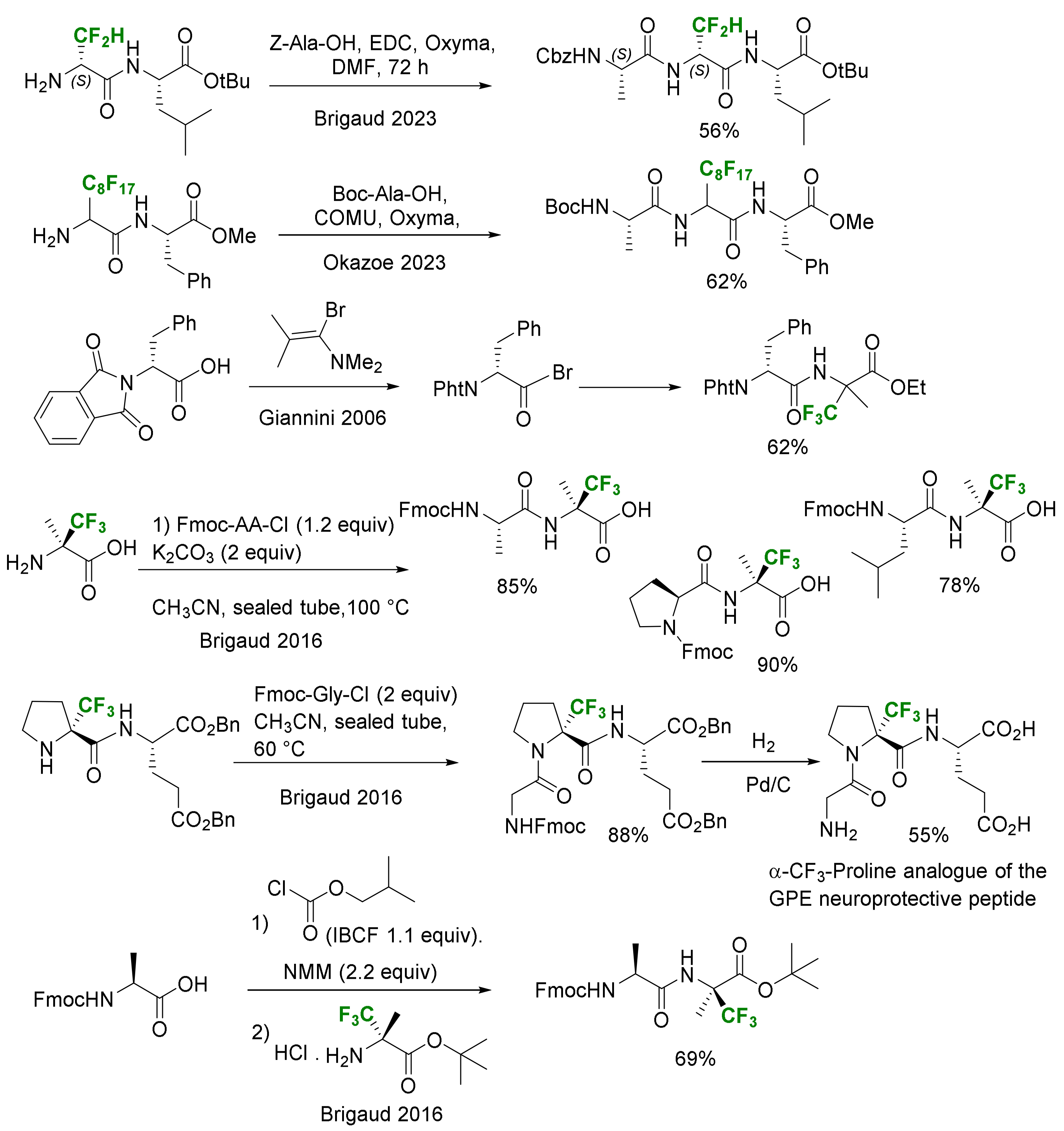
3.2.1. Incorporation in Peptides
- Solution Phase
- Solid Phase Peptide Synthesis (SPPS)
3.2.2. α-Fluoroalkyl Amino Acid-Containing Peptides for Biological Applications
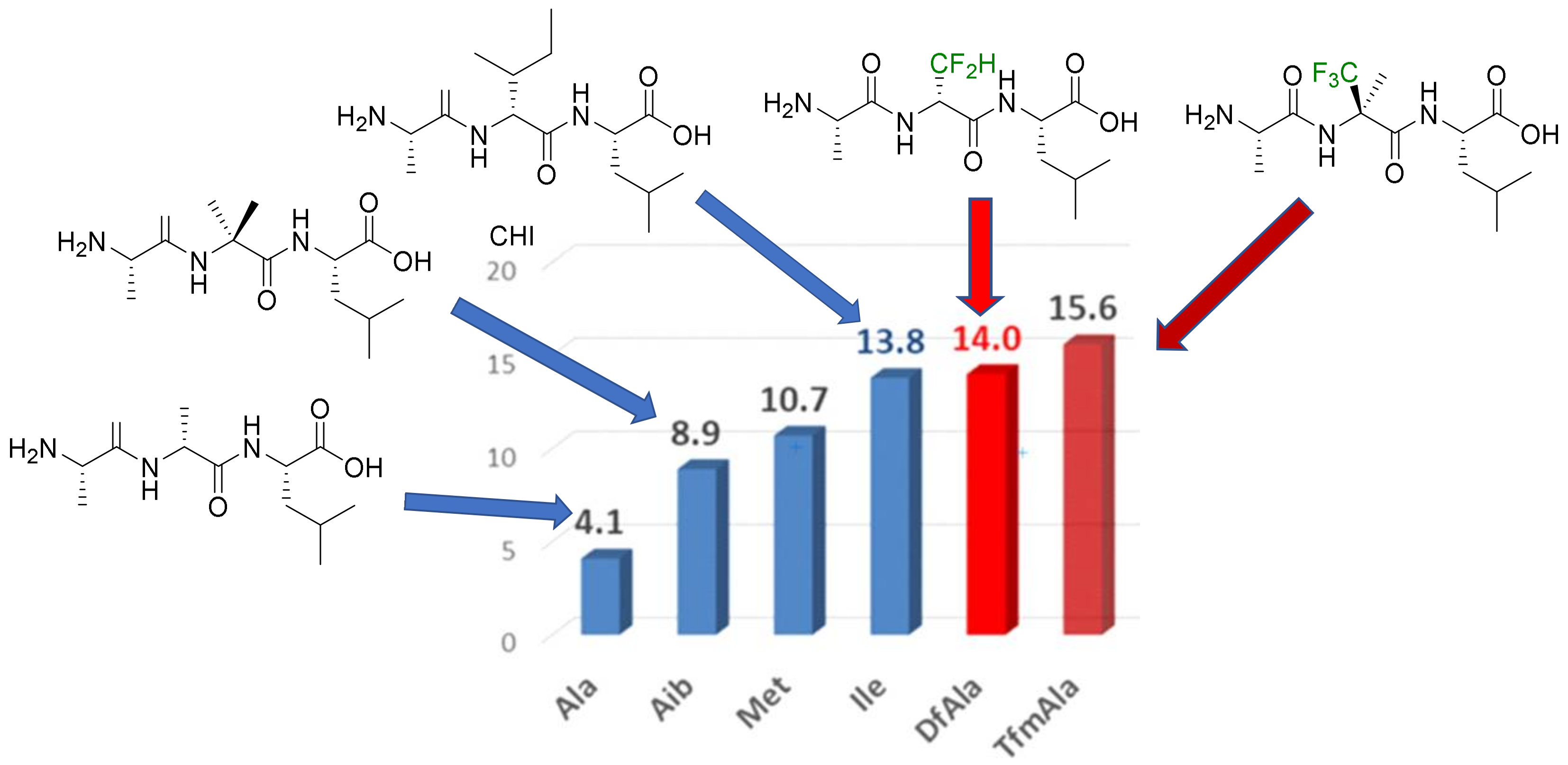
- Hydrophobicity modulation and biological applications
- Increased resistance towards proteases and protease inhibitor design
3.3. Conformationnal Aspects of α-Fluoroalkyl-α-Amino Acids and Their Peptides
3.3.1. α-Fluoroalkyl Amino Acid Conformations
- Acyclic amino acids
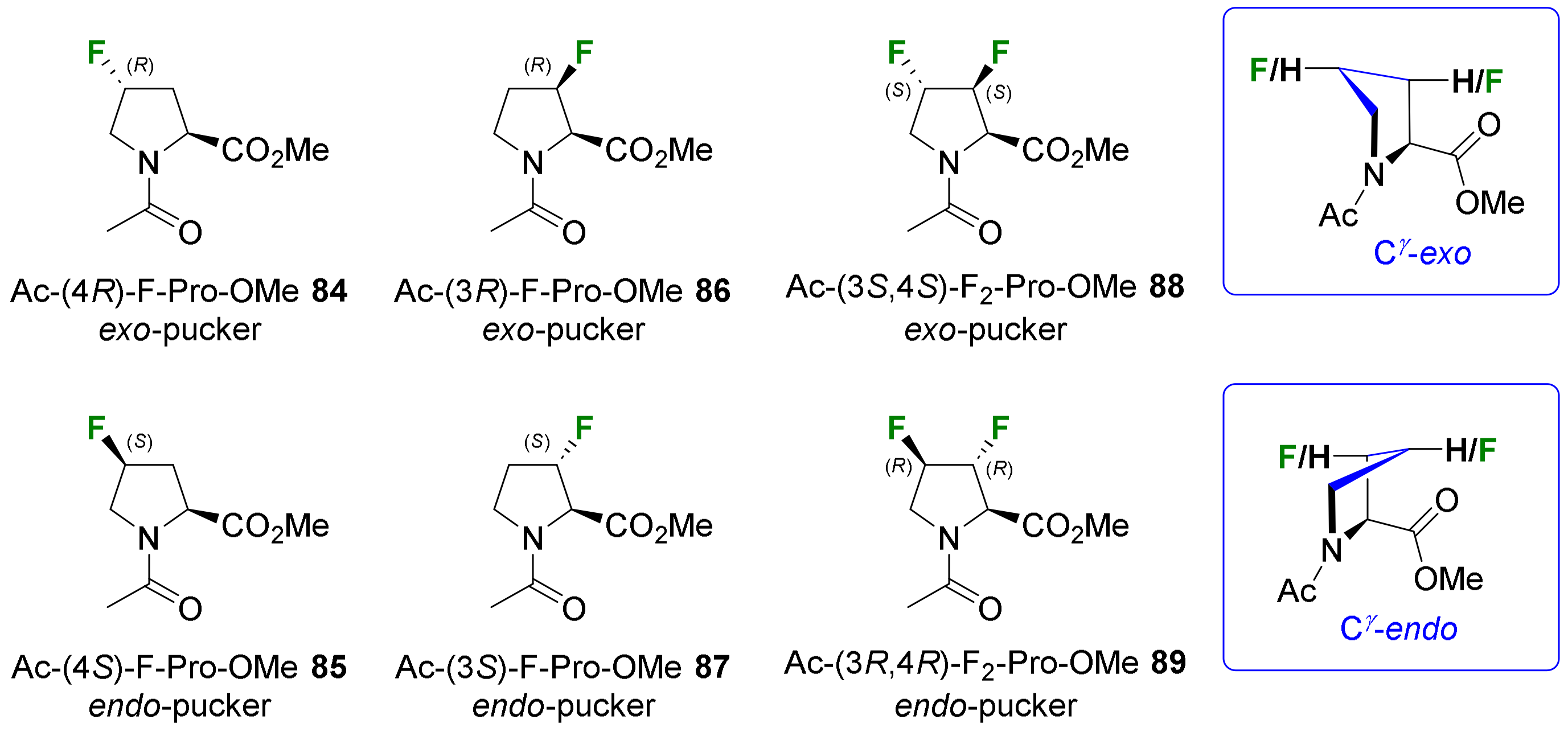
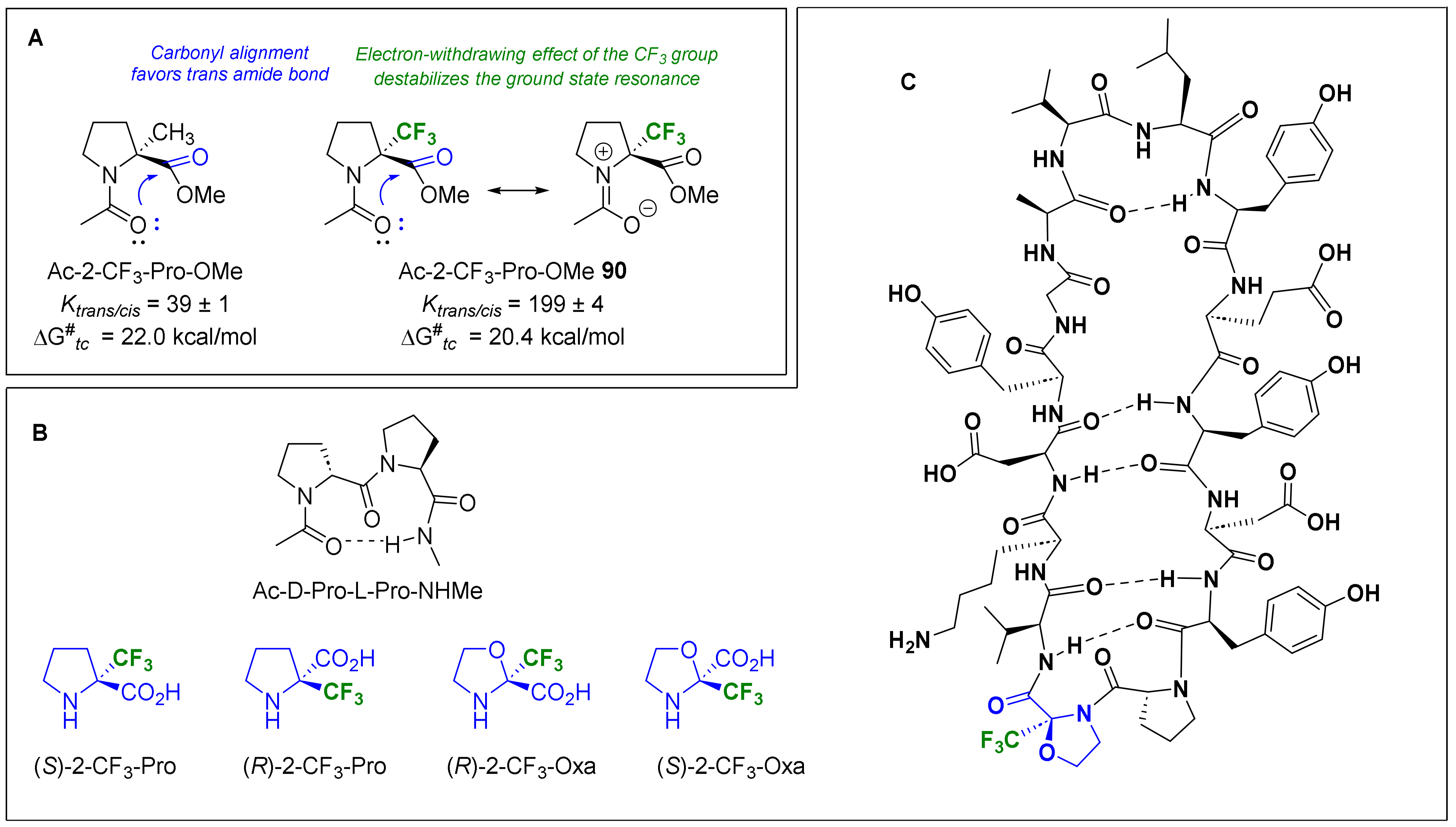
- Cyclic amino acids
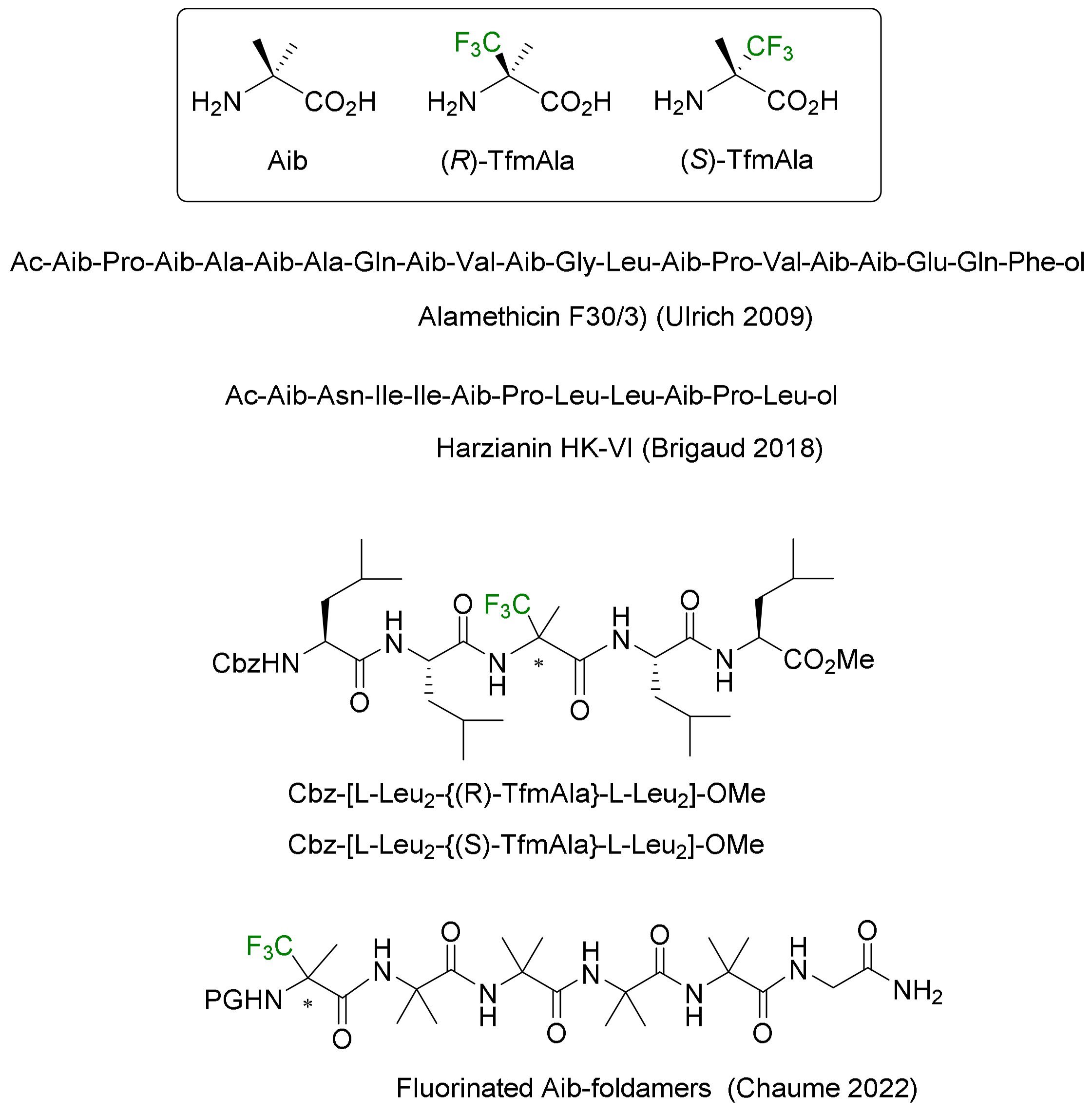
3.3.2. α-Fluoroalkyl Amino Acids as Tools for the Control of Peptide Conformations
3.4. α-Fluoroalkyl-α-Amino Acids as [18F]-PET Tracers
3.5. α-Fluoroalkyl-α-Amino Acids as 19F NMR Probes
3.5.1. NMR Sensitivity of 19F and Its Opportunities
3.5.2. α-Trifluoromethylalanine (α-TfmAla)
3.5.3. 3-Fluorovaline, 3-Fluoroalanine, and (S) 3-Trifluoroalanine
3.5.4. 3-Fluoroprolines, 3,4-Difluoroprolines, 2-Trifluoromethyl Prolines
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moschner, J.; Stulberg, V.; Fernandes, R.; Huhmann, S.; Leppkes, J.; Koksch, B. Approaches to Obtaining Fluorinated α-Amino Acids. Chem. Rev. 2019, 119, 10718–10801. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Feng, Z.; Zhang, X. Recent Advances in the Synthesis of Fluorinated Amino Acids and Peptides. Chem. Commun. 2023, 59, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Brittain, W.D.G.; Lloyd, C.M.; Cobb, S.L. Synthesis of Complex Unnatural Fluorine-Containing Amino Acids. J. Fluor. Chem. 2020, 239, 109630. [Google Scholar] [CrossRef] [PubMed]
- Aceña, J.; Sorochinsky, A.; Soloshonok, V. Recent Advances in the Asymmetric Synthesis of α-(Trifluoromethyl)-Containing α-Amino Acids. Synthesis 2012, 44, 1591–1602. [Google Scholar] [CrossRef]
- Qiu, X.L.; Qing, F.L. Recent Advances in the Synthesis of Fluorinated Amino Acids. Eur. J. Org. Chem. 2011, 2011, 3261–3278. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; da Silva, E.S.; Llop, J.; López-Gallego, F. Sustainable and Continuous Synthesis of Enantiopure L-Amino Acids by Using a Versatile Immobilised Multienzyme System. ChemBioChem 2018, 19, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Bea, H.S.; Lee, S.H.; Yun, H. Asymmetric Synthesis of (R)-3-Fluoroalanine from 3-Fluoropyruvate Using Omega-Transaminase. Biotechnol. Bioproc. E. 2011, 16, 291–296. [Google Scholar] [CrossRef]
- Park, E.S.; Dong, J.Y.; Shin, J.S. ω-Transaminase-Catalyzed Asymmetric Synthesis of Unnatural Amino Acids Using Isopropylamine as an Amino Donor. Org. Biomol. Chem. 2013, 11, 6929–6933. [Google Scholar] [CrossRef]
- Bravo, P.; Cavicchio, G.; Crucianelli, M.; Poggiali, A.; Zanda, M. Stereoselective Synthesis of the Antibacterial 3-Fluoro-D-Alanine. Tetrahedron Asymmetry 1997, 8, 2811–2815. [Google Scholar] [CrossRef]
- Hoveyda, H.R.; Pinault, J.F. (2R)- and (2S)-3-Fluoroalanine and Their N-Methyl Derivatives: Synthesis and Incorporation in Peptide Scaffolds. Org. Lett. 2006, 8, 5849–5852. [Google Scholar] [CrossRef]
- Carpentier, C.; Godbout, R.; Otis, F.; Voyer, N. Synthesis and Use of N-Fmoc-l-Fluoroalanine. Tetrahedron Lett. 2015, 56, 1244–1246. [Google Scholar] [CrossRef]
- Davis, F.A.; Srirajan, V.; Titus, D.D. Efficient Asymmetric Synthesis of β-Fluoro α-Amino Acids. J. Org. Chem. 1999, 64, 6931–6934. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Khangarot, R.K.; Stahl, L.; Veresmortean, C.; Pradhan, P.; Yang, L.; Zajc, B. Generating Stereodiversity: Diastereoselective Fluorination and Highly Diastereoselective Epimerization of α-Amino Acid Building Blocks. Org. Lett. 2018, 20, 3574–3578. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kitamura, Y.; Lectard, S.; Hamashima, Y.; Sodeoka, M. Catalytic Asymmetric Mono-Fluorination of α-Keto Esters: Synthesis of Optically Active β-Fluoro-α-Hydroxy and β-Fluoro-α-Amino Acid Derivatives. Angew. Chem. Int. Ed. 2012, 51, 4581–4585. [Google Scholar] [CrossRef] [PubMed]
- Takahira, Y.; Chen, M.; Kawamata, Y.; Mykhailiuk, P.; Nakamura, H.; Peters, B.K.; Reisberg, S.H.; Li, C.; Chen, L.; Hoshikawa, T.; et al. Electrochemical C(Sp 3)-H Fluorination. Synlett 2019, 30, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Halperin, S.D.; Fan, H.; Chang, S.; Martin, R.E.; Britton, R. A Convenient Photocatalytic Fluorination of Unactivated C-H Bonds. Angew. Chem. Int. Ed. 2014, 53, 4690–4693. [Google Scholar] [CrossRef]
- Kalow, J.A.; Schmitt, D.E.; Doyle, A.G. Synthesis of β-Fluoroamines by Lewis Base Catalyzed Hydrofluorination of Aziridines. J. Org. Chem. 2012, 77, 4177–4183. [Google Scholar] [CrossRef]
