
Discovery of Novel 4-Hydroxyquinazoline Derivatives: In Silico, In Vivo and In Vitro Studies Using Primary PARPi-Resistant Cell Lines

Abstract
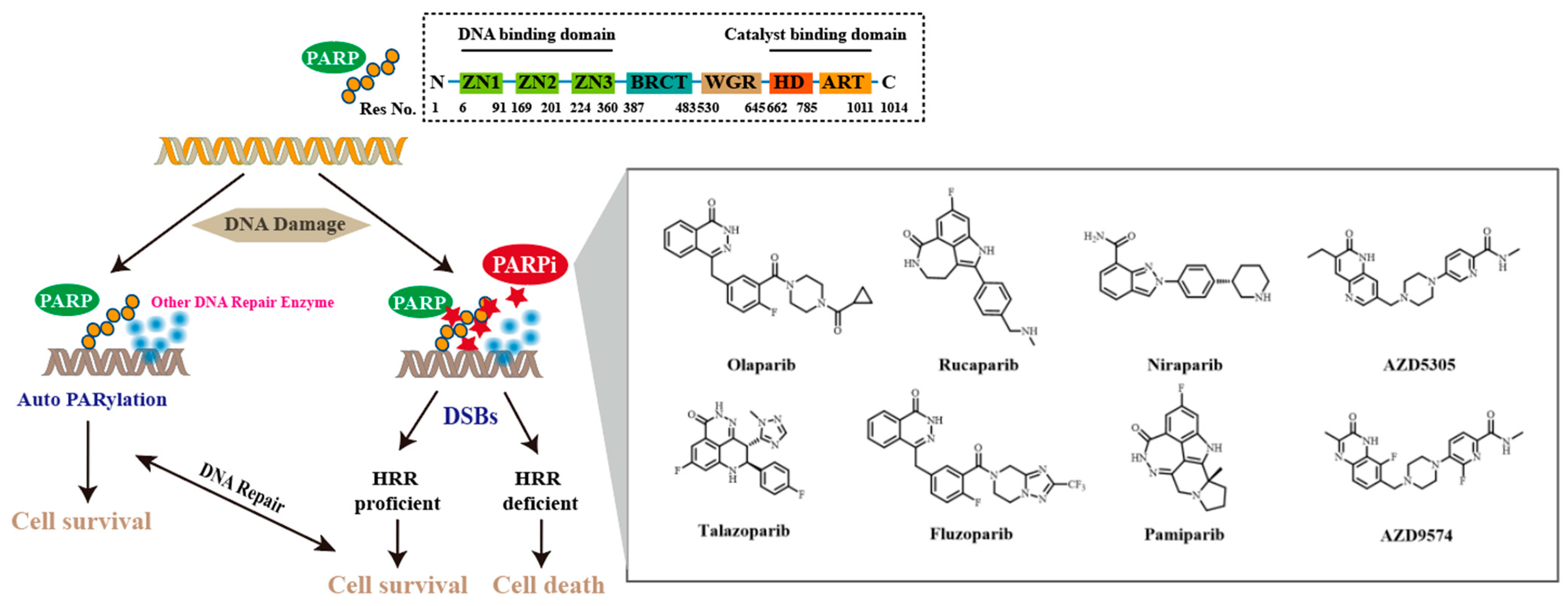
1. Introduction
2. Results and Discussion
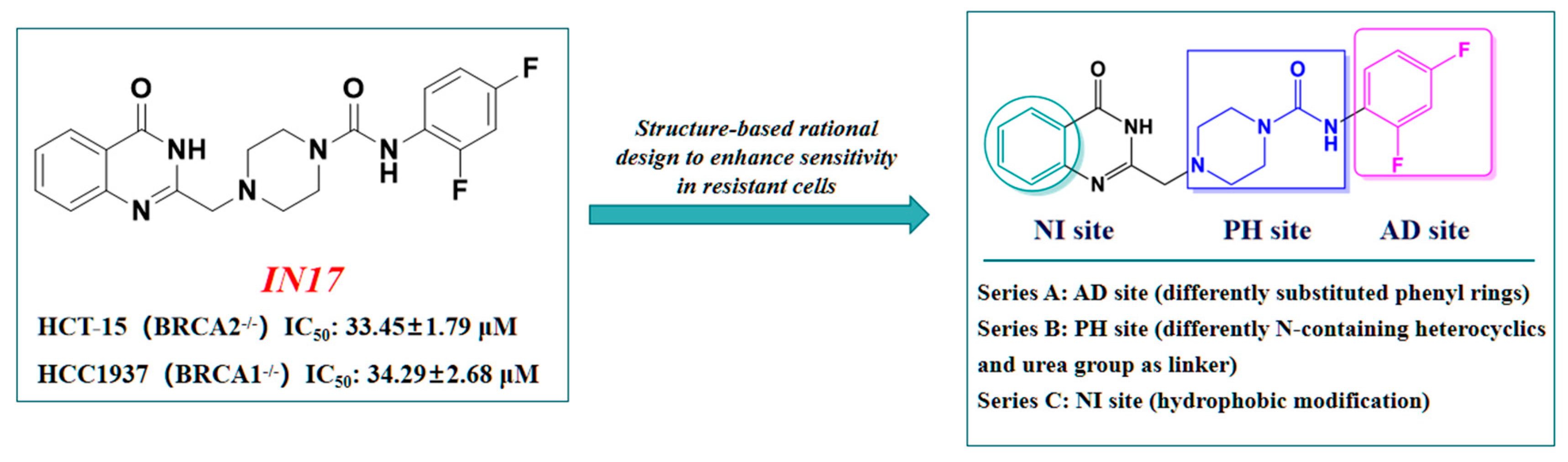
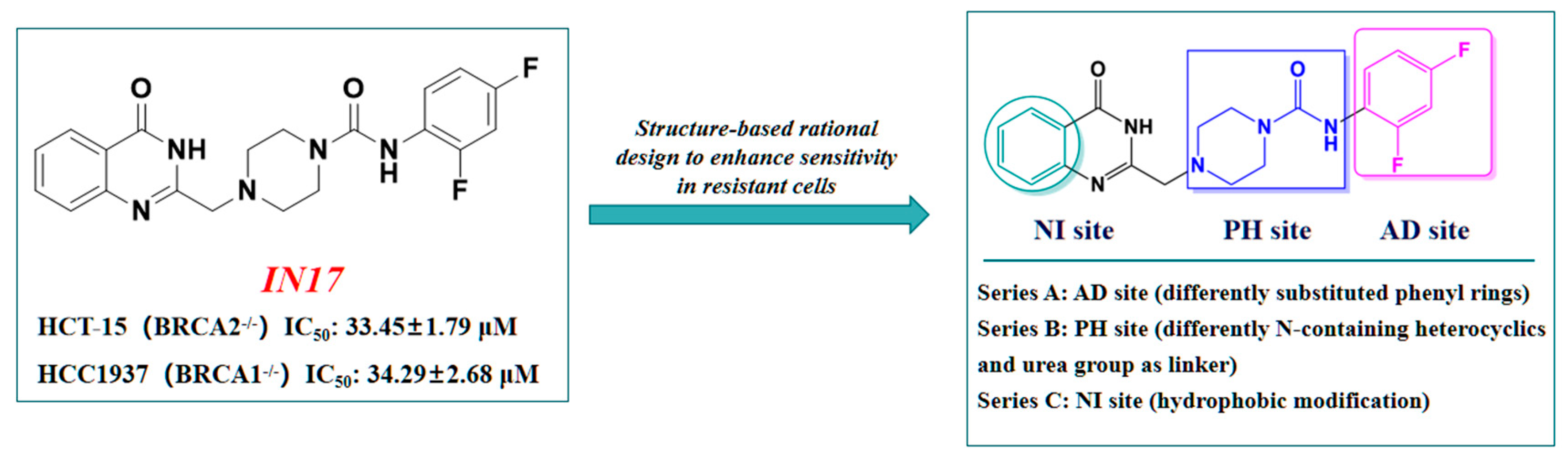
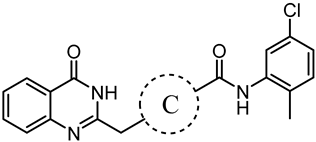
2.1. Design Strategy Based on IN17
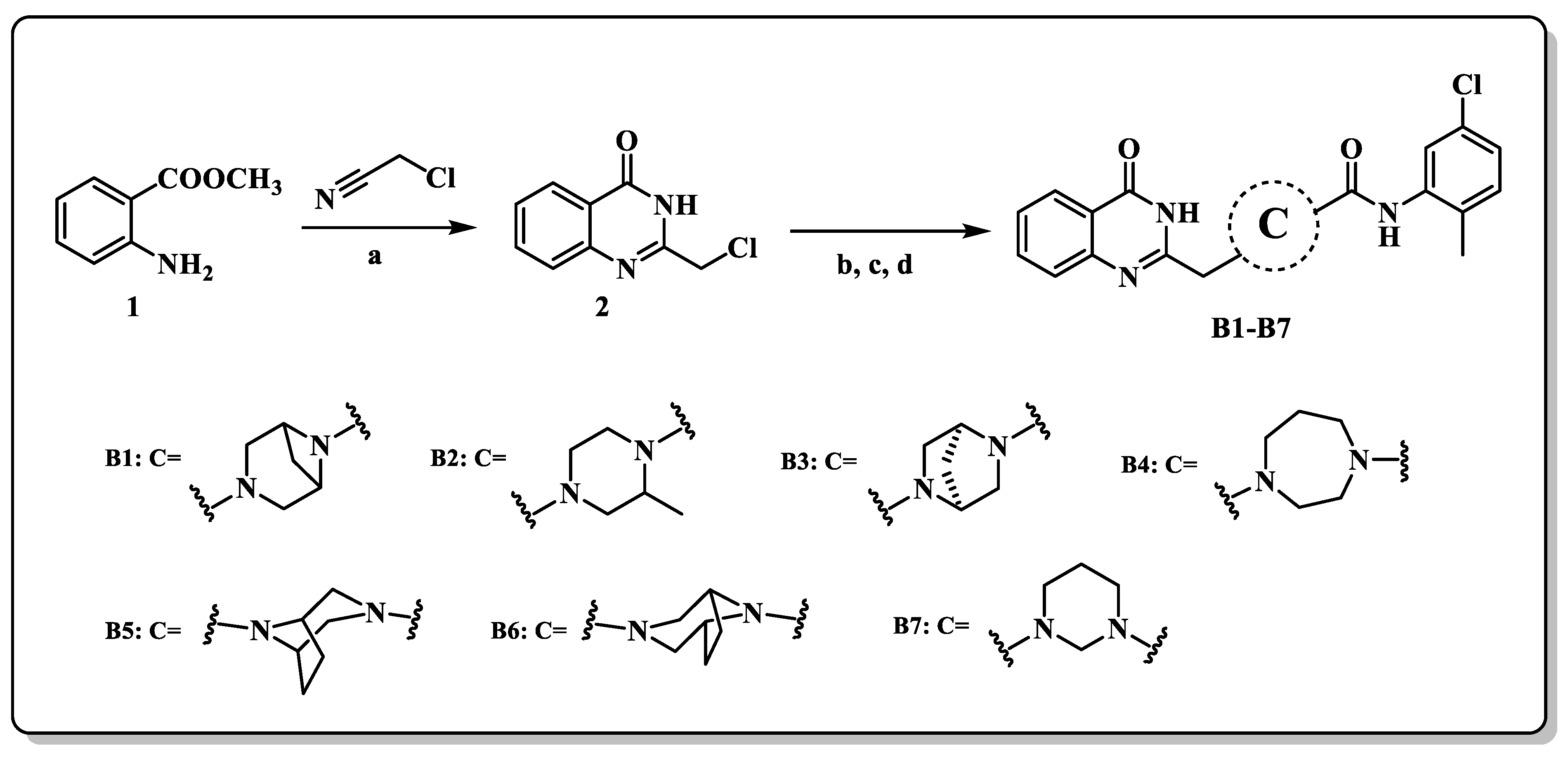
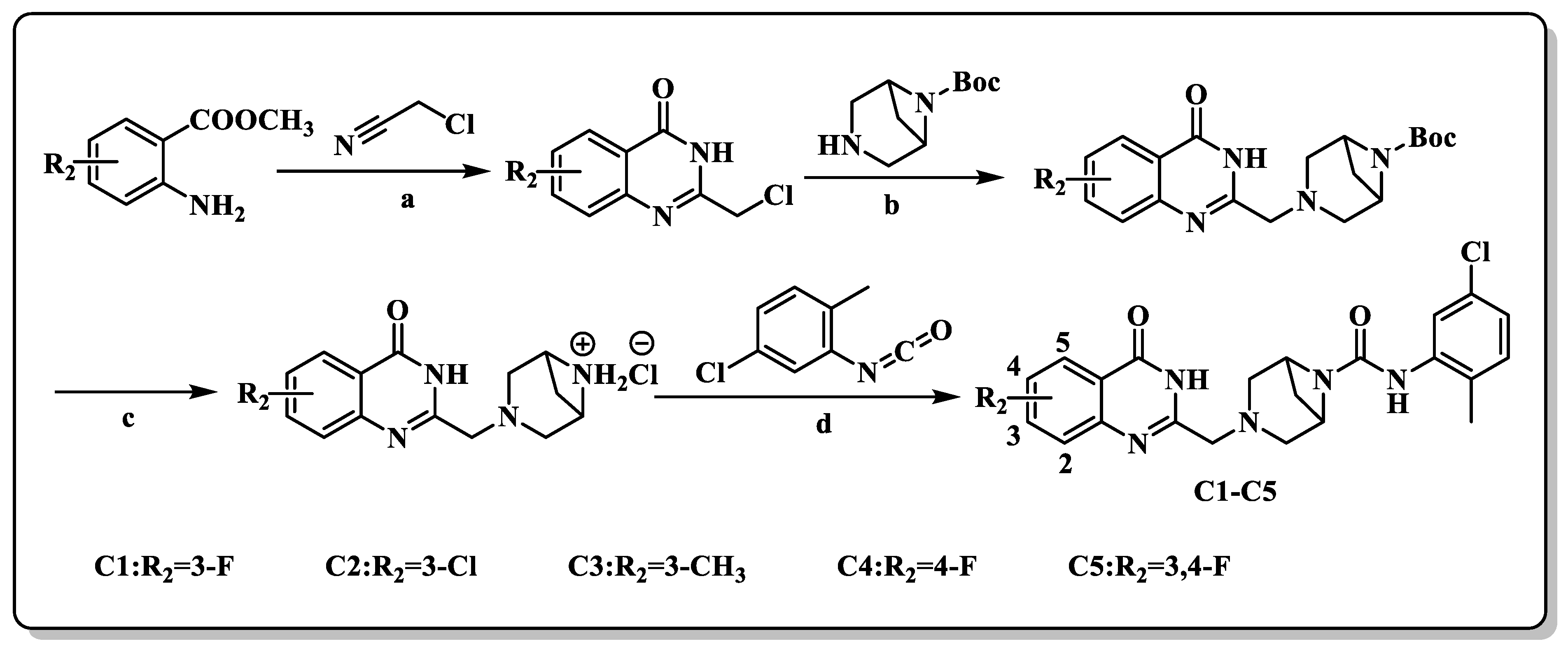
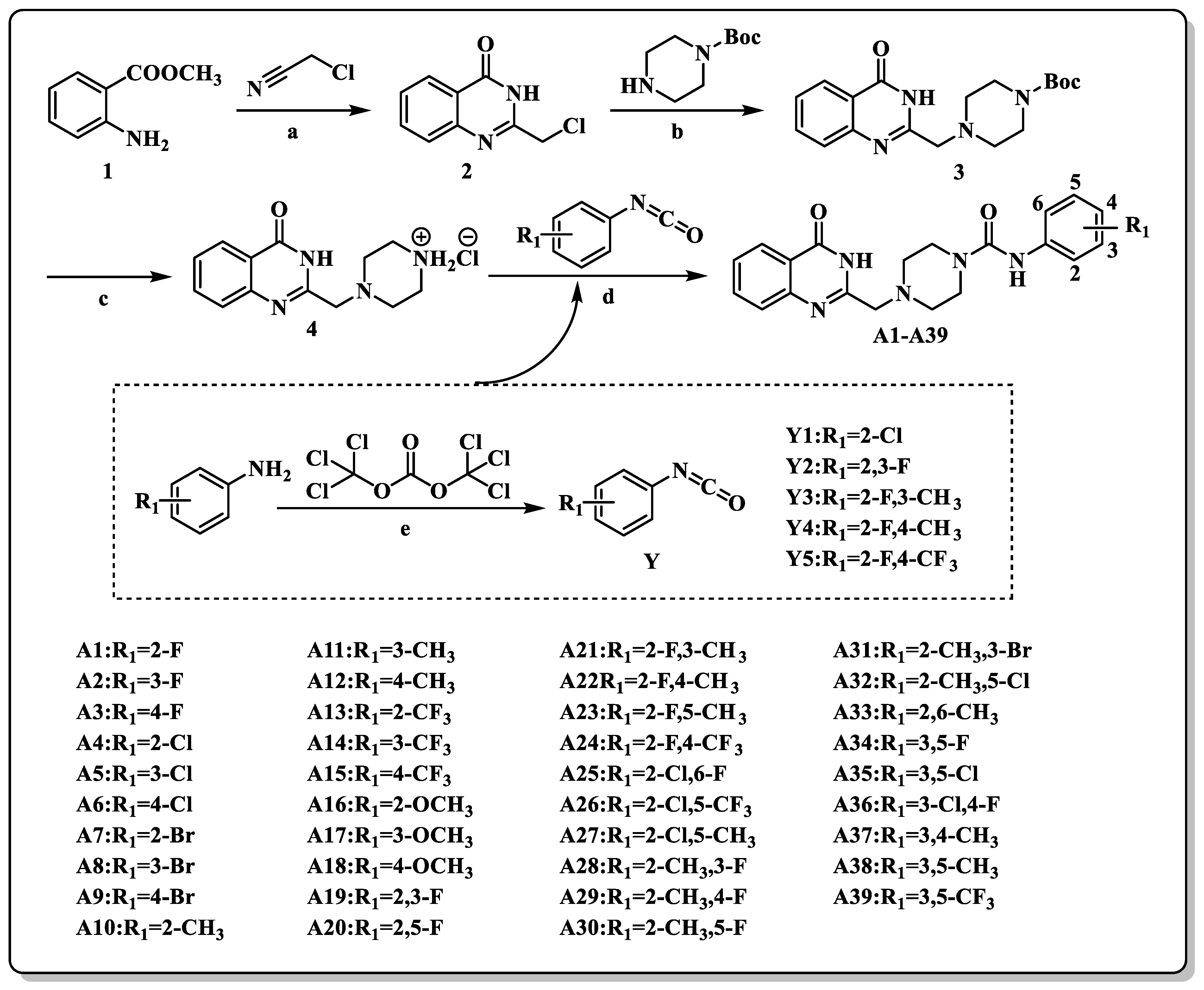
2.2. Chemistry
2.3. Biological Evaluation
2.3.1. In Vitro Anti-Proliferative Activities against HCT-15 and HCC1937 Cell Lines
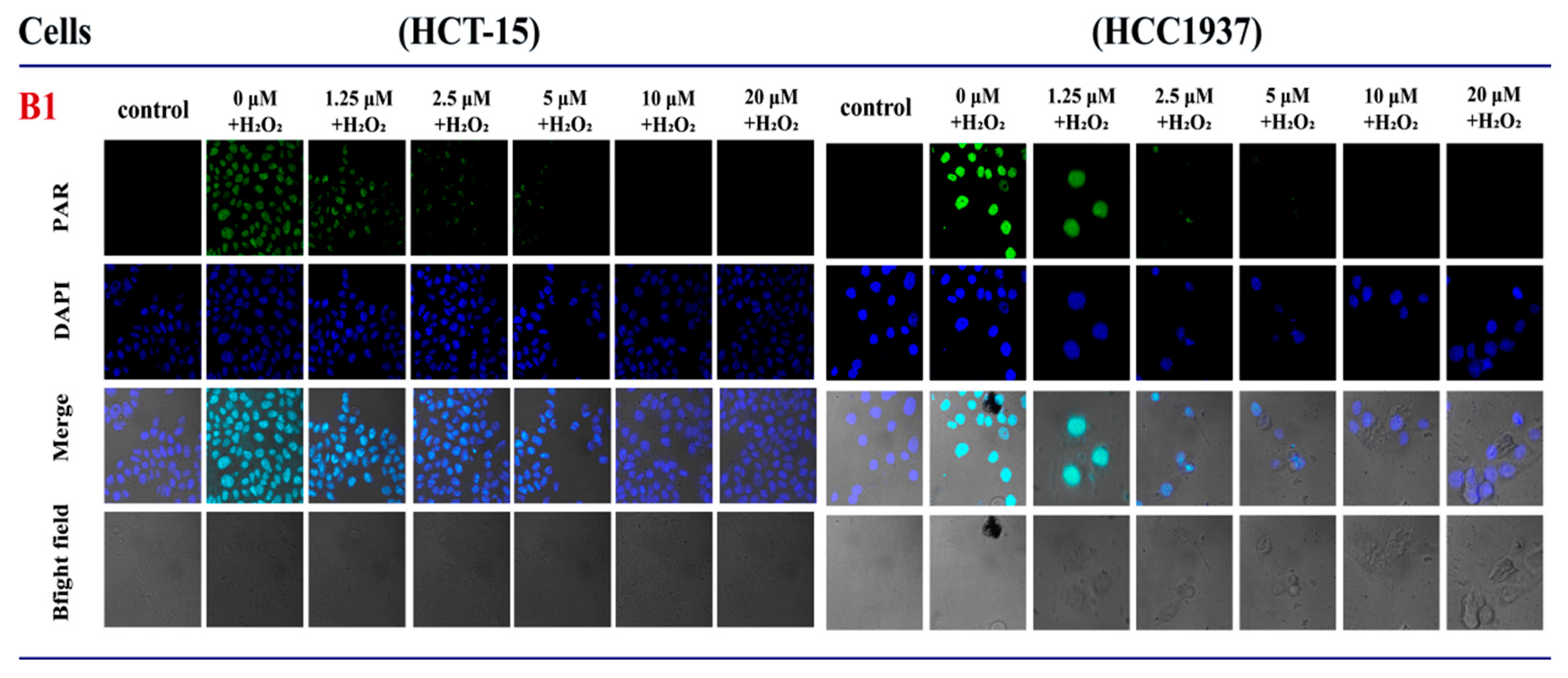
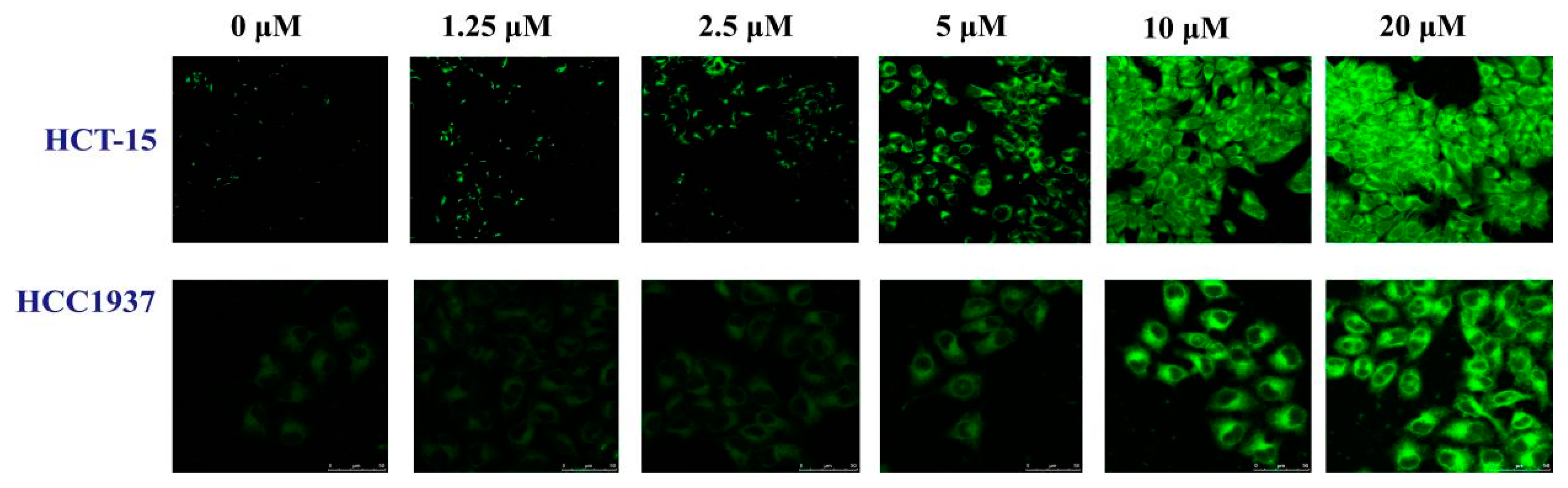
2.3.2. The Influence of B1 on the Expression of PAR in HCT-15 and HCC1937 Cell Lines
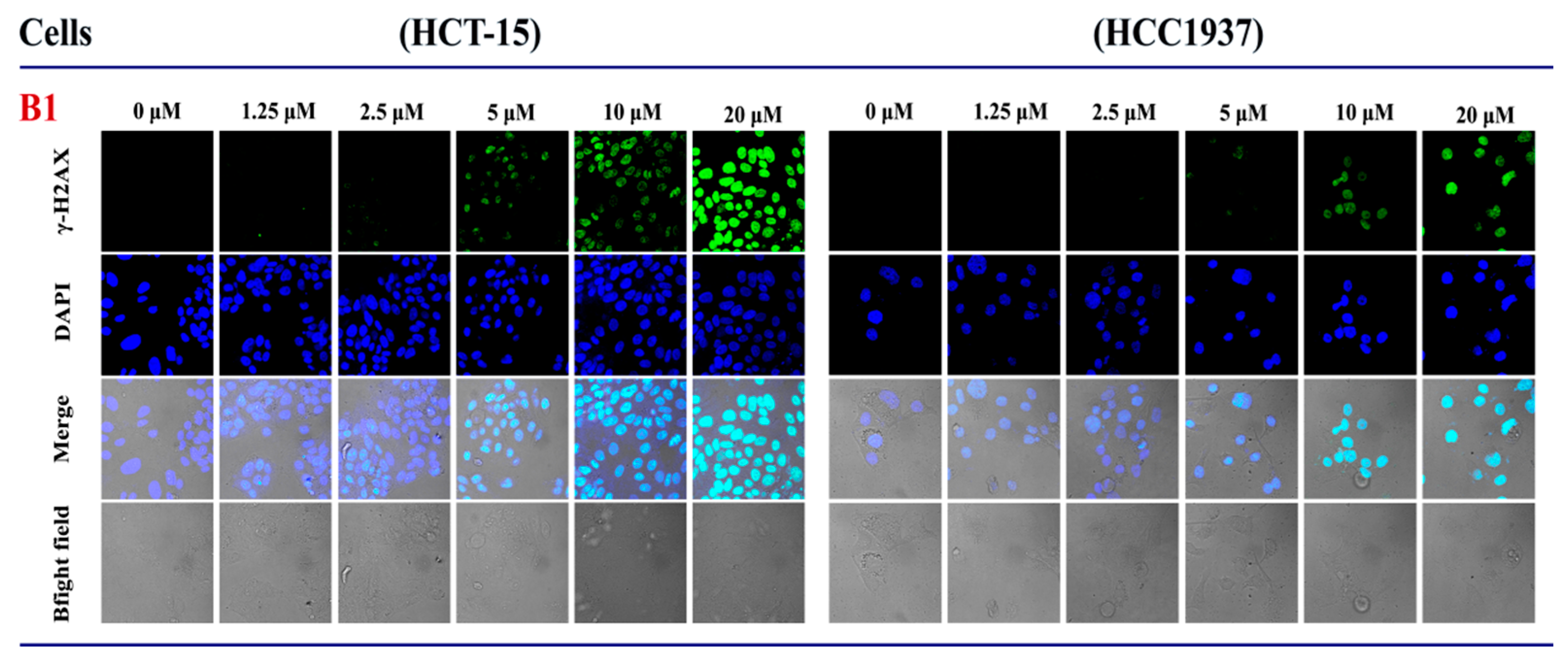
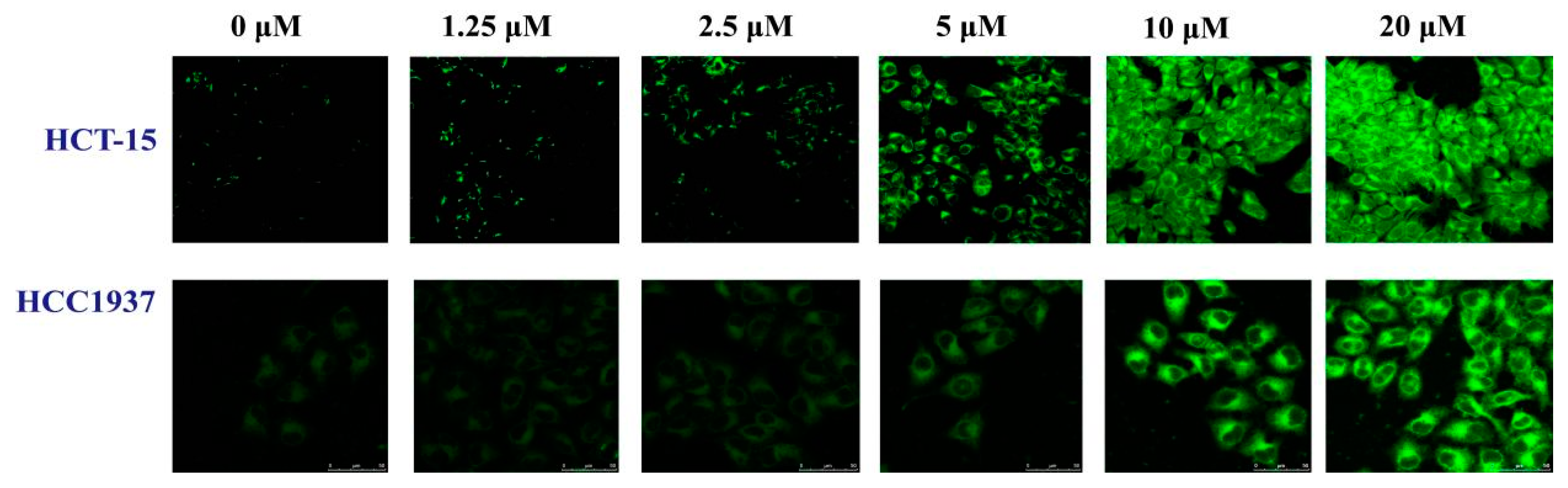
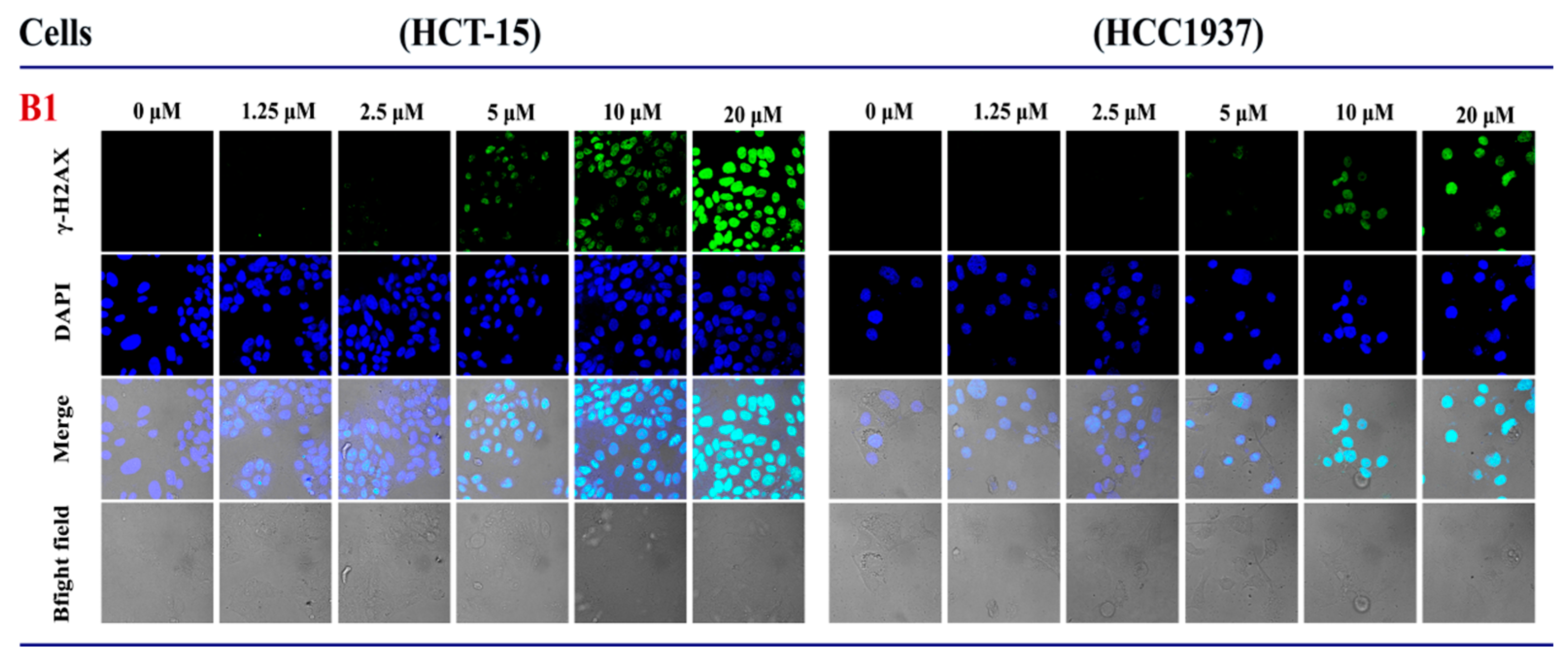
2.3.3. The Influence of B1 on the Expression of γH2AX in HCT-15 and HCC1937 Cell Lines
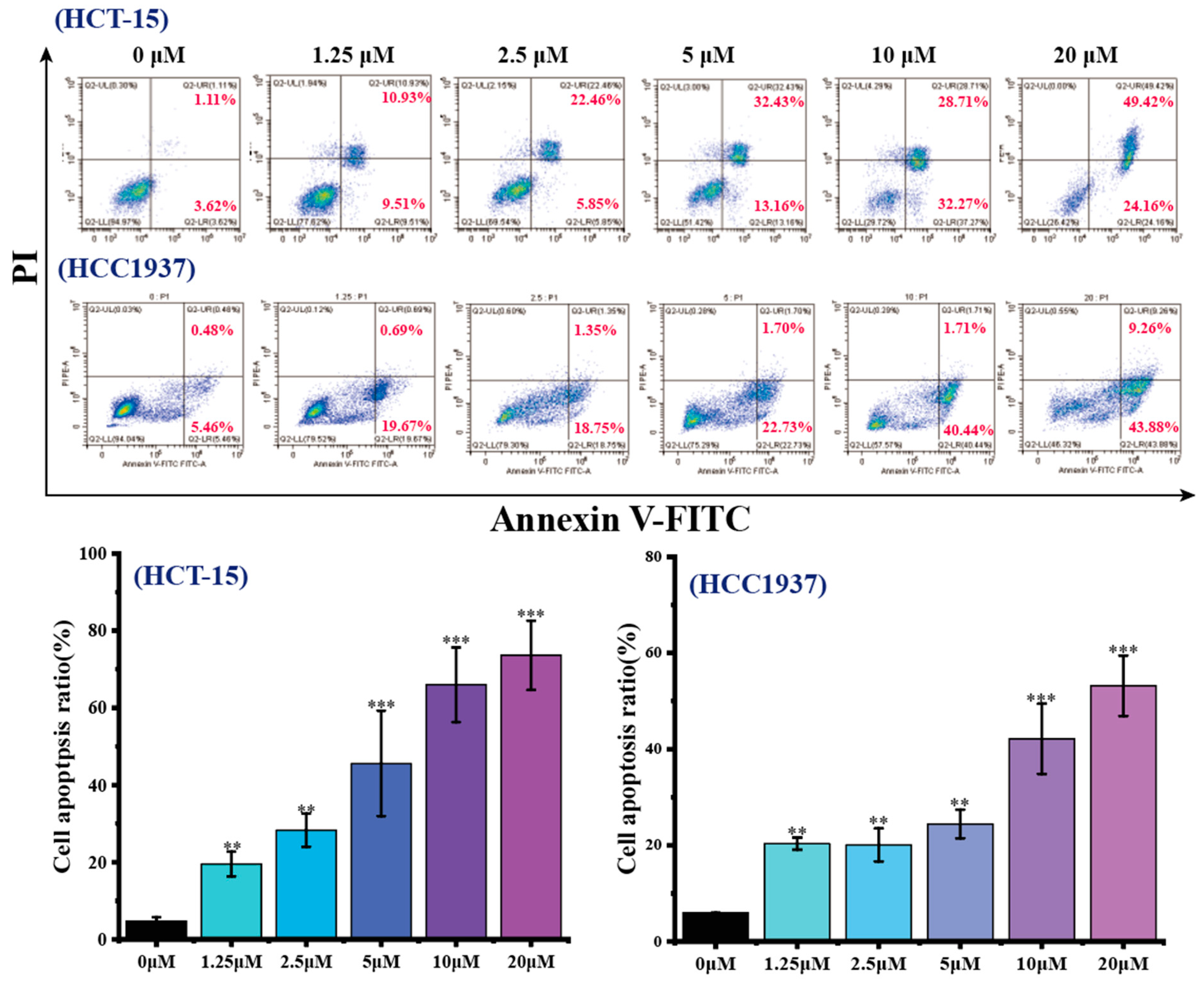
2.3.4. Effects of B1 on Apoptosis in HCT-15 and HCC1937 Cell Lines
2.3.5. Effects of B1 on the Expression of Apoptosis-Related Proteins in HCT-15 and HCC1937 Cell Lines
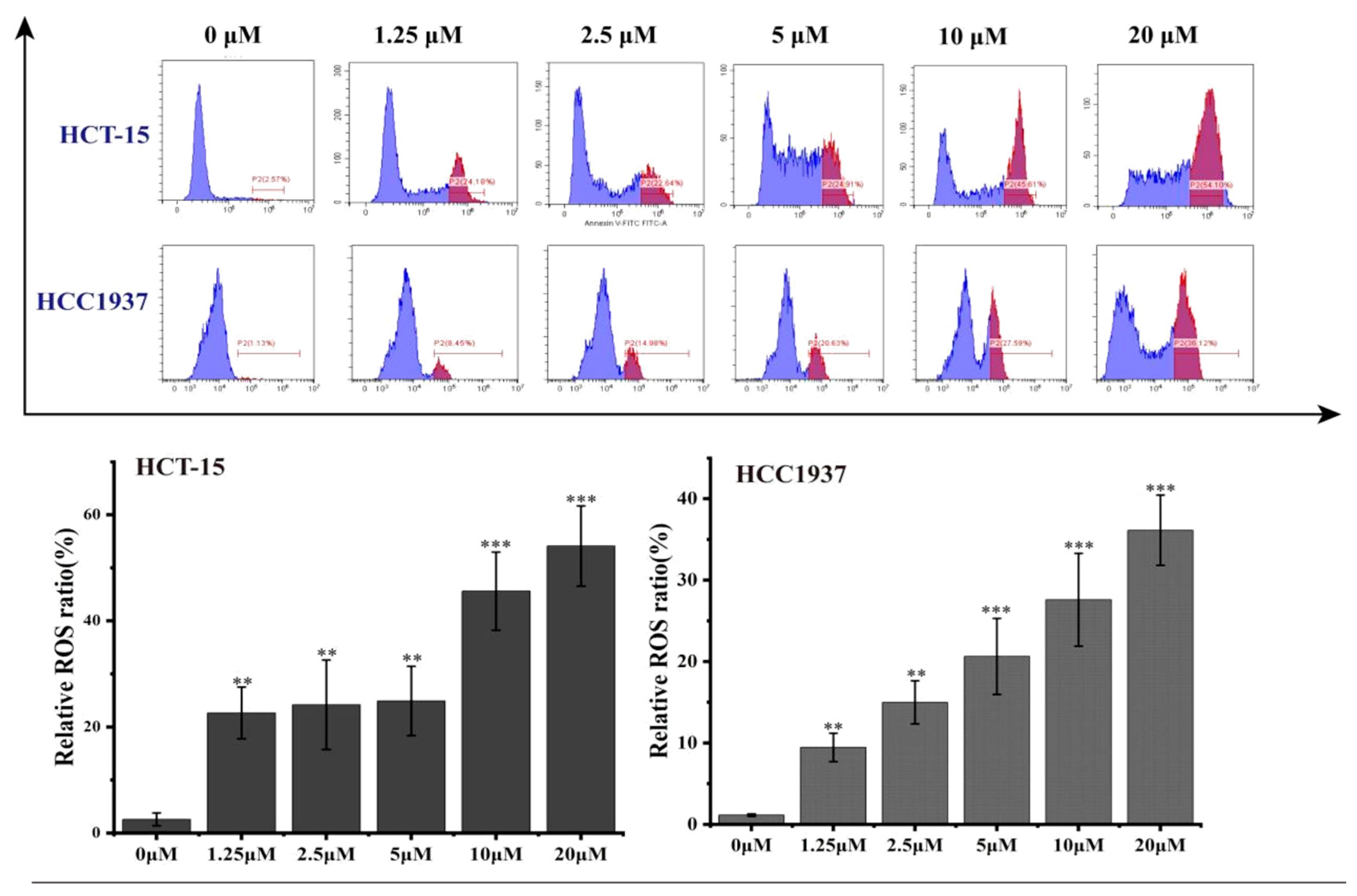
2.3.6. Effects of B1 on Intracellular ROS Levels in HCT-15 and HCC1937 Cell Lines
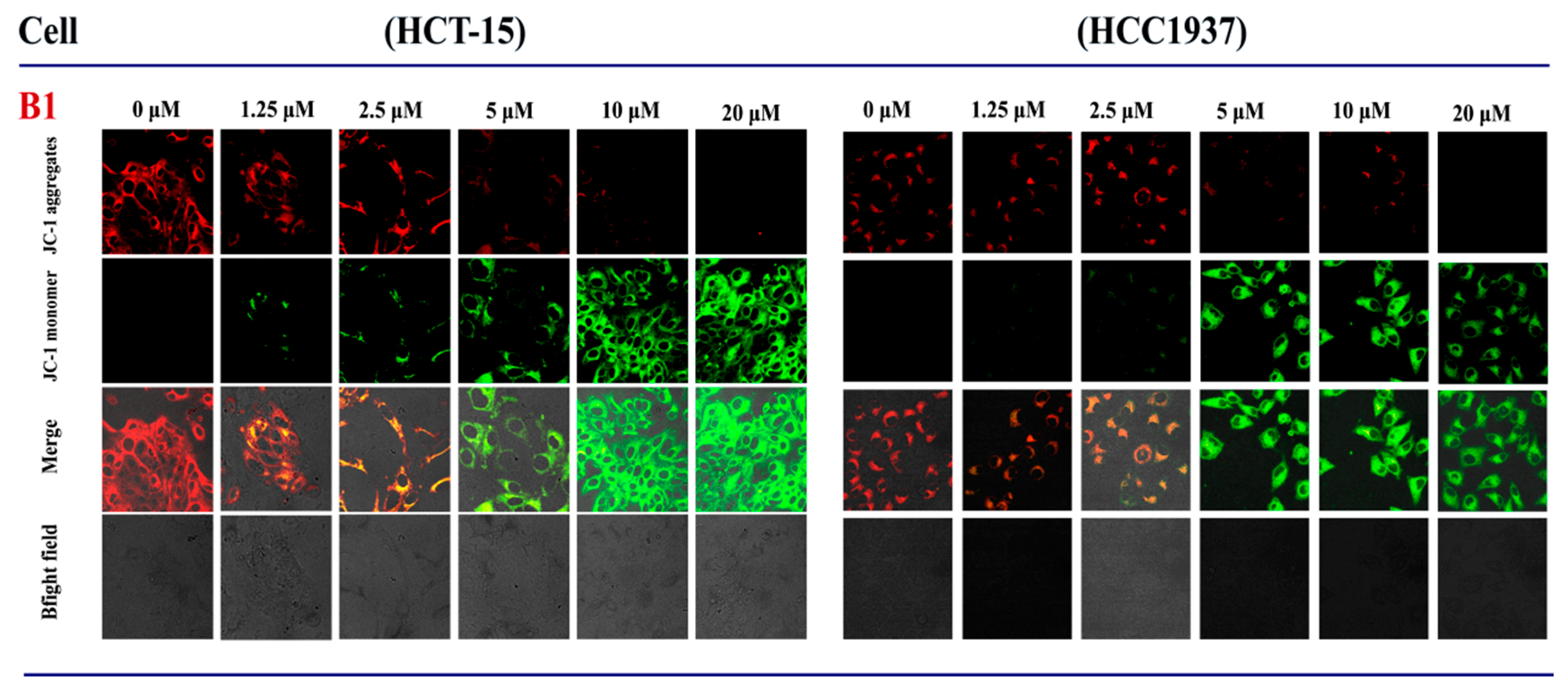
2.3.7. Effects of B1 on Mitochondrial Membrane Potential in HCT-15 and HCC1937 Cell Lines
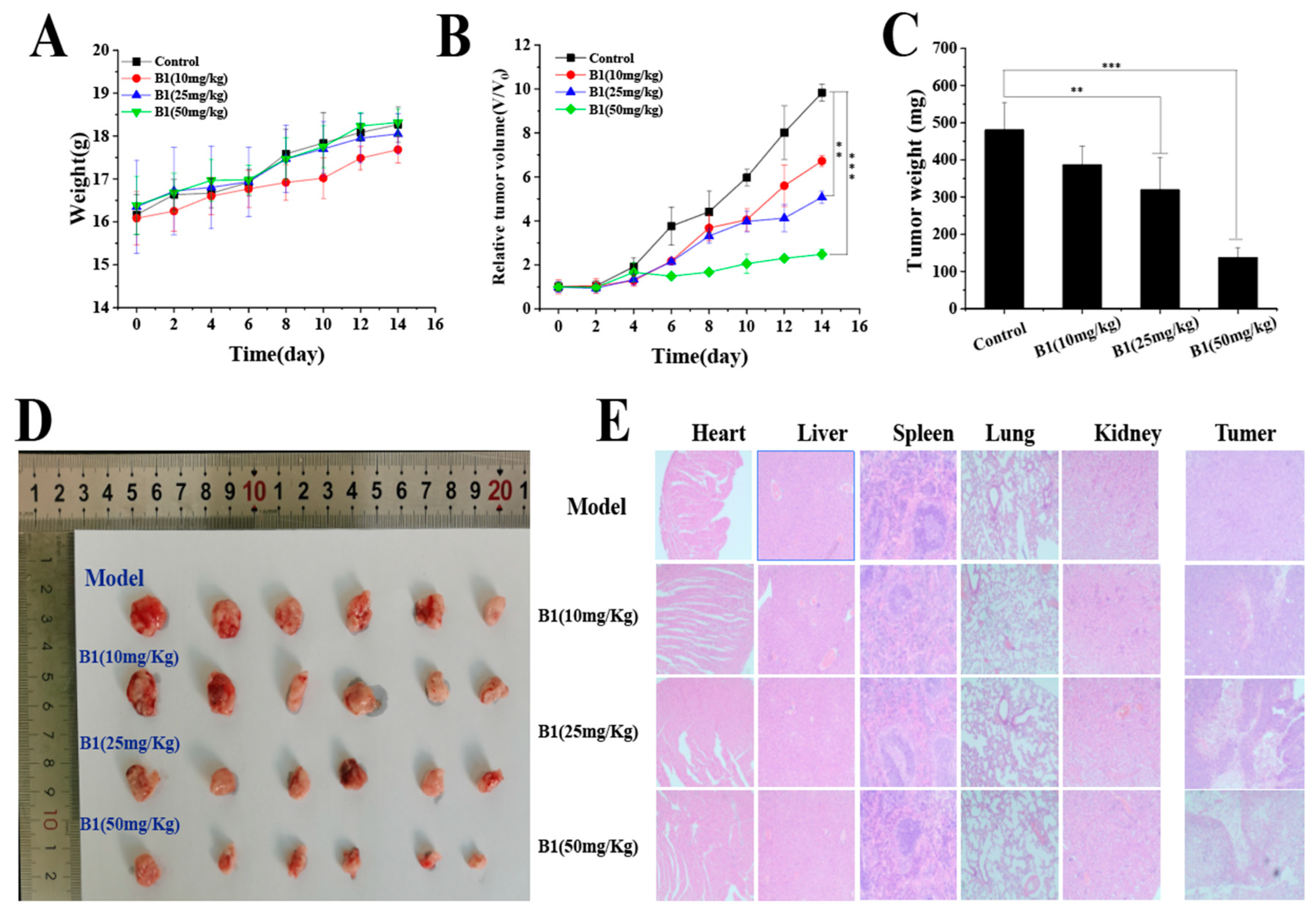
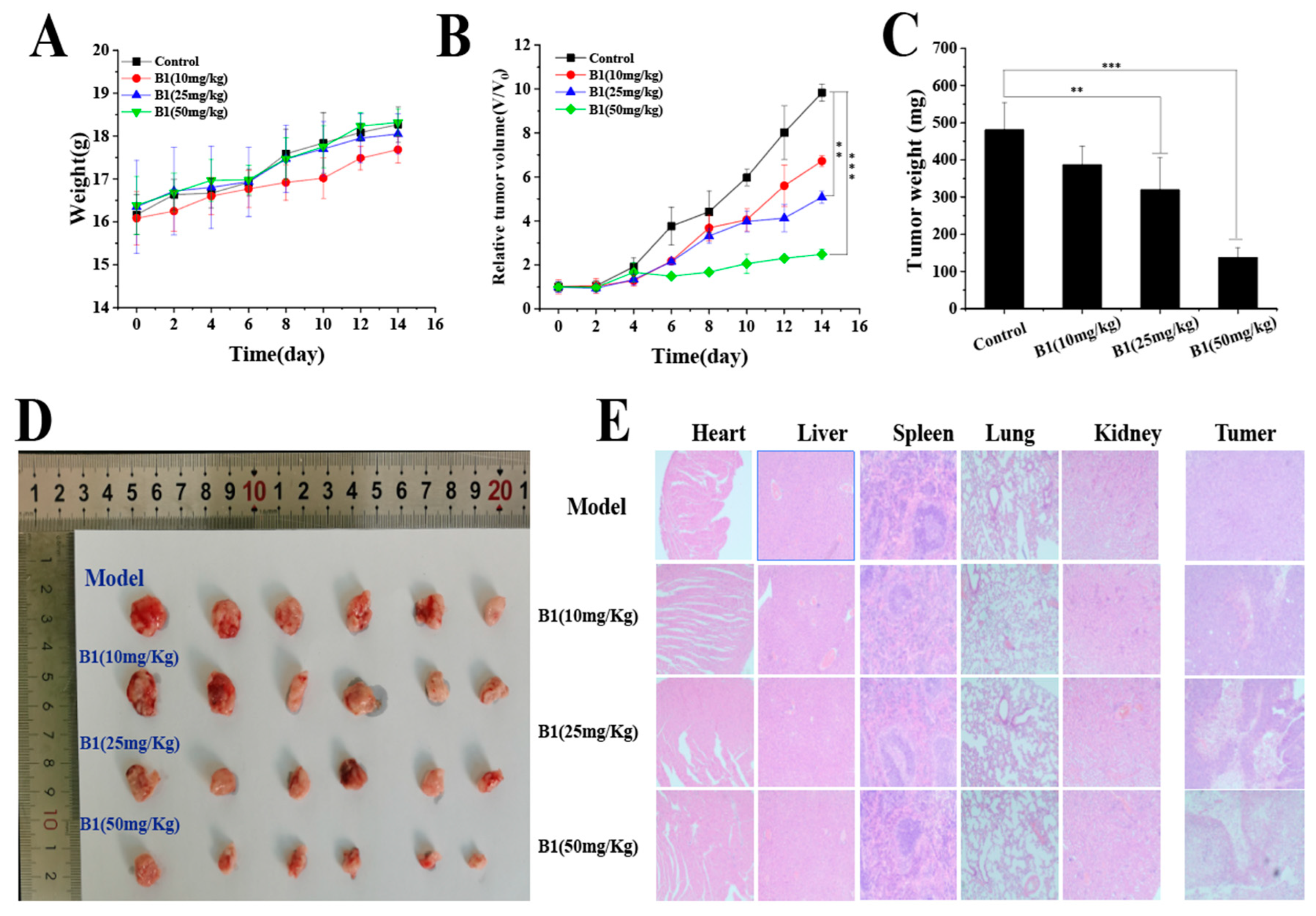
2.3.8. In Vivo Study of B1
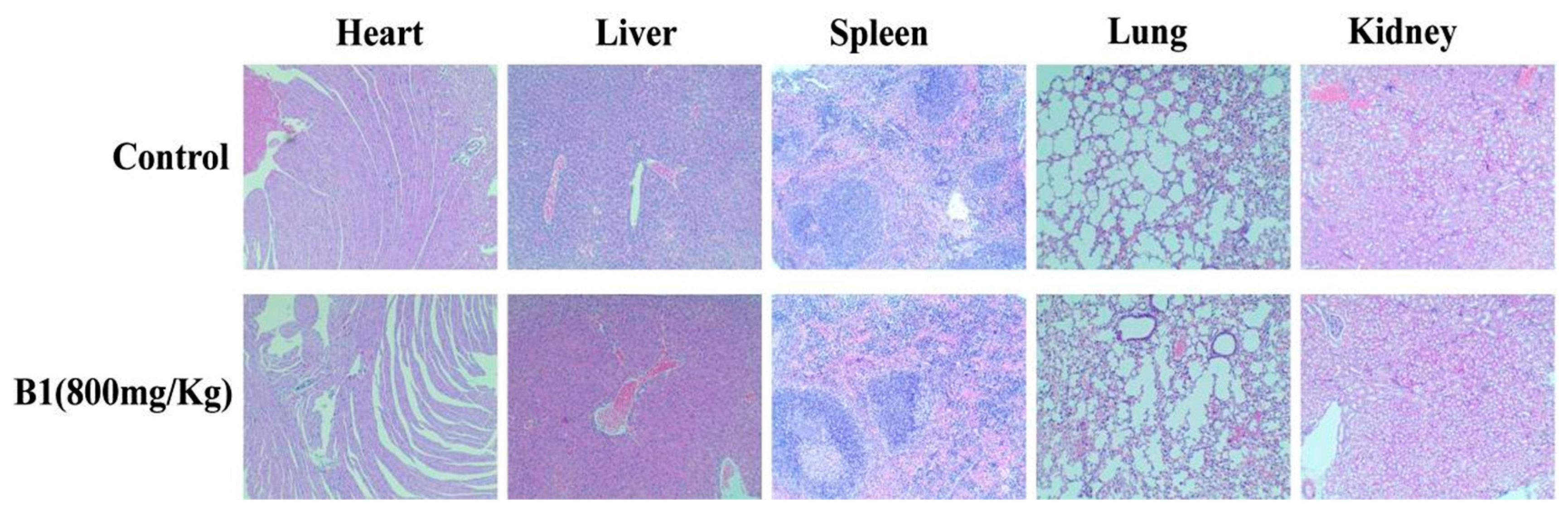
2.3.9. Acute Toxicity Study of B1
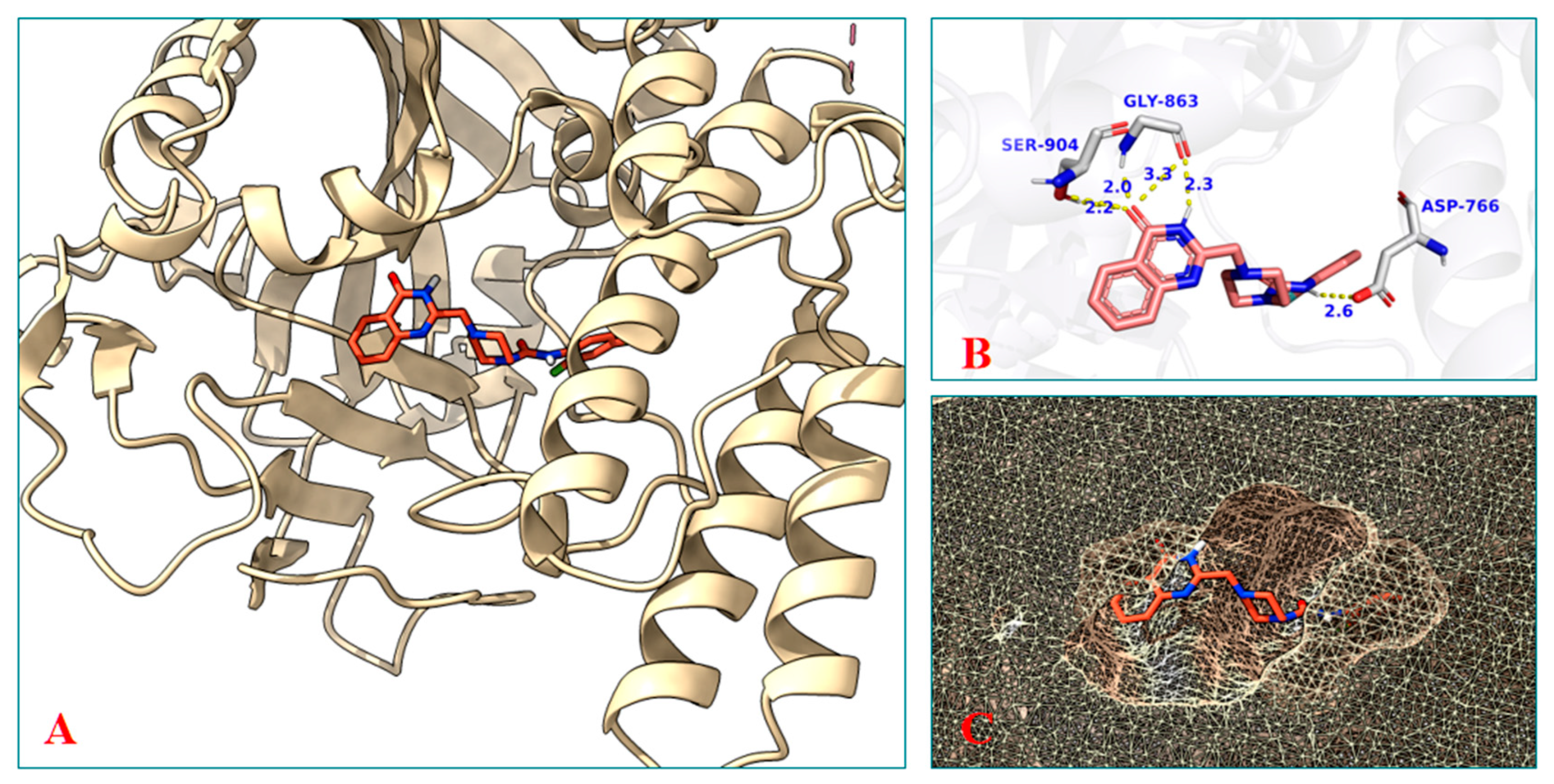
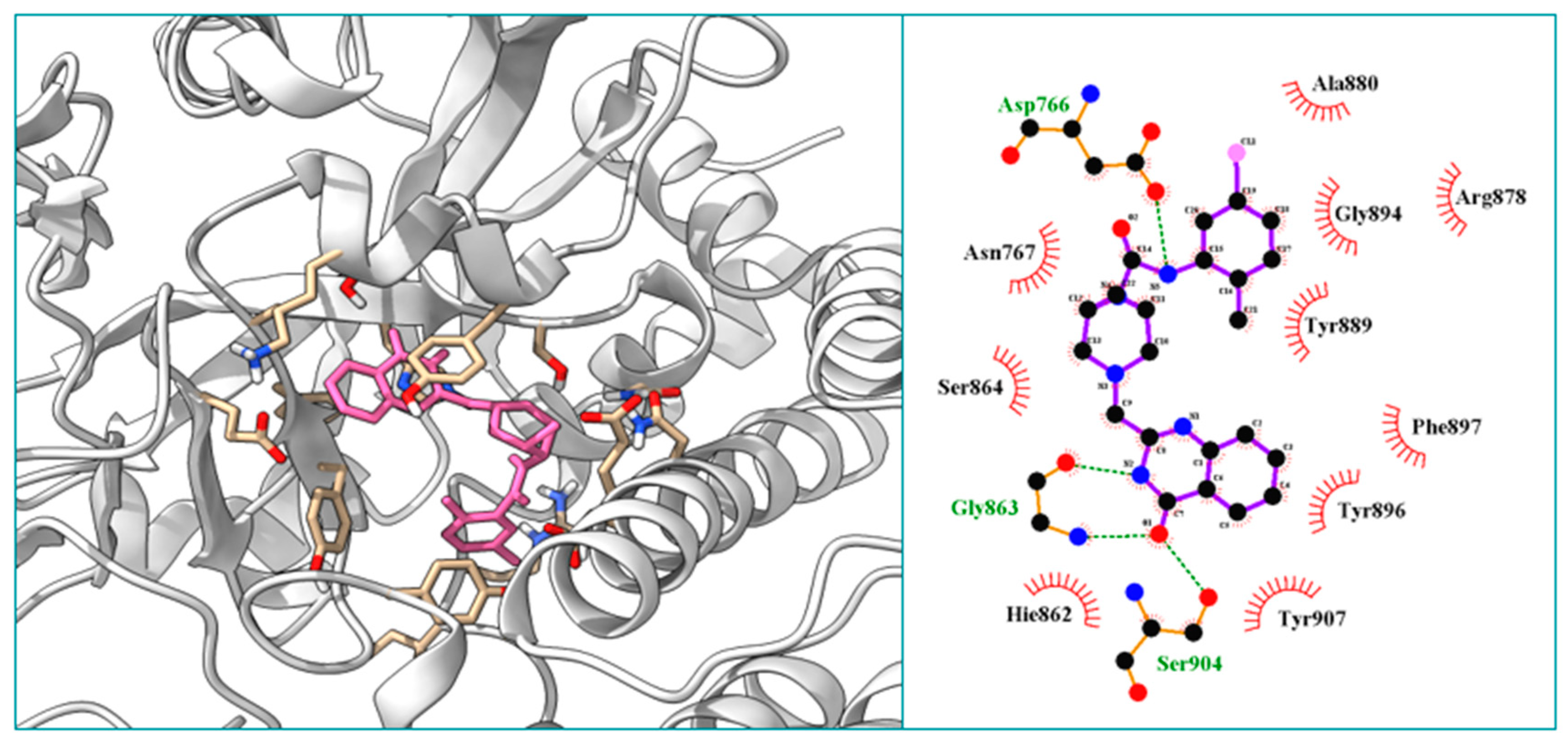
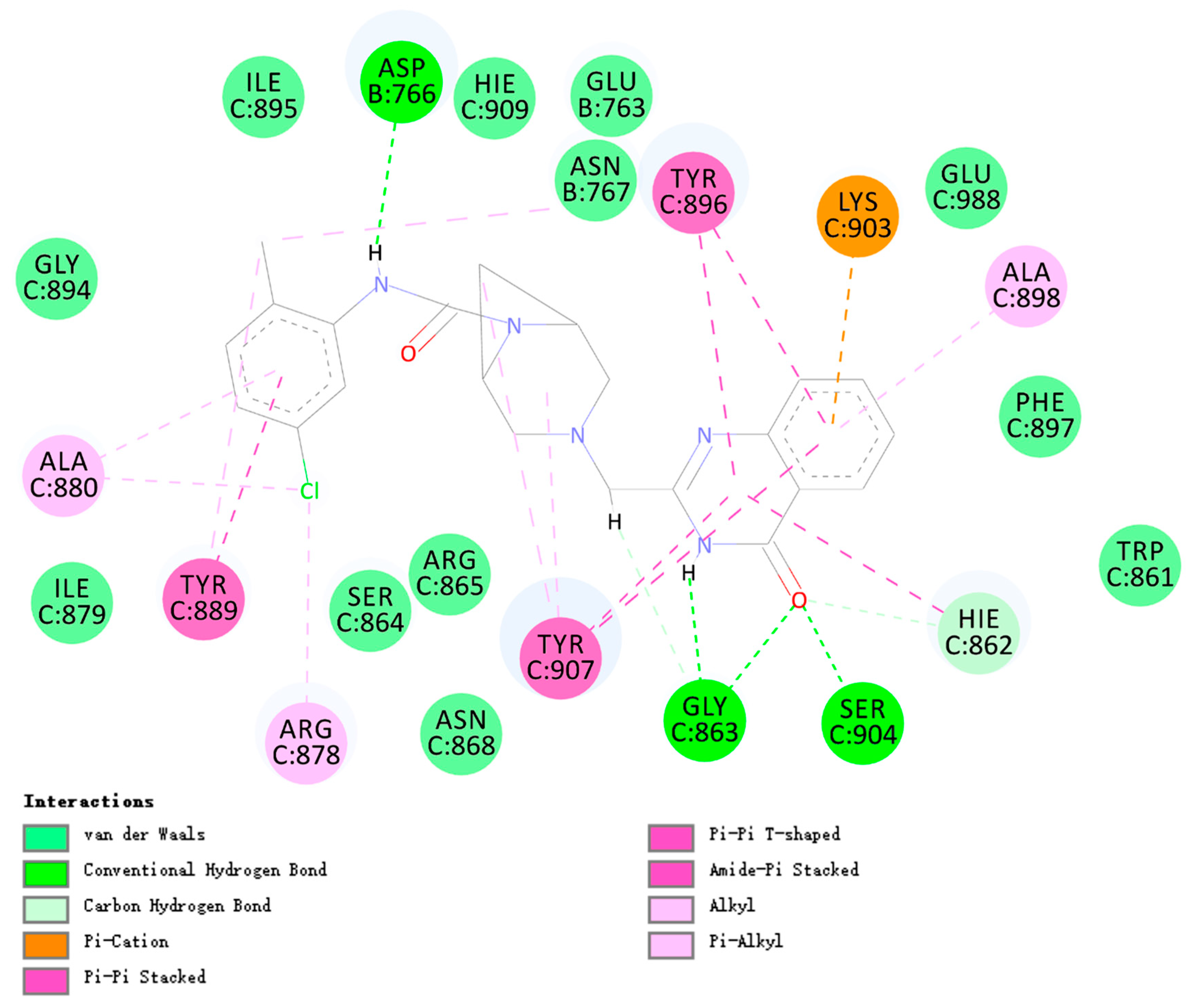
2.3.10. Molecular Docking Study of the Compound B1
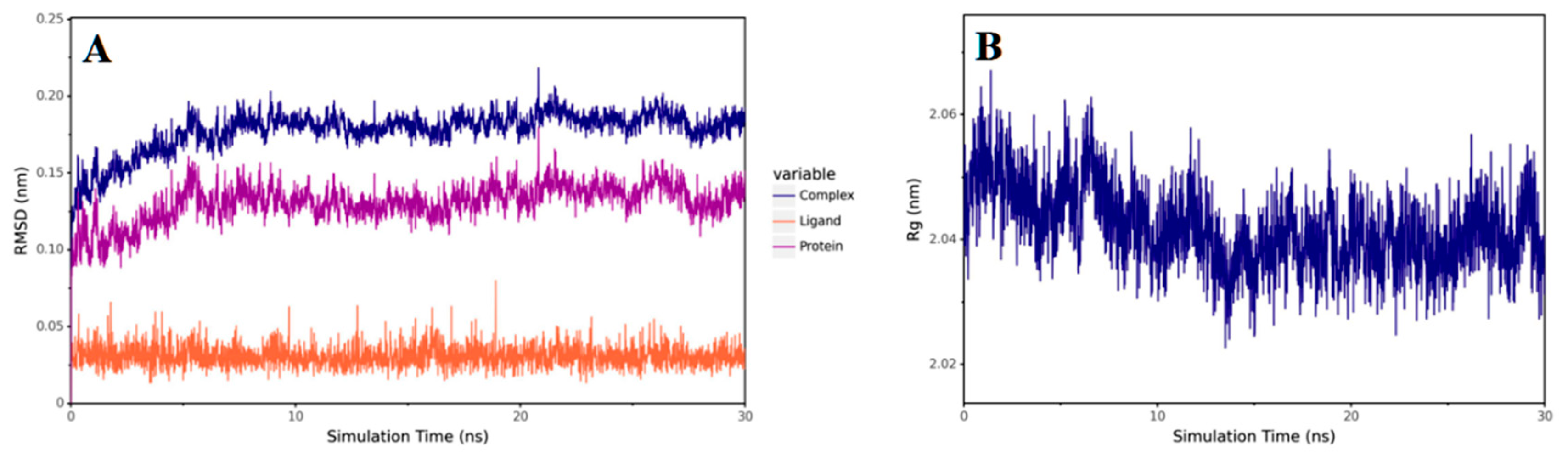
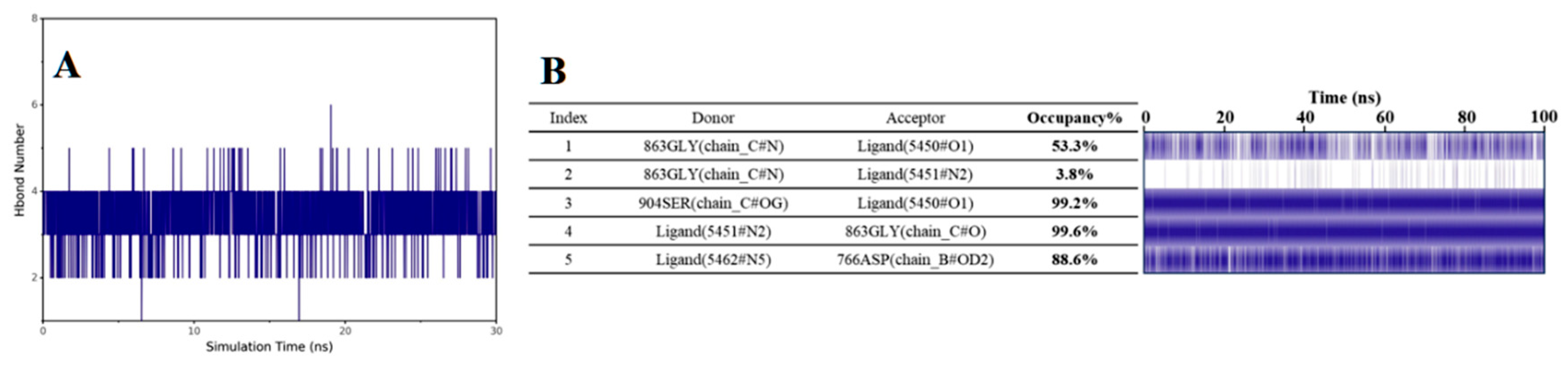
2.3.11. Molecular Dynamics Study of the Compound B1
3. Materials and Method
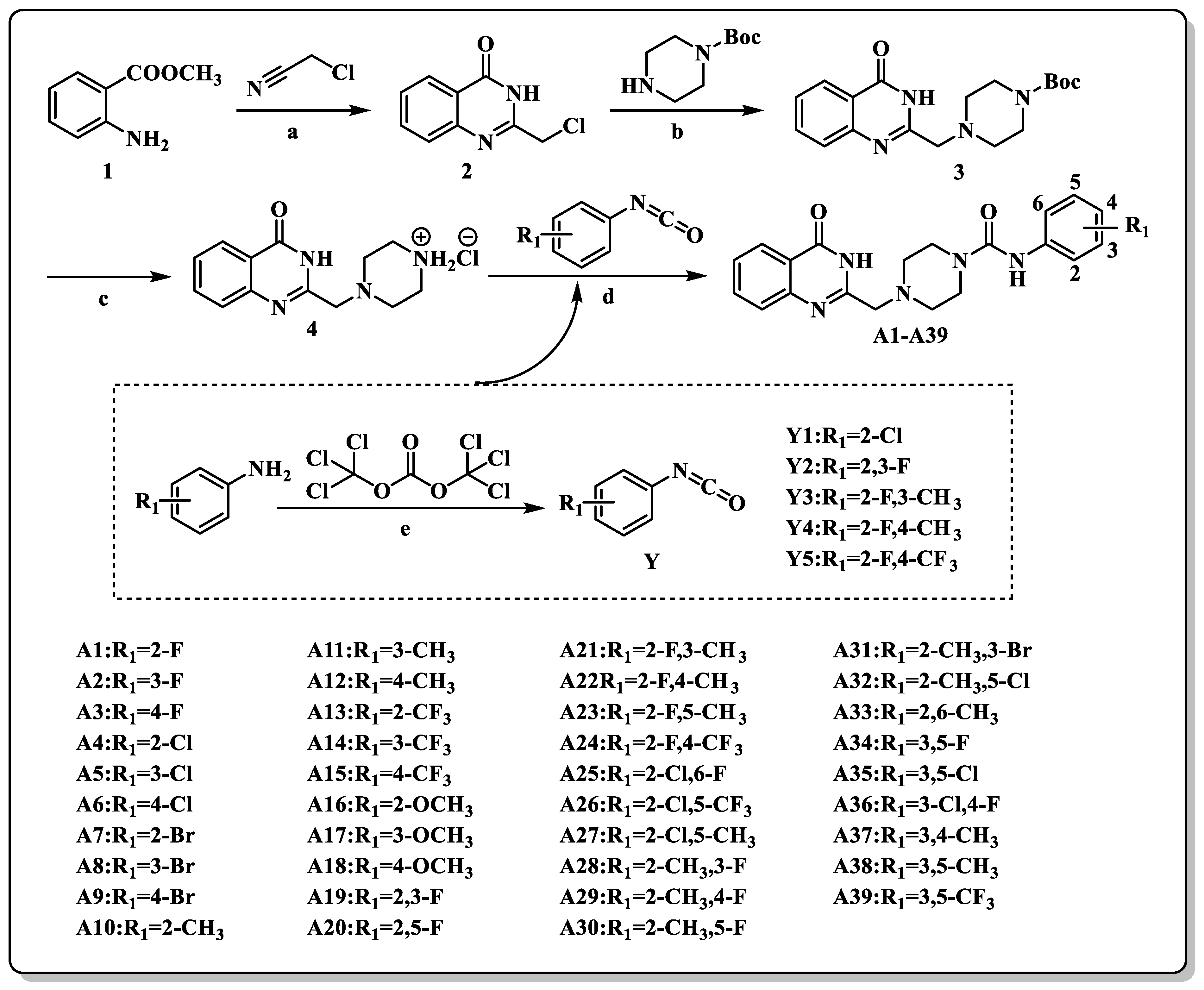
3.1. Chemistry
3.1.1. General Synthetic Procedures for the Synthesis of the Compound 2
3.1.2. General Synthetic Procedures for the Synthesis of the Compound 3
3.1.3. General Synthetic Procedures for the Synthesis of the Compound 4
3.1.4. General Synthetic Procedures for the Synthesis of the Compounds A1–A39
N-(2-fluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A1)
N-(3-fluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A2)
