1. Introduction
Several efforts have been pursued in computational chemistry to determine the acid dissociation constant (Ka) of organic compounds in aqueous solution [
1]. The advantages of the
in silico pKa determination are connected with the development of environment-friendly and rapid approaches that avoid experimental and costly procedures, in particular when dealing with structurally complex molecules [
1,
2]. Achieving high reliability in pKa calculation is still challenging, due to the recurrent discrepancies between experimental and computational results, which sensitively depend on the chosen level of theory, the density functional and the adopted solvation model [
2,
3]. The computational determination of pKa is usually performed through several methodologies, varying from the pioneering “machine learning” approach, to the more traditional physicochemical-inspired models. The former is focused on the quantitative structure–activity relationship (thereafter QSAR), but although it generally requires huge computational costs, it has not yet shown good reliability [
4]. The latter relies on solid computational methods [
5,
6] that can be classified into two categories: (i) the “indirect” approach, which exploits a thermodynamic cycle that takes into consideration the energy at equilibrium of the deprotonated species in the gas and in the solution phase; (ii) the “direct” approach, in which the ionogenic equation in water is explicitly considered [
5,
6]. In computing carboxylic acids pKa, the direct approach is widely used. Indeed, according to the literature, a variable amount of water molecules, ranging from one to eight, can be merged into the reaction center to faithfully mimic the first solvation shell [
2]. In a recent example [
7], authors calculated the pKa of a series of differently substituted carboxylic acids using a linear correlation fit that includes the H-bond length between water molecules and the acid and/or the conjugated base involved at the reaction center. Among the others, B3LYP resulted in the most appropriate functional, coupled with a 6-31G+(d,p) basis set, in the aqueous continuum model. Other approaches have delved into mixed levels of theory, such as DFT, Hartree–Fock (HF) and/or Møller–Plesset (MP), to calculate
in silico carboxylic acids pKa. Indeed, B3LYP was directly compared with M05-2X or HF [
5]. The authors inserted four water molecules at the reaction center and a radii correction factor to pursue more reliable outputs. Starting from these assumptions, additional works investigated the effects of re-shaping the atoms’ radii and the relative position of the involved species, keeping the solvation model constant. Thus, it has been demonstrated that the geometry of the acid moiety played a fundamental role in obtaining consistent calculated pKa values, while the selected continuum model barely affects method reliability [
5,
6,
7]. Nevertheless, such procedures, independently from the applied level of theory, still require mathematical fittings, the use of internal correction coefficients, and/or H
+ experimental energy values [
1,
8].
Recently, we developed an “easy-to-use” method that allowed us to directly predict the pKa of a series of substituted phenols and carboxylic acids with reliable results [
1,
8]. Briefly, our methodology requires the introduction of two explicit water molecules at the reaction center, CAM-B3LYP as functional, 6-311G+(d,p) as the basis set and SMD. Belonging to the RSH-GGA class of functionals [
9], CAM-B3LYP exploits different parameters to describe long- or short-range interactions. In the long-range interaction, a Hartree–Fock contribution equal to 19% is utilized, while in the short one, it amounts to 65%. In both cases, the counterpart of the total is fulfilled with Becke 1988 (B88) exchange interaction [
9]. Hence, CAM-B3LYP requires a higher computational cost with respect to hybrid-GGA or meta-GGA functionals, belonging to the third and fourth rungs of “Jacob’s ladder” (
Figure 1) [
10], which classifies density functionals according to exchange–correlation energy as a function of the electron density.
For instance, PBEPBE belongs to the second rung, the GGAs, where the non-local correlation and exchange are improved with respect to the spin electron density functionals family (first rung, LDSA) [
10,
11]. In the third rung, which includes meta-GGA functionals, as TPSSTPSS, the accuracy in treating chemical bonds and/or weak interactions is improved. In the fourth and fifth rungs, hybrid and meta-hybrid functionals exploit 20% of the HF exchange term [
12]. In particular, the hybrids-GGAs are constructed on the Kohn–Sham orbitals, and they exploit the Laplacian of the spin density, avoiding non-locality. Nevertheless, this system is not more computationally expensive than the GGA ones [
10]. Thus, PBE1PBE and B3PW91 are a revised version of the Kohn–Sham determinant, where angles, system-average exchange and orbitals nodality are partially or completely fixed [
13]. CAM-B3LYP and WB97XD, belonging to the RSH-GGA family, are not included in “Jacob’s ladder” [
14]. They combine an exact exchange term and an exact “partial correlation” term, maintaining a certain degree of approximation [
10,
14,
15], thus boding good accuracy in a wide panel of applications.