- Alluri, S.R.; Riss, P.J. Stereospecific Radiosynthesis of 3-Fluoro Amino Acids: Access to Enantiomerically Pure Radioligands for Positron Emission Tomography. Org. Biomol. Chem. 2018, 16, 2219–2224. [Google Scholar] [CrossRef]
- Chia, P.W.; Livesey, M.R.; Slawin, A.M.Z.; Van Mourik, T.; Wyllie, D.J.A.; O’Hagan, D. 3-Fluoro-N-Methyl-D-Aspartic Acid (3F-NMDA) Stereoisomers as Conformational Probes for Exploring Agonist Binding at NMDA Receptors. Chem. Eur. J. 2012, 18, 8813–8819. [Google Scholar] [CrossRef]
- Fokina, N.A.; Kornilov, A.M.; Kulik, I.B.; Kukhar, V.P. Towards Optically Pure Mono- and Difluorinated Amino Acids: Common Methodology Based on (R)-2,3-O-Isopropylideneglyceraldehyde. Synthesis 2002, 2002, 2589–2596. [Google Scholar] [CrossRef]
- Okuda, K.; Hirota, T.; Kingery, D.A.; Nagasawa, H. Synthesis of a Fluorine-Substituted Puromycin Derivative for Brønsted Studies of Ribosomal-Catalyzed Peptide Bond Formation. J. Org. Chem. 2009, 74, 2609–2612. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.G.; Fletcher, A.M.; Frost, A.B.; Roberts, P.M.; Thomson, J.E. Asymmetric Synthesis of Substituted Anti -β-Fluorophenylalanines. Org. Lett. 2015, 17, 2254–2257. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Liu, C.Y.; Angamuthu, V.; Chen, W.C.; Wen, C.S.; Hou, D.R. Synthesis of Optically Active Organofluorides by Ring Opening of Oxazolidinone-Fused Aziridines. Org. Lett. 2023, 25, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jenkinson, S.F.; Vermaas, T.; Adachi, I.; Wormald, M.R.; Hata, Y.; Kurashima, Y.; Kaji, A.; Yu, C.Y.; Kato, A.; et al. 3-Fluoroazetidinecarboxylic Acids and Trans,Trans-3,4-Difluoroproline as Peptide Scaffolds: Inhibition of Pancreatic Cancer Cell Growth by a Fluoroazetidine Iminosugar. J. Org. Chem. 2015, 80, 4244–4258. [Google Scholar] [CrossRef] [PubMed]
- Testa, A.; Lucas, X.; Castro, G.V.; Chan, K.H.; Wright, J.E.; Runcie, A.C.; Gadd, M.S.; Harrison, W.T.A.; Ko, E.J.; Fletcher, D.; et al. 3-Fluoro-4-Hydroxyprolines: Synthesis, Conformational Analysis, and Stereoselective Recognition by the VHL E3 Ubiquitin Ligase for Targeted Protein Degradation. J. Am. Chem. Soc. 2018, 140, 9299–9313. [Google Scholar] [CrossRef] [PubMed]
- Hofman, G.-J.; Ottoy, E.; Light, M.E.; Kieffer, B.; Kuprov, I.; Martins, J.C.; Sinnaeve, D.; Linclau, B. Minimising Conformational Bias in Fluoroprolines through Vicinal Difluorination. Chem. Commun. 2018, 54, 5118–5121. [Google Scholar] [CrossRef] [PubMed]
- Hofman, G.J.; Ottoy, E.; Light, M.E.; Kieffer, B.; Martins, J.C.; Kuprov, I.; Sinnaeve, D.; Linclau, B. Synthesis and Conformational Properties of 3,4-Difluoro-l-Prolines. J. Org. Chem. 2019, 84, 3100–3120. [Google Scholar] [CrossRef] [PubMed]
- Demange, L.; Cluzeau, J.; Ménez, A.; Dugave, C. Synthesis of Optically Pure N-Boc-Protected (2R,3R)- and (2R,3S)-3-Fluoroprolines. Tetrahedron Lett. 2001, 42, 651–653. [Google Scholar] [CrossRef]
- Hodges, J.A.; Raines, R.T. Stereoelectronic and Steric Effects in the Collagen Triple Helix: Toward a Code for Strand Association. J. Am. Chem. Soc. 2005, 127, 15923–15932. [Google Scholar] [CrossRef]
- Durie, A.J.; Fujiwara, T.; Al-Maharik, N.; Slawin, A.M.Z.; O’Hagan, D. Stepwise Preparation of All- Cis 1,3,4-Trifluoro-2-Phenylcyclohexane, Avoiding a Phenonium Intermediate. J. Org. Chem. 2014, 79, 8228–8233. [Google Scholar] [CrossRef]
- Bykova, T.; Al-Maharik, N.; Slawin, A.M.Z.; Bühl, M.; Lebl, T.; O’Hagan, D. Benzylic Functionalisation of Phenyl All-Cis-2,3,5,6-Tetrafluorocyclohexane Provides Access to New Organofluorine Building Blocks. Chem. Eur. J. 2018, 24, 13290–13296. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Leriche, C.; Liu, H.-W. Synthesis of β-difluorine-containing amino acids. Bioorg. Med. Chem. Lett. 1998, 8, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Van Der Donk, W.A. Efficient Synthesis of Suitably Protected β-Difluoroalanine and γ-Difluorothreonine from L-Ascorbic Acid. Org. Lett. 2007, 9, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wang, H.; Guo, C. Copper-Catalyzed Enantioselective Difluoromethylation of Amino Acids via Difluorocarbene. J. Am. Chem. Soc. 2021, 143, 6376–6381. [Google Scholar] [CrossRef] [PubMed]
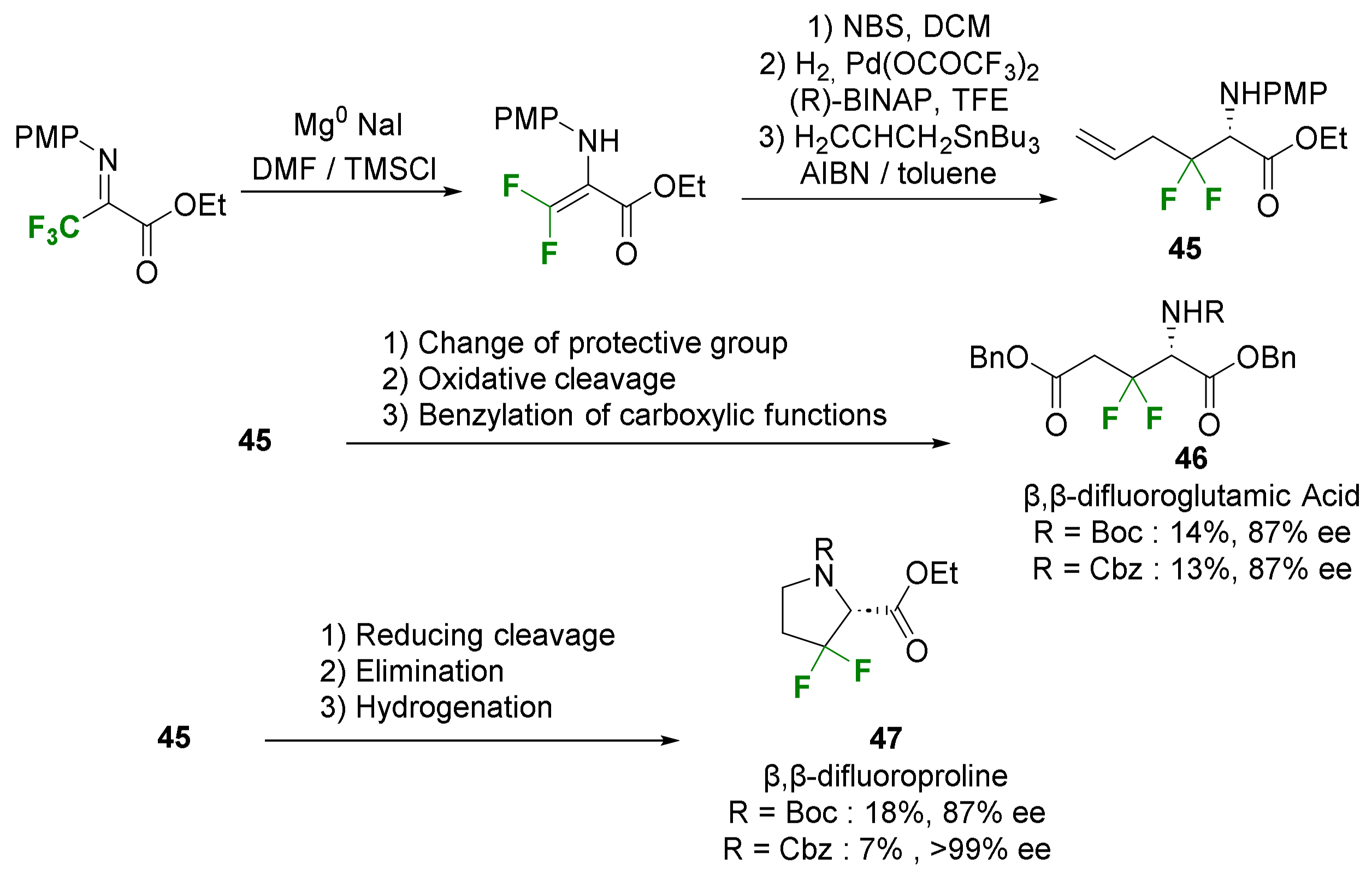
- Suzuki, A.; Mae, M.; Amii, H.; Uneyama, K. Catalytic Route to the Synthesis of Optically Active β,β-Difluoroglutamic Acid and β,β-Difluoroproline Derivatives. J. Org. Chem. 2004, 69, 5132–5134. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Shi, T.D.; Zhou, F.; Zhao, X.L.; Wang, X.; Zhou, J. Organocatalytic Asymmetric Strecker Reaction of Di- and Trifluoromethyl Ketoimines. Remarkable Fluorine Effect. Org. Lett. 2011, 13, 3826–3829. [Google Scholar] [CrossRef] [PubMed]