N-(4-fluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A3)
N-(2-chlorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A4)
N-(3-chlorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A5)
N-(4-chlorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A6)
N-(2-bromophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A7)
N-(3-bromophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A8)
N-(4-bromophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A9)
4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-N-(o-tolyl)piperazine-1-carboxamide (A10)
4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-N-(m-tolyl)piperazine-1-carboxamide (A11)
4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-N-(p-tolyl)piperazine-1-carboxamide (A12)
4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-N-(2-(trifluoromethyl)phenyl) piperazine-1-carboxamide (A13)
4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-N-(3-(trifluoromethyl)phenyl)piperazine-1-carboxamide (A14)
4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-N-(4-(trifluoromethyl)phenyl)piperazine-1-carboxamide (A15)
N-(2-methoxyphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A16)
N-(3-methoxyphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A17)
N-(4-methoxyphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A18)
N-(2,3-difluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A19)
N-(2,5-difluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A20)
N-(2-fluoro-3-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A21)
N-(2-fluoro-4-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A22)
N-(2-fluoro-5-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A23)
N-(2-fluoro-4-(trifluoromethyl)phenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A24)
N-(2-chloro-6-fluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A25)
N-(2-chloro-5-(trifluoromethyl)phenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A26)
N-(2-chloro-5-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A27)
N-(3-fluoro-2-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A28)
N-(4-fluoro-2-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A29)
N-(5-fluoro-2-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A30)
N-(3-bromo-2-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A31)
N-(5-chloro-2-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A32)
N-(2,6-dimethylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A33)