Herein, the reliability of our “easy-to-use method” [
1,
8] for pKa determination was verified, by screening a panel of functionals belonging to different rungs of Jacob’s ladder. The main goal was to propose other functionals that show similar accuracy and reliability to CAM-B3LYP, with lower computational cost, eventually. Hence, PBEPBE, TPSSTPSS, PBE1PBE, B3PW91 and the “outcasts” CAM-B3LYP and WB97XD were exploited for the
in silico pKa determination of a series of benzoic acids, functionalized with different substituents. No correction factors, mathematical fittings or experimental energy values were introduced. The 6-311G+(d,p) basis set and SMD were kept constant for each analysis. In addition, taking into consideration the pivotal role of the final geometry of the reaction center highlighted in the literature, a thorough analysis of the solvation cavities, bond lengths and dihedral angles was carried out. Computational time cost was also estimated for each molecule, to better clarify the performance of the exploited functionals.
2. Results and Discussion
To calculate the pKa of the selected benzoic acid derivatives, our previously optimized method was herein adopted [
1,
8]. In particular, the acid dissociation equilibrium of a generic carboxylic acid (RCO
2H) and the corresponding reaction free energy (ΔG
dep) can be defined according to Equations (1) and (2), respectively [
8].
where G
RCO2−, G
H2O, G
OH− and G
RCO2H are the Gibbs free energies of each species in the presence of two explicit water molecules, in an aqueous solvent. Afterwards, the pKa equation, at 298.15 K, can be calculated according to Equation (3) [
16]:
where R is the gas constant, T is the absolute temperature, and 15.74 is the pKa of water at 298.15 K [
17].
To calculate pKa values, PBEPBE was selected as functional due to its ability to predict non-covalent and weak interactions. It belongs to the second rung of “Jacob’s ladder”, showing a moderate computational cost and providing a fine consistency on a wide range of interactions [
18]. TPSSTPSS well reproduces the geometry of small organic compounds bearing oxygen and/or hydrogen in functional groups [
19]. Further, belonging to the third rung of Jacob’s ladder, it still requires reduced processing time. In the fourth rung, PBE1PBE and B3PW91 were selected to investigate how the HF contribution to exchange interactions affects the system description accuracy. For instance, PBE1PBE is based on PBEPBE, but it includes an HF contribution of 3:1 [
20,
21]. It was selected because of its reliability in describing the geometry of small compounds, as well as bond lengths, van der Waals interactions and hydrogen-bonded complexes, especially when a 6-311G+(d,p) basis set is exploited [
21]. B3PW91 shows a non-local correlation function equal to 20%, producing stunning results in determining the H-bond length between oxygen belonging to medium/small organic compounds and water molecules [
22]. CAM-B3LYP has already proven its accountability and reliability in the pKa determination of small organic molecules [
1,
8], while WB97XD is an RSH-GGA functional, widely used for
in silico prediction of organic molecules properties. It usually ensures even greater reliability with respect to CAM-B3LYP, comprising 22% of HF exchange for short-range interactions and 100% HF for long-range interactions. Further, the two functionals differ in the intermediate region connecting short- and long-range interactions: while in CAM-B3LYP the standard error function, describing the range separation (ω), is approximately 0.33 [
23], in WB97XD it is significantly smaller (ω = 0.2) [
24].
The basis set, namely 6-311 G+(d,p), and the solvation model based on density, i.e., SMD, were kept constant in all the investigations [
1,
8].