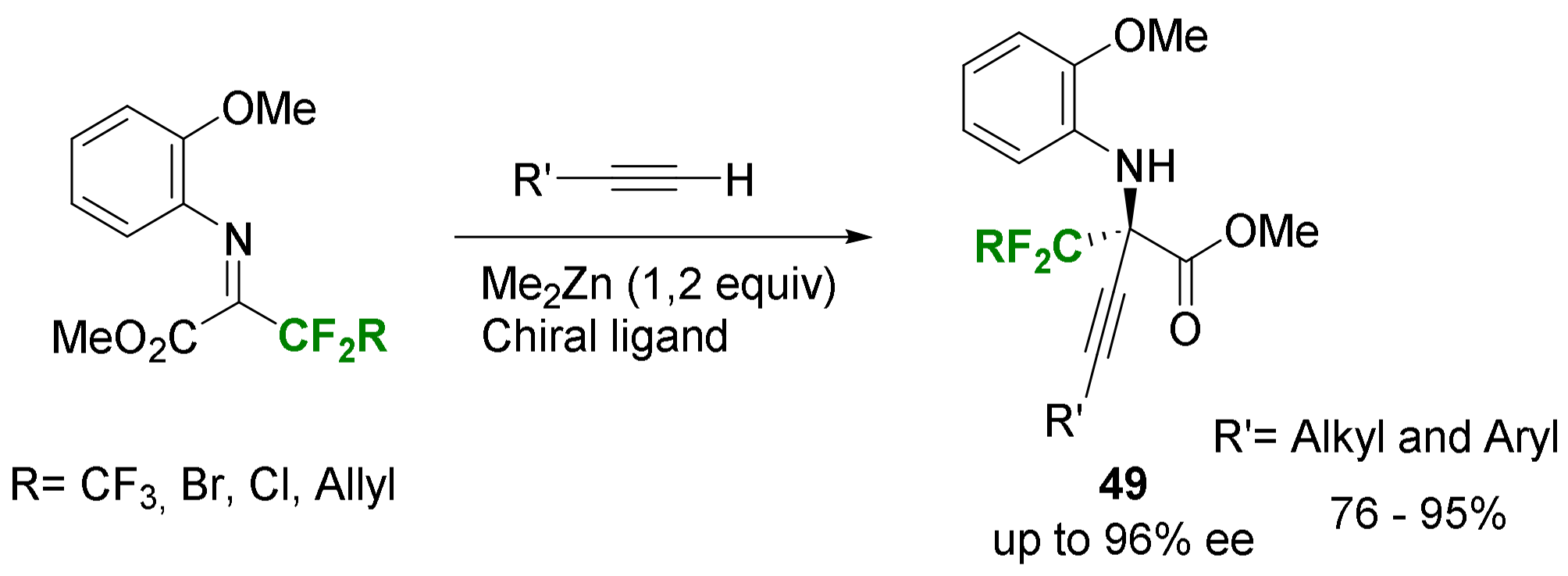
- Huang, G.; Yin, Z.; Zhang, X. Construction of Optically Active Quaternary Propargyl Amines by Highly Enantioselective Zinc/BINOL-Catalyzed Alkynylation of Ketoimines. Chem. Eur. J. 2013, 19, 11992–11998. [Google Scholar] [CrossRef]
- Voznesenskaia, N.G.; Shmatova, O.I.; Khrustalev, V.N.; Nenajdenko, V.G. Enantioselective Synthesis of α-Perfluoroalkylated Prolines, Their 6,7-Membered Homologues and Derivatives. Org. Biomol. Chem. 2018, 16, 7004–7011. [Google Scholar] [CrossRef]
- Fustero, S.; Volonterio, A.; Zanda, M.; Navarro, A.; Pina, B.; Soler, J.G.; Bartolomé, A.; Asensio, A.; Simón, A.; Bravo, P.; et al. Enantioselective Synthesis of Fluorinated α-Amino Acids and Derivatives in Combination with Ring-Closing Metathesis: Intramolecular π-Stacking Interactions as a Source of Stereocontrol. Org. Lett. 2001, 3, 2621–2624. [Google Scholar] [CrossRef]
- Fustero, S.; Sánchez-Roselló, M.; Rodrigo, V.; Del Pozo, C.; Sanz-Cervera, J.F.; Simón, A. Asymmetric Synthesis of New β,β-Difluorinated Cyclic Quaternary α-Amino Acid Derivatives. Org. Lett. 2006, 8, 4129–4132. [Google Scholar] [CrossRef]
- Huguenot, F.; Billac, A.; Brigaud, T.; Portella, C. Umpolung Reactivity of Difluoroenol Silyl Ethers with Amines and Amino Alcohols. Application to the Synthesis of Enantiopure α-Difluoromethyl Amines and Amino Acids. J. Org. Chem. 2008, 73, 2564–2569. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Zhang, F.; Liu, J.T. Asymmetric Synthesis of β,β-Difluoroamino Acids via Cross-Coupling and Strecker Reactions. Tetrahedron 2008, 64, 1731–1735. [Google Scholar] [CrossRef]
- Liu, J.; Hu, J. Highly Diastereoselective Synthesis of α-Difluoromethyl Amines from N-Tert-Butylsulfinyl Ketimines and Difluoromethyl Phenyl Sulfone. Chem. Eur. J. 2010, 16, 11443–11454. [Google Scholar] [CrossRef] [PubMed]
- Boutahri, Y.; Ben Haj Salah, K.; Tisserand, N.; Lensen, N.; Crousse, B.; Brigaud, T. Difluoroalanine: Synthesis, Incorporation in Peptides, and Hydrophobic Contribution Assessment. Org. Lett. 2023, 25, 6937–6941. [Google Scholar] [CrossRef] [PubMed]
- Smits, R.; Cadicamo, C.D.; Burger, K.; Koksch, B. Synthetic Strategies to α–Trifluoromethyl and α–Difluoromethyl Substituted α–Amino Acids. Chem. Soc. Rev. 2008, 37, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Albert, L.; Aceña, J.L.; Sanz-Cervera, J.F.; Asensio, A. An Efficient Entry to Optically Active Anti- and Syn-β-Amino-α- Trifluoromethyl Alcohols. Org. Lett. 2008, 10, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Shchetnikov, G.T.; Peregudov, A.S.; Osipov, S.N. Effective Pathway to the α-CF3-Substituted Azahistidine Analogues. Synlett 2007, 2007, 136–140. [Google Scholar] [CrossRef]
- Shchetnikov, G.T.; Zotova, M.A.; Bruneau, C.; Dixneuf, P.H.; Osipov, S.N. Synthesis of α-Alkynyl-β,β,β-Trifluoroalanine Derivatives by Sonogashira Cross-Coupling Reaction. Eur. J. Org. Chem. 2010, 2010, 1587–1592. [Google Scholar] [CrossRef]
- Zotova, M.A.; Vasil’Eva, T.P.; Osipov, S.N. Intramolecular Cyclization of Acetylene-Containing α-Amino Carboxylates and α-Amino Phosphonates: Synthesis of α-CF 3-Substituted Dehydroprolines and Their P-Analogs. Russ. Chem. Bull. 2013, 62, 792–796. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Karimova, N.M.; Odinets, I.L.; Röschenthaler, G.V.; Osipov, S.N. Click-Chemistry Approach to Isoxazole-Containing α-CF 3-Substituted α-Aminocarboxylates and α-Aminophosphonates. Org. Biomol. Chem. 2011, 9, 7335–7342. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Peregudov, A.S.; Röschenthaler, G.V.; Osipov, S.N. Synthesis of α-CF3-Containing Triazolyl Amino Acids as Potential Neurotransmitters via Click-Reaction. J. Fluor. Chem. 2015, 175, 60–67. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Petropavlovskikh, D.A.; Godovikov, I.A.; Nefedov, S.E.; Osipov, S.N. Rh(III)-Catalyzed C−H Activation/Annulation of Aryl Hydroxamates with CF3-Containing α-Propargyl α-Amino Acid Derivatives. Eur. J. Org. Chem. 2021, 2021, 1883–1890. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Bubnova, A.S.; Godovikov, I.A.; Danshina, A.A.; Osipov, S.N. Rh(III)-Catalyzed [4 + 2]-Annulation of Indoles: Access to Functionalized Pyrimidoindolones Containing α-Amino Acid Framework. Asian J. Org. Chem. 2023, 12, e202200485. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Petropavlovskikh, D.A.; Godovikov, I.A.; Dolgushin, F.M.; Osipov, S.N. Synthesis of Functionalized Isoquinolone Derivatives via Rh(III)-Catalyzed [4+2]-Annulation of Benzamides with Internal Acetylene-Containing α-CF3-α-Amino Carboxylates. Molecules 2022, 27, 8488. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yang, J.; Zhang, X. Highly Enantioselective Zinc/BINOL-Catalyzed Alkynylation of α-Ketoimine Ester: A New Entry to Optically Active Quaternary α-CF3-α-Amino Acids. Chem. Commun. 2011, 47, 5587–5589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.G.; Ma, H.; Zheng, Y.; Ma, J.A. Zinc-Mediated Enantioselective Addition of Terminal 1,3-Diynes to N-Arylimines of Trifluoropyruvates. Tetrahedron 2012, 68, 7663–7669. [Google Scholar] [CrossRef]