N-(3,5-difluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A34)
N-(3,5-dichlorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A35)
N-(3-chloro-4-fluorophenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A36)
N-(3,4-dimethylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A37)
N-(3,5-dimethylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A38)
N-(3,5-bis(trifluoromethyl)phenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (A39)
3.1.5. General Synthetic Procedures for the Synthesis of Compounds B1–B7
N-(5-chloro-2-methylphenyl)-3-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-6-carboxamide (B1)
N-(5-chloro-2-methylphenyl)-2-methyl-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)piperazine-1-carboxamide (B2)
(1S,4S)-N-(5-chloro-2-methylphenyl)-5-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-2,5-diazabicyclo[2.2.1]heptane-2-carboxamide (B3)
N-(5-chloro-2-methylphenyl)-4-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-1,4-diazepane-1-carboxamide (B4)
(1R,5S)-N-(5-chloro-2-methylphenyl)-8-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,8-diazabicyclo[3.2.1]octane-3-carboxamide (B5)
N-(5-chloro-2-methylphenyl)-3-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,8-diazabicyclo[3.2.1]octane-8-carboxamide (B6)
N-(5-chloro-2-methylphenyl)-3-((4-oxo-3,4-dihydroquinazolin-2-yl)methyl)tetrahydropyrimidine-1(2H)-carboxamide (B7)
3.1.6. General Synthetic Procedures for the Synthesis of Compounds C1–C5
N-(5-chloro-2-methylphenyl)-3-((7-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-6-carboxamide (C1)
N-(5-chloro-2-methylphenyl)-3-((7-chloro-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-6-carboxamide (C2)
N-(5-chloro-2-methylphenyl)-3-((7-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-6-carboxamide (C3)
N-(5-chloro-2-methylphenyl)-3-((6-fluoro-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-6-carboxamide (C4)
N-(5-chloro-2-methylphenyl)-3-((6,7-difluoro-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-6-carboxamide (C5)
3.2. Biological Evaluation
3.2.1. Cell Lines and Cell Culture
3.2.2. Cytotoxicity Assay
3.2.3. PARP1 Enzyme Inhibition Assay
3.2.4. Immunofluorescence Analyses of PAR and γH2AX
3.2.5. Cell Apoptosis Assay
3.2.6. Western Blot Analysis
3.2.7. ROS Assay
3.2.8. Mitochondrial Membrane Potential Assay
3.2.9. In Vivo Anti-Tumor Study
3.2.10. Molecular Docking
3.2.11. Molecular Dynamics