Selected compounds are the following: Benzoic acid as the leading compound, 2,6-Dimethylbenzoic acid, where the pKa is affected by the “ortho-effect” of the alkyl substituents [
8], 4-Cyanobenzoic acid, bearing a difficult-to-model electron-withdrawing group (EWG) as -CN, 2-Bromobenzoic and 4-Bromobenzoic acid, 2-, 3- and 4-Chlorobenzoic acids and 2-, 3- and 4-Methoxybenzoic acids, bearing a halogen atom or a methoxy group in different positions of the ring, thus implying different inductive and/or conjugative effects.
The initial geometry of benzoic acid and benzoate in the presence of 2 explicit water molecules was drawn as close as possible to the final ones, according to the literature [
1,
3,
17]. Geometry optimization of the other target molecules was then performed, starting from the leading compounds and adding the appropriate substituent(s) on the ring. Hence, following our model, for each set of compounds (i.e., the acid and the conjugated base), two water molecules were placed at the reaction center [
1,
8], as shown in
Figure 2.
After geometry optimization, the pKa values of the abovementioned molecules were calculated according to Equations (2) and (3). The results are listed in
Table 1.
CAM-B3LYP confirmed its reliability in determining the pKa of carboxylic acids, with an MAE well below 1 unit (MAE = 0.23), which is the threshold limit for reliable methods [
1,
8]. WB97XD, despite belonging to the same class of functionals, showed an MAE value higher than 1 (MAE = 1.30). Due to the differences between the two functionals, namely the HF contribution to the long-range interactions and the ω value, this finding suggests that those parameters affect the WB97XD capability to correctly describe ionogenic reactions. Remarkably, B3PW91 and PBE1PBE show an MAE value slightly higher than the one obtained with CAM-B3LYP, even with a simpler level of theory (MAE = 0.38 for the former and MAE = 0.34 for the latter). The worst results were obtained with PBEPBE (MAE = 1.88), while TPSSTPSS showed a slightly lower MAE value (1.42).
Correlation plot analysis confirmed the overall reliability of CAM-B3LYP with a slope close to the theoretical one (slope = 1.05 ± 0.02), a Pearson’s r value (R) and R-Square (COD) of 0.99, (
Figure S1, Supplementary Information). B3PW91 showed a COD of 0.97 and a slope value nearly coincident with the unit, while PBE1PBE was less accurate (slope = 0.94), even with a lower MAE.
En masse, the results suggested that these two functionals marginally underestimated pKas, in a systemic manner (R > 0.99 and COD > 0.98 in both cases). Functionals belonging to rungs 2 and 3 (
Figure 1, namely PBEPBE and TPSSTPSS) pointed out overestimated pKa values, leading to slopes higher than 1 unit (slope
PBEPBE = 1.51 and slope
TPSSTPSS = 1.14, respectively). Further, while PBEPBE R and COD values suggested a systematic internal error (R > 0.99 and COD > 0.99), TPSSTPSS showed less accuracy in predicting pKas (R > 0.93 and COD > 0.87).
A more detailed analysis identified that CAM-B3LYP well predicted the pKa values of bromo- and chloro-substituted benzoic acids, with negligible ΔpKa, while a slightly lower precision in predicting the pKa of compounds affected by the “ortho” effect was obtained. Indeed, the major discrepancy in ΔpKa was in the case of 2,6-Dimethylbenzoic acid (
Table S1, Supplementary Information). Conversely, B3PW91 well depicted such an effect, showing the lowest ΔpKa among the screened functionals. It also well predicted the pKa of 3- and 4-Chlorobenzoic acids, as well as methoxy-substituted benzoic acids. Conversely, unsatisfactory results were obtained when -Br and -CN were considered, partially in agreement with what was reported in the literature [
1]. To note, PBE1PBE proved to be remarkable in predicting the same compounds, with a trivial discrepancy.