- Morisaki, K.; Sawa, M.; Nomaguchi, J.Y.; Morimoto, H.; Takeuchi, Y.; Mashima, K.; Ohshima, T. Rh-Catalyzed Direct Enantioselective Alkynylation of α-Ketiminoesters. Chem. Eur. J. 2013, 19, 8417–8420. [Google Scholar] [CrossRef]
- Morisaki, K.; Sawa, M.; Yonesaki, R.; Morimoto, H.; Mashima, K.; Ohshima, T. Mechanistic Studies and Expansion of the Substrate Scope of Direct Enantioselective Alkynylation of α-Ketiminoesters Catalyzed by Adaptable (Phebox)Rhodium(III) Complexes. J. Am. Chem. Soc. 2016, 138, 6194–6203. [Google Scholar] [CrossRef]
- Fustero, S.; Bello, P.; Miró, J.; Sánchez-Roselló, M.; Maestro, M.A.; González, J.; del Pozo, C. Gold Catalyzed Stereoselective Tandem Hydroamination-Formal Aza-Diels-Alder Reaction of Propargylic Amino Esters. Chem. Commun. 2013, 49, 1336–1338. [Google Scholar] [CrossRef]
- Sánchez-Roselló, M.; Miró, J.; Del Pozo, C.; Fustero, S. Differential Reactivity of Fluorinated Homopropargylic Amino Esters vs Gold(I) Salts. the Role of the Nitrogen Protecting Group. J. Fluor. Chem. 2015, 171, 60–66. [Google Scholar] [CrossRef]
- Sun, X.S.; Ou-Yang, Q.; Xu, S.M.; Wang, X.H.; Tao, H.Y.; Chung, L.W.; Wang, C.J. Asymmetric Synthesis of Quaternary α-Trifluoromethyl α-Amino Acids by Ir-Catalyzed Allylation Followed by Kinetic Resolution. Chem. Commun. 2020, 56, 3333–3336. [Google Scholar] [CrossRef] [PubMed]
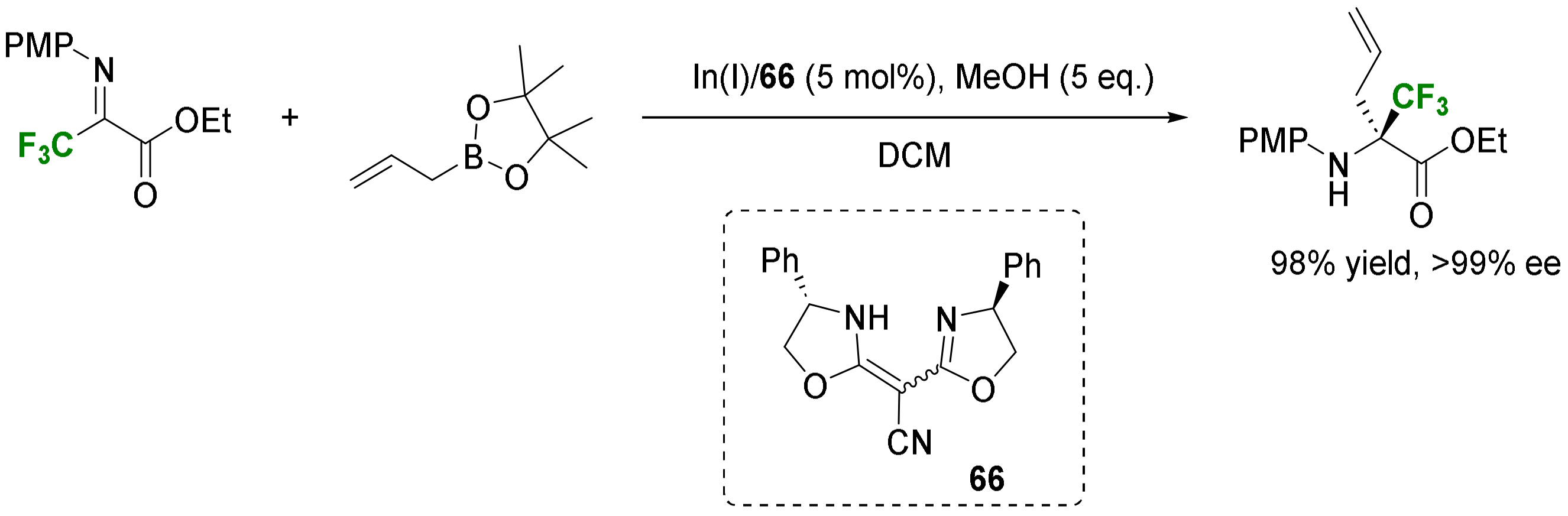
- Bhakta, U.; Kattamuri, P.V.; Siitonen, J.H.; Alemany, L.B.; Kürti, L. Enantioselective Catalytic Allylation of Acyclic Ketiminoesters: Synthesis of α-Fully-Substituted Amino Esters. Org. Lett. 2019, 21, 9208–9211. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Kim, H.; Waser, M. Pd-Catalyzed Allylation of Imines to Access α-CF3-Substituted α-Amino Acid Derivatives. Eur. J. Org. Chem. 2019, 2019, 7122–7127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Samanta, S.S.; Michieletto, J.; Roche, S.P. A Broad Substrate Scope of Aza-Friedel-Crafts Alkylation for the Synthesis of Quaternary α-Amino Esters. Org. Lett. 2020, 22, 5822–5827. [Google Scholar] [CrossRef] [PubMed]
- Yonesaki, R.; Kondo, Y.; Akkad, W.; Sawa, M.; Morisaki, K.; Morimoto, H.; Ohshima, T. 3-Mono-Substituted BINOL Phosphoric Acids as Effective Organocatalysts in Direct Enantioselective Friedel–Crafts-Type Alkylation of N-Unprotected α-Ketiminoester. Chem. Eur. J. 2018, 24, 15211–15214. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Liu, B.; Zhou, H.B.; Dong, C. Enhanced Efficiency of Recyclable C 3-Symmetric Cinchonine-Squaramides in the Asymmetric Friedel-Crafts Reaction of Indoles with Alkyl Trifluoropyruvate. Tetrahedron Asymmetry 2012, 23, 1332–1337. [Google Scholar] [CrossRef]
- Han, X.; Wu, H.; Wang, W.; Dong, C.; Tien, P.; Wu, S.; Zhou, H.B. Synthesis and SARs of Indole-Based α-Amino Acids as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Org. Biomol. Chem. 2014, 12, 8308–8317. [Google Scholar] [CrossRef]
- Waki, M.; Katagiri, T.; Matsuno, K.; Miyachi, H. Synthesis of β-Amino-α-Trifluoromethyl-α-Amino Acids Exhibiting Intramolecular Interaction of CF3 with NHβ. Tetrahedron Lett. 2014, 55, 6915–6918. [Google Scholar] [CrossRef]
- Katagiri, T.; Katayama, Y.; Taeda, M.; Ohshima, T.; Iguchi, N.; Uneyama, K. Preparation of Optically Pure α-Trifluoromethyl-α-Amino Acids from N-Tosyl-2-Trifluoromethyl-2-Alkyloxycarbonyl Aziridine. J. Org. Chem. 2011, 76, 9305–9311. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Bezgubenko, L.V.; Stanko, O.V.; Rusanov, E.B.; Kulik, I.B.; Onys’ko, P.P. Diastereoselective Cycloaddition of (S)-N-(1-Phenylethylimino)Trifluoropropionate and Trifluoroethylphosphonate with Diazomethane. Tetrahedron Asymmetry 2017, 28, 555–560. [Google Scholar] [CrossRef]
- Marsini, M.A.; Reeves, J.T.; Desrosiers, J.N.; Herbage, M.A.; Savoie, J.; Li, Z.; Fandrick, K.R.; Sader, C.A.; McKibben, B.; Gao, D.A.; et al. Diastereoselective Synthesis of α-Quaternary Aziridine-2-Carboxylates via Aza-Corey-Chaykovsky Aziridination of N-Tert-Butanesulfinyl Ketimino Esters. Org. Lett. 2015, 17, 5614–5617. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Faust, K.; Himmelsbach, M.; Waser, M. Synthesis of α-CF3-Proline Derivatives by Means of a Formal (3 + 2)-Cyclisation between Trifluoropyruvate Imines and Michael Acceptors. Org. Biomol. Chem. 2019, 17, 5731–5735. [Google Scholar] [CrossRef] [PubMed]