3.2.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Langelier, M.F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes: Regulation and catalysis of the poly (ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, function, and biotechnological aspects. Biotechnol. Appl. Biochem. 2022, 69, 1633–1645. [Google Scholar] [CrossRef]
- Demény, M.A.; Virág, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 1. Cell Intrinsic Hallmarks. Cancers 2021, 13, 2042. [Google Scholar] [CrossRef]
- Langelier, M.F.; Zandarashvili, L.; Aguiar, P.M.; Black, B.E.; Pascal, J.M. NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat. Commun. 2018, 9, 13. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A. PARP inhibitors as therapeutics: Beyond modulation of PARylation. Cancers 2020, 12, 16. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wilkerson, P.M.; Wetterskog, D.; Weigelt, B.; Ashworth, A.; Reis-Filho, J.S. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011, 10, 1192–1199. [Google Scholar] [CrossRef]
- Yi, M.; Dong, B.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 12. [Google Scholar] [CrossRef]
- Hu, X.Y.; Zhang, J.F.; Zhang, Y.; Jiao, F.; Wang, J.; Chen, H.; Ouyang, L.; Wang, Y. Dual-target inhibitors of poly (ADP-ribose) polymerase-1 for cancer therapy: Advances, challenges, and opportunities. Eur. J. Med. Chem. 2022, 230, 18. [Google Scholar] [CrossRef]
- Cheng, B.B.; Pan, W.; Xing, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem 2022, 230, 21. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, L.X.; Jiang, T.; Long, J.; Ma, Z.-Y.; Lu, A.-P.; Cheng, Y.; Cao, D.-S. The ups and downs of Poly (ADP-ribose) Polymerase-1 inhibitors in cancer therapy-Current progress and future direction. Eur. J. Med. Chem. 2020, 203, 17. [Google Scholar] [CrossRef]
- Zhang, N.; Tian, Y.N.; Zhou, L.N.; Li, M.Z.; Chen, H.D.; Song, S.S.; Huan, X.J.; Bao, X.B.; Zhang, A.; Miao, Z.H.; et al. Glycogen synthase kinase 3β inhibition synergizes with PARP inhibitors through the induction of homologous recombination deficiency in colorectal cancer. Cell Death Dis. 2021, 12, 18. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.-Y.; Wu, N.; Chen, Y.-C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 16. [Google Scholar] [CrossRef]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors resistance: Mechanisms and perspectives. Cancers 2022, 14, 15. [Google Scholar] [CrossRef]
- Fu, X.Y.; Li, P.; Zhou, Q.; He, R.; Wang, G.; Zhu, S.; Bagheri, A.; Kupfer, G.; Pei, H.; Li, J. Mechanism of PARP inhibitor resistance and potential overcoming strategies. Genes Dis. 2024, 11, 306–320. [Google Scholar] [CrossRef]