Realistic modeling of the shape of the solvation cavity is a major goal in obtaining consistent computed pKa values [
8]. Indeed, it was previously demonstrated that coarse models of the first solvation shell could cause huge inaccuracy in determining pKa [
5]. Further, several attempts were made in re-shaping the “reaction centre” with encouraging results [
1,
2,
3,
4,
5,
8]. Therefore, to better understand if the difference in pKa values obtained with different functionals could be related with a significant modification of the solvation cavity, an extensive analysis was performed. However, no remarkable discrepancy among CAM-B3LYP and the other functionals was observed in the reaction center geometry, nor in the shape of the overall solvation cavity around the molecules (
Figure S2, Supplementary Information). Thus, a punctual estimation of each species involved in the reaction center was performed, through the bond length determination and dihedral evaluation. Due to the trustworthy MAE value [
1,
8], the bond lengths returned by CAM-B3LYP were considered as the standard ones, assuming its ability to mimic the solvation cage geometry similar to the “real” one. Thus, the differences were referred to its bond values (thereafter Δ
length). For each acid species, the distance among the moieties describing the reaction center was considered, as reported in
Figure 3.
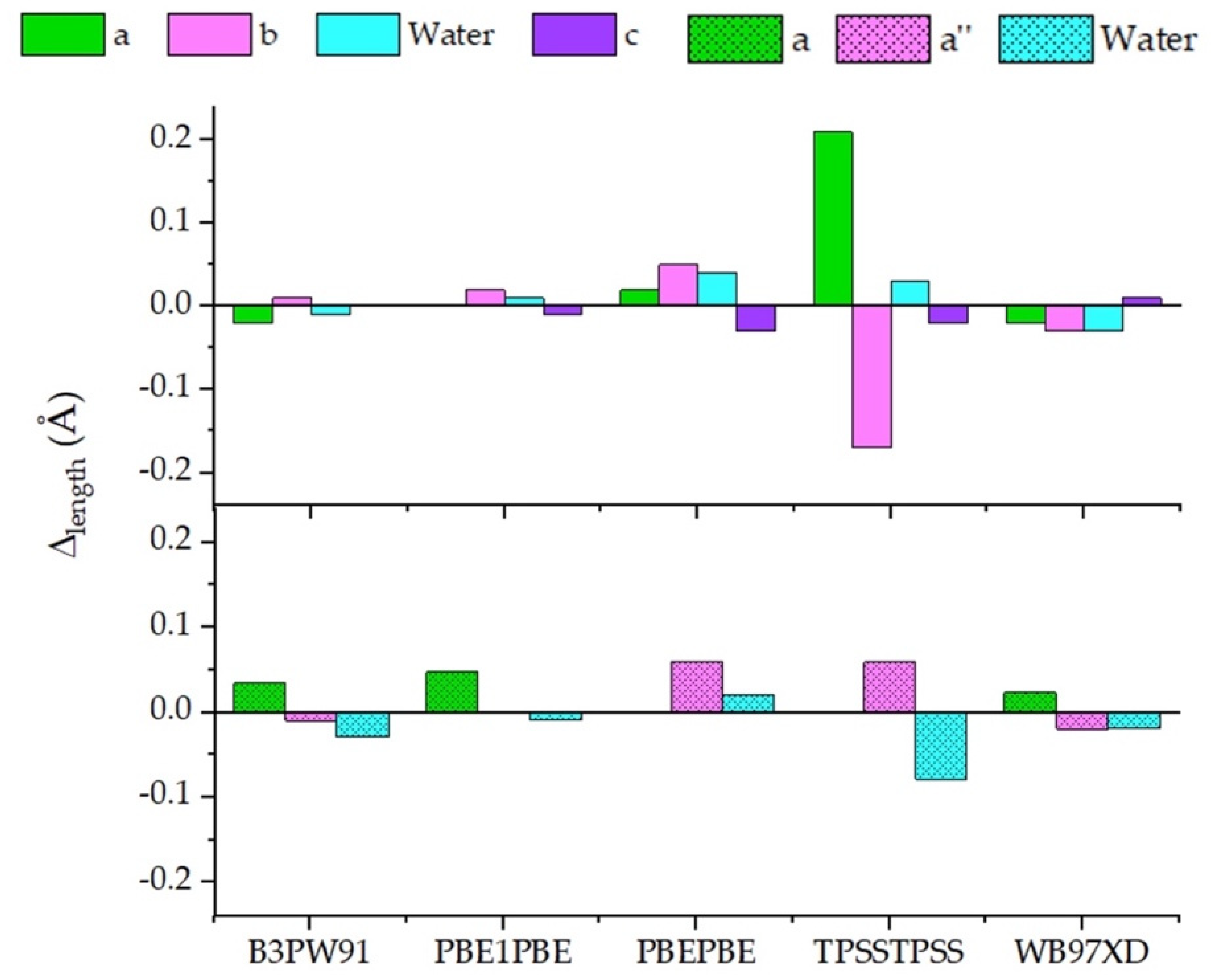
Overall, in our computational settings, B3PW91 reported more similar bond lengths compared to the CAM-B3LYP ones. Remarkably, this functional showed the highest discrepancy (0.07 Å) in predicting the
c length of 4-Cyanobenzoic acid, although returning an accurate pKa value. This behavior was shared with the other functionals, where the same reliability in pKa determination was not pursued. Similarly, no significant changes with respect to CAM-B3LYP emerged in the bond length analysis with PBE1PBE. Further, in the conjugated bases, the bond length discrepancy was always below 0.02 Å, even when the 2-Chlorobenzoic acid was considered. PBEPBE, together with TPSSTPSS, and WB97XD showed the most scattered values, and the larger gap was approximately 0.06 Å by PBEPBE in describing 2-Chlorobenzoate and 2-Methoxybenzoate. Unlike what was observed for other systems, where even minimal variations in bond length were significative [
22,
24], in our conditions not a clear correlation between Δ
length and calculated pKa emerged.
Thus, dihedral angles in the acid and conjugated bases were also analyzed (