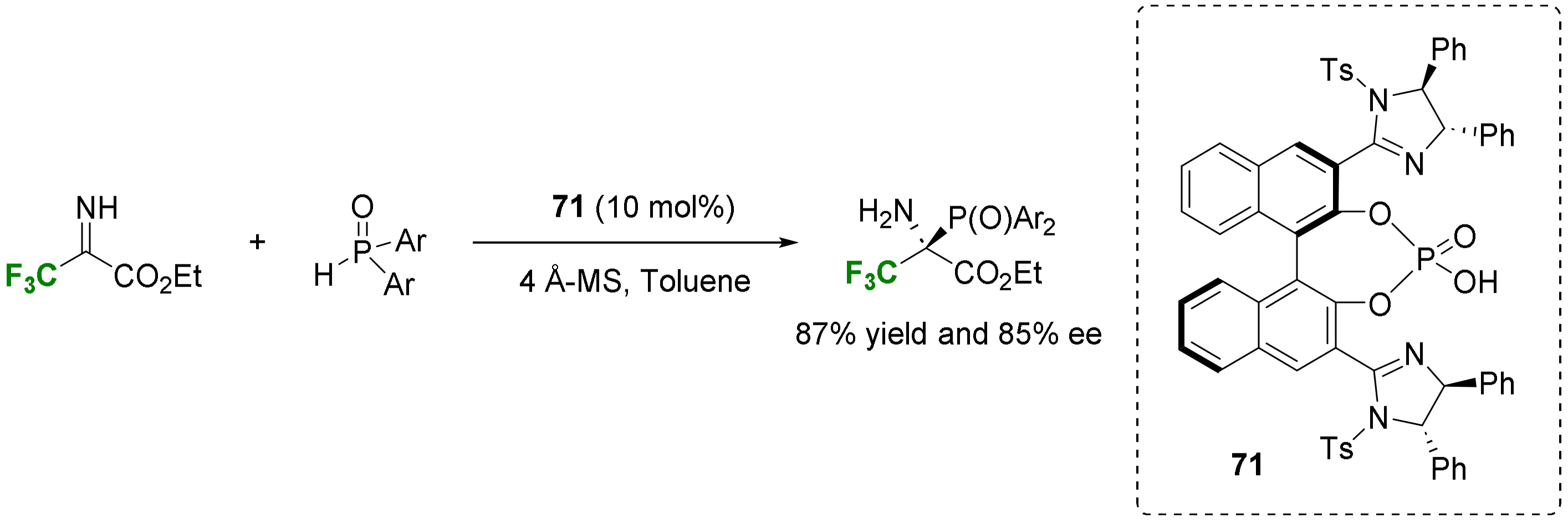
- Ogura, K.; Isozumi, I.; Takehara, T.; Suzuki, T.; Nakamura, S. Enantioselective Reaction of N-Unprotected Activated Ketimines with Phosphine Oxides Catalyzed by Chiral Imidazoline-Phosphoric Acids. Org. Lett. 2022, 24, 8088–8092. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liu, Z.J.; Wu, F. Highly Regio- and Diastereoselective Addition of Organolithium Reagents to Chiral Fluoroalkyl α,β-Unsaturated n-Tert-Butanesulfinyl Ketimines: A General and Efficient Access to α-Tertiary Fluoroalkyl Allylic Amines and α-Fluoroalkyl α-Amino Acids. Adv. Synth. Catal. 2015, 357, 818–822. [Google Scholar] [CrossRef]
- Sawa, M.; Morisaki, K.; Kondo, Y.; Morimoto, H.; Ohshima, T. Direct Access to N-Unprotected α- and/or β-Tetrasubstituted Amino Acid Esters via Direct Catalytic Mannich-Type Reactions Using N-Unprotected Trifluoromethyl Ketimines. Chem. Eur. J. 2017, 23, 17022–17028. [Google Scholar] [CrossRef] [PubMed]
- Cherednichenko, A.S.; Bezgubenko, L.V.; Rusanov, E.B.; Onys’ko, P.P.; Rassukana, Y.V. Enantiomeric N-Tert-Butylsulfinyl Imines of Methyl Trifluoropyruvate: Promising Building Blocks in Asymmetric Synthesis of α-Trifluoromethylated Amino Acids and Derivatives. ChemistrySelect 2020, 5, 13569–13574. [Google Scholar] [CrossRef]
- Ouerfelli, O.; Simon, J.; Chelain, E.; Pytkowicz, J.; Besbes, R.; Brigaud, T. Enantiopure α-Trifluoromethylated Aziridine-2-Carboxylic Acid (α-TfmAzy): Synthesis and Peptide Coupling. Org. Lett. 2020, 22, 2946–2949. [Google Scholar] [CrossRef]
- Lensen, N.; Marais, J.; Brigaud, T. Straightforward Synthesis of Novel Enantiopure α-Trifluoromethylated Azetidine 2-Carboxylic Acid and Homoserines. Org. Lett. 2015, 17, 342–345. [Google Scholar] [CrossRef]
- Sanchez, C.A.; Gadais, C.; Diarra, S.; Bordessa, A.; Lensen, N.; Chelain, E.; Brigaud, T. Synthesis of Enantiopure α-Tfm-Proline and α-Tfm-Pipecolic Acid from Oxazolo-Pyrrolidines and -Piperidines. Org. Biomol. Chem. 2021, 19, 6771–6775. [Google Scholar] [CrossRef]
- Nam, D.; Tinoco, A.; Shen, Z.; Adukure, R.D.; Sreenilayam, G.; Khare, S.D.; Fasan, R. Enantioselective Synthesis of α-Trifluoromethyl Amines via Biocatalytic N-H Bond Insertion with Acceptor-Acceptor Carbene Donors. J. Am. Chem. Soc. 2022, 144, 2590–2602. [Google Scholar] [CrossRef]
- LoGiudice, N.; Le, L.; Abuan, I.; Leizorek, Y.; Roberts, S.C. Alpha-Difluoromethylornithine, an Irreversible Inhibitor of Polyamine Biosynthesis, as a Therapeutic Strategy against Hyperproliferative and Infectious Diseases. Med. Sci. 2018, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Levin, V.A.; Ictech, S.E.; Hess, K.R. Clinical Importance of Eflornithine (α-Difluoromethylornithine) for the Treatment of Malignant Gliomas. CNS Oncol. 2018, 7, CNS16. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B.; Abeles, R.H. Inactivation of Pyridoxal Phosphate Dependent Enzymes by Mono- and Polyhaloalanines. Biochemistry 1976, 15, 4718–4723. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, B.W.; Bey, P.; Danzin, C.; Jung, M.J.; Casara, P.; Vevert, J.P. Catalytic Irreversible Inhibition of Mammalian Ornithine Decarboxylase (E.C.4.1.1.17) by Substrate and Product Analogs. J. Am. Chem. Soc. 1978, 100, 2551–2553. [Google Scholar] [CrossRef]
- Seo, Y.M.; Khang, Y.H.; Yun, H. Kinetic Resolution of 3-Fluoroalanine Using a Fusion Protein of D-Amino Acid Oxidase with Vitroscilla Hemoglobin. Biosci. Biotechnol. Biochem. 2011, 75, 820–822. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phillips, R.S.; Dua, R.K. Indole Protects Tryptophan Indole-Lyase, but Not Tryptophan Synthase, from Inactivation by Trifluoroalanine. Arch. Biochem. Biophys. 1992, 296, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, D.; Baxter, R.L.; Campopiano, D.J.; Kerbarh, O.; Sawyer, L.; Tomczyk, N.; Watt, R.; Webster, S.P. Suicide Inhibition of α-Oxamine Synthases: Structures of the Covalent Adducts of 8-Amino-7-Oxononanoate Synthase with Trifluoroalanine. Org. Biomol. Chem. 2006, 4, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Tysoe, C.; Withers, S.G. Fluorinated Mechanism-Based Inhibitors: Common Themes and Recent Developments. Curr. Top. Med. Chem. 2014, 14, 865–874. [Google Scholar] [CrossRef]
- Sinisi, R.; Sani, M.; Candiani, G.; Parente, R.; Pecker, F.; Bellosta, S.; Zanda, M. Synthesis of α-Trifluoromethyl-α-Amino-β-Sulfone Hydroxamates: Novel Nanomolar Inhibitors of Matrix Metalloproteinases. Tetrahedron Lett. 2005, 46, 6515–6518. [Google Scholar] [CrossRef]