- Keung, M.Y.; Wu, Y.Y.; Badar, F.; Vadgama, J.V. Response of breast cancer cells to PARP inhibitors is independent of BRCA status. J. Clin. Med. 2020, 9, 15. [Google Scholar] [CrossRef]
- Osoegawa, A.; Gills, J.J.; Kawabata, S.; Dennis, P.A. Rapamycin sensitizes cancer cells to growth inhibition by the PARP inhibitor olaparib. Oncotarget 2017, 8, 87044–87053. [Google Scholar] [CrossRef]
- Yuan, B.; Ye, N.; Song, S.S.; Wang, Y.T.; Song, Z.; Chen, H.D.; Chen, C.H.; Huan, X.J.; Wang, Y.Q.; Su, Y.; et al. Poly (ADP-ribose) polymerase (PARP) inhibition and anticancer activity of simmiparib, a new inhibitor undergoing clinical trials. Cancer Lett. 2017, 386, 47–56. [Google Scholar] [CrossRef]
- Eskiler, G.G.; Ozman, Z.; Haciefendi, A.; Cansaran-Duman, D. Novel combination treatment of CDK 4/6 inhibitors with PARP inhibitors in triple negative breast cancer cells. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 11, 1031–1041. [Google Scholar] [CrossRef]
- El Hassab, M.A.; El-Hafeez, A.A.A.; Almahli, H.; Elsayed, Z.M.; Eldehna, W.M.; Hassan, G.S.; Abou-Seri, S.M. Phthalimide-tethered isatins as novel poly (ADP-ribose) polymerase inhibitors: Design, synthesis, biological evaluations, and molecular modeling investigations. Arch. Pharm. 2023, 357, e2300599. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, H.; Yuan, J.; Wang, L.; Song, S.; Chen, R.; Bao, X.; Jia, L.; Yang, T.; Zhang, X.; et al. YCH1899, a highly effective phthalazin-1(2H)-one derivative that overcomes resistance to prior PARP inhibitors. J. Med. Chem. 2023, 66, 12284–12303. [Google Scholar] [CrossRef]
- Chang, X.; Sun, D.; Shi, D.; Wang, G.; Chen, Y.; Zhang, K.; Tan, H.; Liu, J.; Liu, B.; Ouyang, L. Design, synthesis, and biological evaluation of quinazolin-4(3H)-one derivatives co-targeting poly (ADP-ribose) polymerase-1 and bromodomain containing protein 4 for breast cancer therapy. Acta Pharm. Sin. B 2021, 11, 156–180. [Google Scholar] [CrossRef]
- Zheng, L.; Ren, R.; Sun, X.; Zou, Y.; Shi, Y.; Di, B.; Niu, M.M. Discovery of a dual Tubulin and Poly (ADP-ribose) polymerase-1 inhibitor by structure-based pharmacophore modeling, virtual screening, molecular docking, and biological evaluation. J. Med. Chem. 2021, 64, 15702–15715. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Singh, S.; Shah, J.R.; Low, W.