Table S2, Supplementary Information). The results showed that the twist angle formed by the carboxylic/carboxylate moiety and the aromatic ring marginally affected the reliability in pKa determination. For instance, in benzoic acid, the dihedral angle was in CAM-B3LYP, PBE1PBE and PBEPBE, despite the ΔpKa varying up to 1.87 units. In 2-Bromobenzoic acid, a change of only two degrees was related with a pKa variation up to 1.5, as shown in PBEPBE. Similar outcomes were obtained comparing 4-Metoxybenzoic acid: PBE1PBE and WB97XD returned similar dihedral values, although substantial differences in pKas were observed. Even for the conjugated bases, the differences in the dihedral angles were blurred. For instance, 3-Chlorobenzoate, 4-Chlorobenzoate and 2-Methoxybenzoate showed similar dihedral values in contempt of massive ΔpKa variations, with PBEPBE and TPSSTPSS. Since functionals include self-correction factors that might shield meaningful dihedral differences between the acid and the conjugated base, the average twist occurring between the carboxylic moiety and the aromatic one was considered.
Table 2 summarizes the average value in the angles formed by the carboxylic group with respect to the aromatic ring, in the acids and conjugated bases.
Overall, such data suggested no strict correlation between the geometry organization of the solvation cage and the reliability in returning pKa values. Therefore, a large contribution of the self-correction error of the functional itself, coupled with the adopted solvation model, can be anticipated.
For each functional, the computational cost (CC) was defined as the average time, in minutes, for computing the acid and the conjugated base of all compounds. Specifically, the number of shared processors and the memory (CPU) limit were kept constant for each functional. Thus, in the present work, values obtained with CAM-B3LYP belong to a new set of optimizations, confirming previous massive results [
7].
It should be noted that WB97XD and TPSSTPSS showed a considerable computational cost (49 and 51 min, respectively) (
Figure S4, Supplementary Information). PBEPBE, PBE1PBE and B3PW91 implied a machine working time in the range of half an hour, while CAM-B3LYP required the highest computational cost, equal to 52 min, which is compensated for by its high accuracy in predicting pKas. Interestingly, B3PW91 and PBE1PBE pointed out very interesting results in terms of the machine-costs/reliability ratio, since they led to slightly less accurate results than CAM-B3LYP, but in shorter processing time.









{kind=link}
{kind=link}
{kind=link}
{kind=link}