- Török, B.; Sood, A.; Bag, S.; Kulkarni, A.; Borkin, D.; Lawler, E.; Dasgupta, S.; Landge, S.; Abid, M.; Zhou, W.; et al. Structure-Activity Relationships of Organofluorine Inhibitors of β-Amyloid Self-Assembly. ChemMedChem 2012, 7, 910–919. [Google Scholar] [CrossRef]
- Margiotta, N.; Papadia, P.; Lazzaro, F.; Crucianelli, M.; De Angelis, F.; Pisano, C.; Vesci, L.; Natile, G. Platinum-Based Antitumor Drugs Containing Enantiomerically Pure α-Trifluoromethyl Alanine as Ligand. J. Med. Chem. 2005, 48, 7821–7828. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, V.B.; Aksinenko, A.Y.; Sokolov, A.V.; Gabrelian, A.; Efimova, A.D.; Grigoriev, V.V. Modification of Biologically Active Amides and Amines with Fluorine containing Heterocycles 12. Endo- and Exocyclic Modifications of the Drug Riluzole with Trifluoromethyllcontaining Heterocycles. Russ. Chem. Bul. 2017, 68, 2241–2244. [Google Scholar] [CrossRef]
- Wehner, V.; Stilz, H.U.; Osipov, S.N.; Golubev, A.S.; Sieler, J.; Burger, K. Trifluoromethyl-Substituted Hydantoins, Versatile Building Blocks for Rational Drug Design. Tetrahedron 2004, 60, 4295–4302. [Google Scholar] [CrossRef]
- Brown, M.L.; Eidam, H.A.; Paige, M.; Jones, P.J.; Patel, M.K. Comparative Molecular Field Analysis and Synthetic Validation of a Hydroxyamide-Propofol Binding and Functional Block of Neuronal Voltage-Dependent Sodium Channels. Bioorg. Med. Chem. 2009, 17, 7056–7063. [Google Scholar] [CrossRef] [PubMed]
- Rogacki, M.K.; Pitta, E.; Balabon, O.; Huss, S.; Lopez-Roman, E.M.; Argyrou, A.; Blanco-Ruano, D.; Cacho, M.; Vande Velde, C.M.L.; Augustyns, K.; et al. Identification and Profiling of Hydantoins—A Novel Class of Potent Antimycobacterial DprE1 Inhibitors. J. Med. Chem. 2018, 61, 11221–11249. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, T.; Chowdhary, S.; Ataka, K.; Er, J.; Dreyhsig, G.H.; Heberle, J.; Koksch, B. Introducing Aliphatic Fluoropeptides: Perspectives on Folding Properties, Membrane Partition and Proteolytic Stability. Chem. Eur. J. 2023, 29, e202203860. [Google Scholar] [CrossRef] [PubMed]
- Chaume, G.; Lensen, N.; Caupène, C.; Brigaud, T. Convenient Synthesis of N-Terminal Tfm-Dipeptides from Unprotected Enantiopure α-Tfm-Proline and α-Tfm-Alanine. Eur. J. Org. Chem. 2009, 2009, 5717–5724. [Google Scholar] [CrossRef]
- Devillers, E.; Pytkowicz, J.; Chelain, E.; Brigaud, T. Synthesis of Protected Enantiopure (R) and (S)-α-Trifluoromethylalanine Containing Dipeptide Building Blocks Ready to Use for Solid Phase Peptide Synthesis. Amino Acids 2016, 48, 1457–1468. [Google Scholar] [CrossRef]
- Kadota, K.; Mikami, T.; Kohata, A.; Morimoto, J.; Sando, S.; Aikawa, K.; Okazoe, T. Synthesis of Short Peptides with Perfluoroalkyl Side Chains and Evaluation of Their Cellular Uptake Efficiency. ChemBioChem 2023, 24, e202300374. [Google Scholar] [CrossRef]
- Dal Pozzo, A.; Ni, M.; Muzi, L.; De Castiglione, R.; Mondelli, R.; Mazzini, S.; Penco, S.; Pisano, C.; Castorina, M.; Giannini, G. Incorporation of the Unusual Cα-Fluoroalkylamino Acids into Cyclopeptides: Synthesis of Arginine-Glycine-Aspartate (RGD) Analogues and Study of Their Conformational and Biological Behavior. J. Med. Chem. 2006, 49, 1808–1817. [Google Scholar] [CrossRef]
- Simon, J.; Pytkowicz, J.; Lensen, N.; Chaume, G.; Brigaud, T. Incorporation of Trifluoromethylated Proline and Surrogates into Peptides: Application to the Synthesis of Fluorinated Analogues of the Neuroprotective Glycine-Proline-Glutamate (GPE) Tripeptide. J. Org. Chem. 2016, 81, 5381–5392. [Google Scholar] [CrossRef]
- Devillers, E.; Chelain, E.; Dalvit, C.; Brigaud, T.; Pytkowicz, J. (R)-α-Trifluoromethylalanine as a 19F NMR Probe for the Monitoring of Protease Digestion of Peptides. ChemBioChem 2022, 23, e202100470. [Google Scholar] [CrossRef] [PubMed]
- Grage, S.L.; Kara, S.; Bordessa, A.; Doan, V.; Rizzolo, F.; Putzu, M.; Kubař, T.; Papini, A.M.; Chaume, G.; Brigaud, T.; et al. Orthogonal19F-Labeling for Solid-State NMR Spectroscopy Reveals the Conformation and Orientation of Short Peptaibols in Membranes. Chem. Eur. J. 2018, 24, 4328–4335. [Google Scholar] [CrossRef] [PubMed]
- Maisch, D.; Wadhwani, P.; Afonin, S.; Böttcher, C.; Koksch, B.; Ulrich, A.S. Chemical Labeling Strategy with (R)- and (S)-Trifluoromethylalanine for Solid State 19F NMR Analysis of Peptaibols in Membranes. J. Am. Chem. Soc. 2009, 131, 15596–15597. [Google Scholar] [CrossRef] [PubMed]
- Robalo, J.R.; Vila Verde, A. Unexpected Trends in the Hydrophobicity of Fluorinated Amino Acids Reflect Competing Changes in Polarity and Conformation. Phys. Chem. Chem. Phys. 2019, 21, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W.; Langenhan, J.; Huhmann, S.; Moschner, J.; Chang, R.; Accorsi, M.; Seo, J.; Rademann, J.; Meijer, G.; Koksch, B.; et al. An Intrinsic Hydrophobicity Scale for Amino Acids and Its Application to Fluorinated Compounds. Angew. Chem. Int. Ed. 2019, 58, 8216–8220. [Google Scholar] [CrossRef] [PubMed]