-K.; Talele, T.T. Synthesis and SAR optimization of quinazolin-4(3H)-ones as poly (ADP-ribose)polymerase-1 inhibitors. Eur. J. Med. Chem. 2012, 50, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Madbouly, E.A.; Lashine, E.S.M.; Al-Karmalawy, A.A.; Sebaiy, M.M.; Pratsinis, H.; Kletsas, D.; Metwally, K. Design and synthesis of novel quinazolinone-chalcone hybrids as potential apoptotic candidates targeting caspase-3 and PARP-1: In vitro, molecular docking, and SAR studies. New J. Chem. 2022, 46, 22013–22029. [Google Scholar] [CrossRef]
- Ruan, B.; Zhang, Y.; Tadesse, S.; Preston, S.; Taki, A.C.; Jabbar, A.; Hofmann, A.; Jiao, Y.; Garcia-Bustos, J.; Harjani, J.; et al. Synthesis and structure-activity relationship study of pyrrolidine-oxadiazoles as anthelmintics against Haemonchus contortus. Eur. J. Med. Chem. 2020, 190, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.; Aoyagi-Scharber, M.; Post, L.E. Discovery and characterization of (8S,9R)-5-fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN673, Talazoparib), a novel, highly potent, and orally efficacious poly (ADP-ribose) polymerase-1/2 inhibitor, as an anticancer agent. J. Med. Chem. 2016, 59, 335–357. [Google Scholar] [PubMed]
- Páhi, Z.G.; Borsos, B.N.; Pantazi, V.; Ujfaludi, Z.; Pankotai, T. PARylation during transcription: Insights into the fine-tuning mechanism and regulation. Cancers 2020, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Chen, X.; Yin, F.; Li, S.; Zhang, Y.; Luo, H.; Luo, Z.; Cui, N.; Chen, Y.; Li, X.; et al. Indirubin derivatives as bifunctional molecules inducing DNA damage and targeting PARP for the treatment of cancer. Eur. J. Med. Chem. 2023, 261, 18. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Lu, G.; Nie, W.; Xin, M.; Meng, Y.; Gu, J.; Miao, H.; Cheng, X.; Chan, A.S.; Zou, Y. Design, synthesis, biological evaluation and molecular docking study of novel urea-based benzamide derivatives as potent poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors. Eur. J. Med. Chem. 2022, 243, 19. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.J.; Li, X.Y.; Huang, J.Q.; Zhou, X.; Deng, F.A.; Li, Y.M.; Liu, L.S.; Li, S.Y.; Cheng, H. A self-delivery photodynamic sensitizer for enhanced DNA damage by PARP inhibition. Biomater. Sci. 2022, 11, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.R.; Wang, F.; Shi, X.W.; Tan, Y.X.; Zhao, J.Y.; Zhang, J.W.; Li, Q.H.; Lin, G.Q.; Gao, D.; Tian, P. Design, synthesis and biological evaluation of novel potent STAT3 inhibitors based on BBI608 for cancer therapy. Eur. J. Med. Chem. 2020, 201, 112428. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.L.; Feng, K.R.; Liu, J.X.; Ren, Y. Molecular modeling studies of novel naphthyridine and isoquinoline derivatives as CDK8 inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 6355–6369. [Google Scholar] [CrossRef]
- Yang, J.J.; Yang, Y.; Wang, L.; Jin, Q.; Pan, M. Nobiletin selectively inhibits oral cancer cell growth by promoting apoptosis and DNA damage in vitro. Oral Med. Oral Pathol. Oral Radiol. 2020, 130, 419–427. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| No. | R1 | IC50 (μM) a | |
| HCT-15 | HCC1937 | ||
| A1 | 2-F | 18.39 ± 0.91 | 16.63 ± 1.12 |
| A2 | 3-F | 34.89 ± 2.23 | >50 |
| A3 | 4-F | >50 | >50 |
| A4 | 2-Cl | 19.12 ± 1.12 | 14.65 ± 1.67 |
| A5 | 3-Cl | 19.05 ± 0.79 | 25.89 ± 0.96 |
| A6 | 4-Cl | 21.35 ± 0.86 | >50 |
| A7 | 2-Br | 29.61 ± 1.37 | 40.28 ± 3.32 |
| A8 | 3-Br | 27.65 ± 2.56 | 47.41 ± 2.96 |
| A9 | 4-Br | >50 | >50 |
| A10 | 2-CH3 | 15.89 ± 0.85 | 14.73 ± 1.03 |
| A11 | 3-CH3 | 25.57 ± 1.17 | >50 |
| A12 | 4-CH3 | >50 | >50 |
| A13 | 2-CF3 | 20.23 ± 0.78 | >50 |
| A14 | 3-CF3 | 20.02 ± 1.15 | 17.67 ± 0.77 |
| A15 | 4-CF3 | 15.56 ± 0.56 | >50 |
| A16 | 2-OCH3 | 30.65 ± 2.13 | >50 |
| A17 | 3-OCH3 | >50 | >50 |
| A18 | 4-OCH3 | >50 | >50 |
| A19 | 2,3-F | 19.07 ± 0.59 | >50 |
| A20 | 2,5-F | 33.13 ± 2.16 | 41.67 ± 1.89 |
| A21 | 2-F,3-CH3 | 28.29 ± 1.55 | >50 |
| A22 | 2-F,4-CH3 | 21.73 ± 1.16 | >50 |
| A23 | 2-F,5-CH3 | 26.02 ± 1.67 | 44.20 ± 3.47 |
| A24 | 2-F,4-CF3 | 22.77 ± 1.82 | 44.09 ± 2.19 |
| A25 | 2-Cl,6-F | 41.24 ± 1.44 | >50 |
| A26 | 2-Cl,5-CF3 | 37.29 ± 2.76 | >50 |
| A27 | 2-Cl,5-CH3 | 25.37 ± 1.48 | 17.52 ± 0.66 |
| A28 | 2-CH3,3-F | 30.31 ± 3.30 | >50 |
| A29 | 2-CH3,4-F | 29.83 ± 1.57 | >50 |
| A30 | 2-CH3,5-F | 18.31 ± 0.44 | 39.46 ± 1.84 |
| A31 | 2-CH3,3-Br | >50 | >50 |
| A32 | 2-CH3,5-Cl | 10.93 ± 0.71 | 11.35 ± 0.73 |
| A33 | 2,6-CH3 | 35.01 ± 2.28 | 39.58 ± 1.35 |
| A34 | 3,5-F | 47.25 ± 3.35 | >50 |
| A35 | 3,5-Cl | 21.15 ± 0.89 | >50 |
| A36 | 3-Cl,4-F | 25.20 ± 1.66 | >50 |
| A37 | 3,4-CH3 | 22.49 ± 1.49 | 46.41 ± 2.96 |
| A38 | 3,5-CH3 | 18.02 ± 0.88 | 17.85 ± 1.32 |
| A39 | 3,5-CF3 | 20.99 ± 2.33 | 42.16 ± 2.77 |
| Olaparib b | 45.53 ± 3.13 | 37.07 ± 1.89 | |
 | |||
|---|---|---|---|
| No. | C | IC50 (μM) a | |
| HCT-15 | HCC1937 | ||
| B1 | 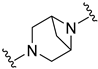 | 2.89 ± 0.78 | 3.26 ± 0.38 |
| B2 |  | 6.37 ± 2.83 | 5.89 ± 0.50 |