- Kubyshkin, V.; Pridma, S.; Budisa, N. Comparative Effects of Trifluoromethyl- and Methyl-Group Substitutions in Proline. New J. Chem. 2018, 42, 13461–13470. [Google Scholar] [CrossRef]
- Plass, M.; Valko, K.; Abraham, M.H. Determination of Solute Descriptors of Tripeptide Derivatives Based on High-Throughput Gradient High-Performance Liquid Chromatography Retention Data. J. Chromatogr. A 1998, 803, 51–60. [Google Scholar] [CrossRef]
- Gadais, C.; Devillers, E.; Gasparik, V.; Chelain, E.; Pytkowicz, J.; Brigaud, T. Probing the Outstanding Local Hydrophobicity Increases in Peptide Sequences Induced by Incorporation of Trifluoromethylated Amino Acids. ChemBioChem 2018, 19, 1026–1030. [Google Scholar] [CrossRef]
- Botz, A.; Gasparik, V.; Devillers, E.; Hoffmann, A.R.F.; Caillon, L.; Chelain, E.; Lequin, O.; Brigaud, T.; Khemtemourian, L. (R)-α-Trifluoromethylalanine Containing Short Peptide in the Inhibition of Amyloid Peptide Fibrillation. Biopolymers 2015, 104, 601–610. [Google Scholar] [CrossRef]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Jlalia, I.; Lensen, N.; Chaume, G.; Dzhambazova, E.; Astasidi, L.; Hadjiolova, R.; Bocheva, A.; Brigaud, T. Synthesis of an MIF-1 Analogue Containing Enantiopure (S)-α- Trifluoromethyl-Proline and Biological Evaluation on Nociception. Eur. J. Med. Chem. 2013, 62, 122–129. [Google Scholar] [CrossRef]
- Oliver, M.; Gadais, C.; García-Pindado, J.; Teixidó, M.; Lensen, N.; Chaume, G.; Brigaud, T. Trifluoromethylated Proline Analogues as Efficient Tools to Enhance the Hydrophobicity and to Promote Passive Diffusion Transport of the L-Prolyl-l-Leucyl Glycinamide (PLG) Tripeptide (Sup Inf). RSC Adv. 2018, 8, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Aikawa, K.; Okazoe, T.; Morimoto, J.; Sando, S. Methyl to Trifluoromethyl Substitution as a Strategy to Increase the Membrane Permeability of Short Peptides. Org. Biomol. Chem. 2021, 19, 9386–9389. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, J.; Dimos, N.; Loll, B.; Hohmann, T.; Dyrks, M.; Wieseke, A.; Keller, B.G.; Koksch, B. Fluorine-Induced Polarity Increases Inhibitory Activity of BPTI towards Chymotrypsin. RSC Chem. Biol. 2022, 3, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Huhmann, S.; Koksch, B. Fine-Tuning the Proteolytic Stability of Peptides with Fluorinated Amino Acids. Eur. J. Org. Chem. 2018, 2018, 3667–3679. [Google Scholar] [CrossRef]
- Koksch, B.; Sewald, N.; Burger, K.; Jakubke, H.-D. Peptide Modification by Incorporation of A-Trifluoromethyl Substituted Amino Acids. Amino Acids 1996, 11, 425–434. [Google Scholar] [CrossRef]
- Koksch, B.; Sewald, N.; Hofmann, H.J.; Burger, K.; Jakubke, H.D. Proteolytically Stable Peptides by Incorporation of Alpha-Tfm Amino Acids. J. Pept. Sci. 1997, 3, 157–167. [Google Scholar] [CrossRef]
- Terzani, F.; Belhattab, S.; Le Guern, A.; Guitot, K.; Monasson, O.; Zanato, C.; Chelain, E.; Leroy-Dudal, J.; Pytkowicz, J. Synthesis and Biological Evaluation of Selective Pepstatin Based Trifluoromethylated Inhibitors of Cathepsin D. Eur. J. Med. Chem. 2024, 267, 116178. [Google Scholar] [CrossRef]
- Kim, W.; Hardcastle, K.I.; Conticello, V.P. Fluoroproline Flip-Flop: Regiochemical Reversal of a Stereoelectronic Effect on Peptide and Protein Structures. Angew. Chem. Int. Ed. 2006, 45, 8141–8145. [Google Scholar] [CrossRef]
- Thomas, C.A.; Talaty, E.R.; Bann, J.G. 3S-Fluoroproline as a Probe to Monitor Proline Isomerization during Protein Folding by 19F-NMR. Chem. Commun. 2009, 3366–3368. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, L.E.; Jenkins, C.L.; Taylor, K.M.; DeRider, M.L.; Raines, R.T. Conformational Stability of Collagen Relies on a Stereoelectronic Effect. J. Am. Chem. Soc. 2001, 123, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Verhoork, S.J.M.; Killoran, P.M.; Coxon, C.R. Fluorinated Prolines as Conformational Tools and Reporters for Peptide and Protein Chemistry. Biochemistry 2018, 57, 6132–6143. [Google Scholar] [CrossRef] [PubMed]
- Newberry, R.W.; Raines, R.T. The N→π* Interaction. Acc. Chem. Res. 2017, 50, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Gadais, C.; Van holsbeeck, K.; Moors, S.L.C.; Buyst, D.; Fehér, K.; Van Hecke, K.; Tourwé, D.; Brigaud, T.; Martin, C.; De Proft, F.; et al. Trifluoromethylated Proline Surrogates as Part of “Pro–Pro” Turn-Inducing Templates. ChemBioChem 2019, 20, 2513–2518. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Ikeda, M.; Kasae, T.; Doi, M.; Demizu, Y.; Oba, M.; Tanaka, M. Synthesis of Chiral α-Trifluoromethyl α,α-Disubstituted α-Amino Acids and Conformational Analysis of L-Leu-Based Peptides with (R)- or (S)-α-Trifluoromethylalanine. ChemistrySelect 2020, 5, 10882–10886. [Google Scholar] [CrossRef]