| B3 | 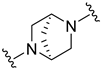 | 12.50 ± 1.49 | 16.46 ± 0.94 |
| B4 | 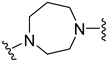 | 5.65 ± 1.29 | 5.77 ± 0.99 |
| B5 |  | 10.95 ± 2.40 | 9.61 ± 1.35 |
| B6 | 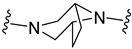 | 7.27 ± 1.28 | 7.52 ± 1.37 |
| B7 |  | 13.72 ± 1.21 | 57.91 ± 3.33 |
| Olaparib b | 45.53 ± 3.13 | 37.07 ± 1.89 | |
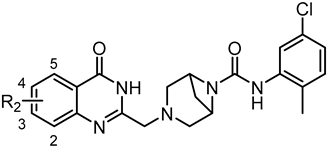 | |||
|---|---|---|---|
| No. | R2 | IC50 (μM) a | |
| HCT-15 | HCC1937 | ||
| C1 | 3-F | 24.12 ± 2.25 | 9.22 ± 1.19 |
| C2 | 3-Cl | 23.01 ± 1.93 | 32.38 ± 2.66 |
| C3 | 3-CH3 | 28.36 ± 2.87 | 30.14 ± 3.12 |
| C4 | 4-F | 9.84 ± 0.63 | 8.49 ± 0.71 |
| C5 | 3,4-F | 29.57 ± 2.16 | 54.98 ± 4.28 |
| Olaparib b | 45.53 ± 3.13 | 37.07 ± 1.89 | |
| No. | IC50 (μM) a | PARP1 IC50 (nM) a | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCT-15 | HCC1937 | MDA-MB-231 | LO2 | NCM460 | |||||
| 3-Days | 7-Days | 3-Days | 7-Days | 3-Days | 7-Days | 3-Days | 3-Days | ||
| B1 | 2.89 ± 0.78 | 0.13 ± 0.03 | 3.26 ± 0.38 | 0.17 ± 0.04 | 11.29 ± 1.93 | 1.33 ± 0.51 | >100 | >100 | 63.81 ± 2.12 |
| Olaparib b | 45.53 ± 3.13 | 19.25 ± 0.75 | 37.07 ± 1.89 | 15.25 ± 0.87 | 33.93 ± 2.57 | 9.22 ± 1.12 | 90.83 ± 2.61 | >100 | 7.30 ± 1.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Liu, B.; Jin, F.; Cao, W.; Xu, G.; Zhang, X.; Peng, P.; Gao, D.; Wang, B.; Feng, K. Discovery of Novel 4-Hydroxyquinazoline Derivatives: In Silico, In Vivo and In Vitro Studies Using Primary PARPi-Resistant Cell Lines. Molecules 2024, 29, 1407. https://doi.org/10.3390/molecules29061407
Zhu L, Liu B, Jin F, Cao W, Xu G, Zhang X, Peng P, Gao D, Wang B, Feng K. Discovery of Novel 4-Hydroxyquinazoline Derivatives: In Silico, In Vivo and In Vitro Studies Using Primary PARPi-Resistant Cell Lines. Molecules. 2024; 29(6):1407. https://doi.org/10.3390/molecules29061407
Chicago/Turabian StyleZhu, Lijie, Binzhuo Liu, Feng Jin, Weilong Cao, Guangzhao Xu, Xinwei Zhang, Peng Peng, Dingding Gao, Bin Wang, and Kairui Feng. 2024. "Discovery of Novel 4-Hydroxyquinazoline Derivatives: In Silico, In Vivo and In Vitro Studies Using Primary PARPi-Resistant Cell Lines" Molecules 29, no. 6: 1407. https://doi.org/10.3390/molecules29061407
APA StyleZhu, L., Liu, B., Jin, F., Cao, W., Xu, G., Zhang, X., Peng, P., Gao, D., Wang, B., & Feng, K. (2024). Discovery of Novel 4-Hydroxyquinazoline Derivatives: In Silico, In Vivo and In Vitro Studies Using Primary PARPi-Resistant Cell Lines. Molecules, 29(6), 1407. https://doi.org/10.3390/molecules29061407