- Bodero, L.; Guitot, K.; Lensen, N.; Lequin, O.; Brigaud, T.; Ongeri, S.; Chaume, G. Introducing the Chiral Constrained α-Trifluoromethylalanine in Aib Foldamers to Control, Quantify and Assign the Helical Screw-Sense. Chem. Eur. J. 2022, 28, e202103887. [Google Scholar] [CrossRef]
- Shukla, A.K.; Kumar, U. Positron emission tomography: An overview. J. Med. Phys. 2006, 31, 13–21. [Google Scholar] [CrossRef]
- Schwenck, J.; Sonanini, D.; Cotton, J.M.; Rammensee, H.G.; la Fougère, C.; Zender, L.; Pichler, B.J. Advances in PET Imaging of Cancer. Nat. Rev. Cancer 2023, 23, 474–490. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjug. Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef]
- Surasi, D.S.; Bhambhvani, P.; Baldwin, J.A.; Almodovar, S.E.; O’Malley, J.P. 18F-FDG PET and PET/CT Patient Preparation: A Review of the Literature. J. Nucl. Med. Technol. 2014, 42, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Bural, G.; Torigian, D.; Houseni, M.; Alavi, A. PET Radiotracers in Oncology Other than 18F-FDG. J. Nucl. Med. 2020, 61, 1177. [Google Scholar]
- Rong, J.; Haider, A.; Jeppesen, T.E.; Josephson, L.; Liang, S.H. Radiochemistry for Positron Emission Tomography. Nat. Commun. 2023, 14, 3257. [Google Scholar] [CrossRef]
- Wang, C.; Lin, R.; Yao, S. Recent Advances in 18F-Labeled Amino Acids Synthesis and Application. Pharmaceutics 2022, 14, 2207. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Huang, X.; Placzek, M.S.; Krska, S.W.; McQuade, P.; Hooker, J.M.; Groves, J.T. Site-Selective 18F Fluorination of Unactivated C-H Bonds Mediated by a Manganese Porphyrin. Chem. Sci. 2018, 9, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Nodwell, M.B.; Yang, H.; Čolović, M.; Yuan, Z.; Merkens, H.; Martin, R.E.; Bénard, F.; Schaffer, P.; Britton, R. 18F-Fluorination of Unactivated C-H Bonds in Branched Aliphatic Amino Acids: Direct Synthesis of Oncological Positron Emission Tomography Imaging Agents. J. Am. Chem. Soc. 2017, 139, 3595–3598. [Google Scholar] [CrossRef] [PubMed]
- Gronenborn, A.M. Small, but Powerful and Attractive: 19F in Biomolecular NMR. Structure 2022, 30, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, N.G.; Gronenborn, A.M. 19F-Modified Proteins and 19F-Containing Ligands as Tools in Solution NMR Studies of Protein Interactions. Methods Enzymol. 2015, 565, 67–95. [Google Scholar] [CrossRef]
- Kubyshkin, V.S.; Komarov, I.V.; Afonin, S.; Mykhailiuk, P.K.; Grage, S.L.; Ulrich, A.S. Trifluoromethyl-Substituted α-Amino Acids as Solid-State 19F NMR Labels for Structural Studies of Membrane-Bound Peptides. In Fluorine in Pharmaceutical and Medicinal Chemistry; Molecular Medicine and Medicinal Chemistry; Imperial College Press: London, UK, 2011; pp. 91–138. [Google Scholar] [CrossRef]
- Miles, S.A.; Nillama, J.A.; Hunter, L. Tinker, Tailor, Soldier, Spy: The Diverse Roles That Fluorine Can Play within Amino Acid Side Chains. Molecules 2023, 28, 6192. [Google Scholar] [CrossRef]
- Maleckis, A.; Abdelkader, E.H.; Herath, I.D.; Otting, G. Synthesis of Fluorinated Leucines, Valines and Alanines for Use in Protein NMR. Org. Biomol. Chem. 2022, 20, 2424–2432. [Google Scholar] [CrossRef]
- Grage, S.L.; Dürr, U.H.N.; Afonin, S.; Mikhailiuk, P.K.; Komarov, I.V.; Ulrich, A.S. Solid State 19F NMR Parameters of Fluorine-Labeled Amino Acids. Part II: Aliphatic Substituents. J. Magn. Reson. 2008, 191, 16–23. [Google Scholar] [CrossRef]






























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picois, N.; Boutahri, Y.; Milbeo, P.; Zanato, C.; Lensen, N.; Chaume, G.; Brigaud, T. Asymmetric α-Fluoroalkyl-α-Amino Acids: Recent Advances in Their Synthesis and Applications. Molecules 2024, 29, 1408. https://doi.org/10.3390/molecules29061408
Picois N, Boutahri Y, Milbeo P, Zanato C, Lensen N, Chaume G, Brigaud T. Asymmetric α-Fluoroalkyl-α-Amino Acids: Recent Advances in Their Synthesis and Applications. Molecules. 2024; 29(6):1408. https://doi.org/10.3390/molecules29061408
Chicago/Turabian StylePicois, Nathan, Yazid Boutahri, Pierre Milbeo, Chiara Zanato, Nathalie Lensen, Grégory Chaume, and Thierry Brigaud. 2024. "Asymmetric α-Fluoroalkyl-α-Amino Acids: Recent Advances in Their Synthesis and Applications" Molecules 29, no. 6: 1408. https://doi.org/10.3390/molecules29061408
APA StylePicois, N., Boutahri, Y., Milbeo, P., Zanato, C., Lensen, N., Chaume, G., & Brigaud, T. (2024). Asymmetric α-Fluoroalkyl-α-Amino Acids: Recent Advances in Their Synthesis and Applications. Molecules, 29(6), 1408. https://doi.org/10.3390/molecules29061408