
Optically Pure Calixarenyl Phosphine via Stereospecific Alkylation on Evans’ Oxazolidinone Moiety


Abstract

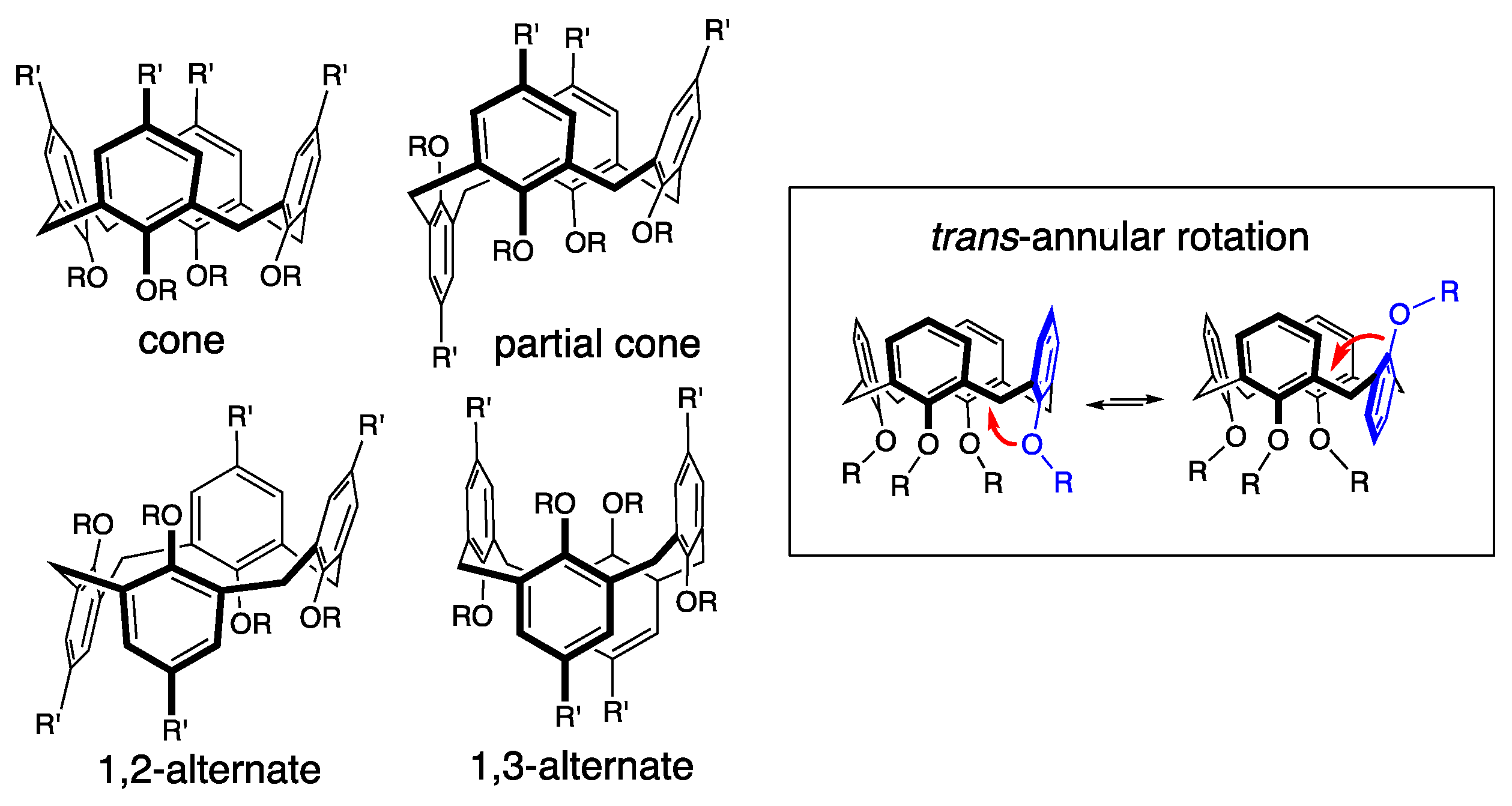
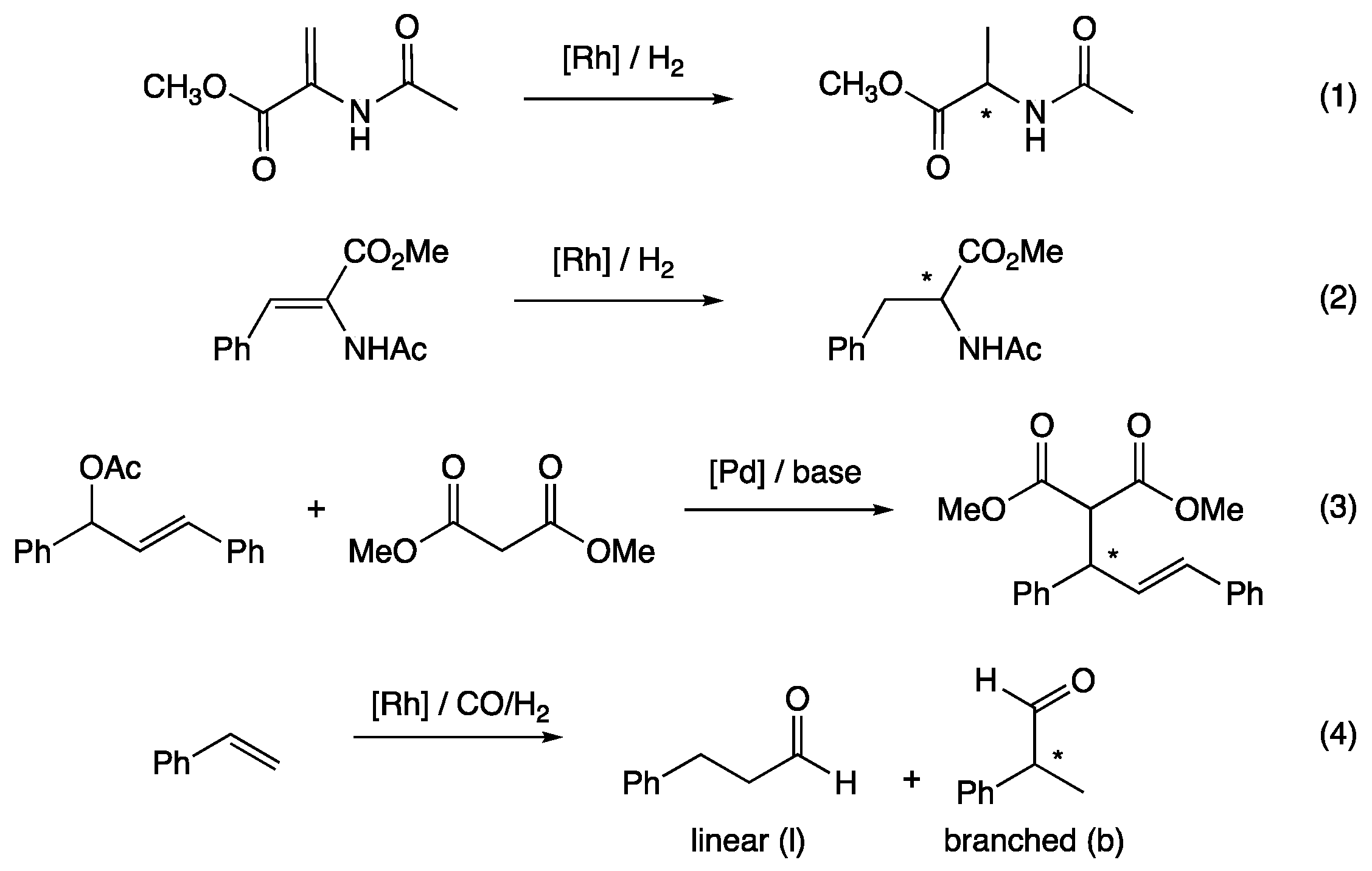
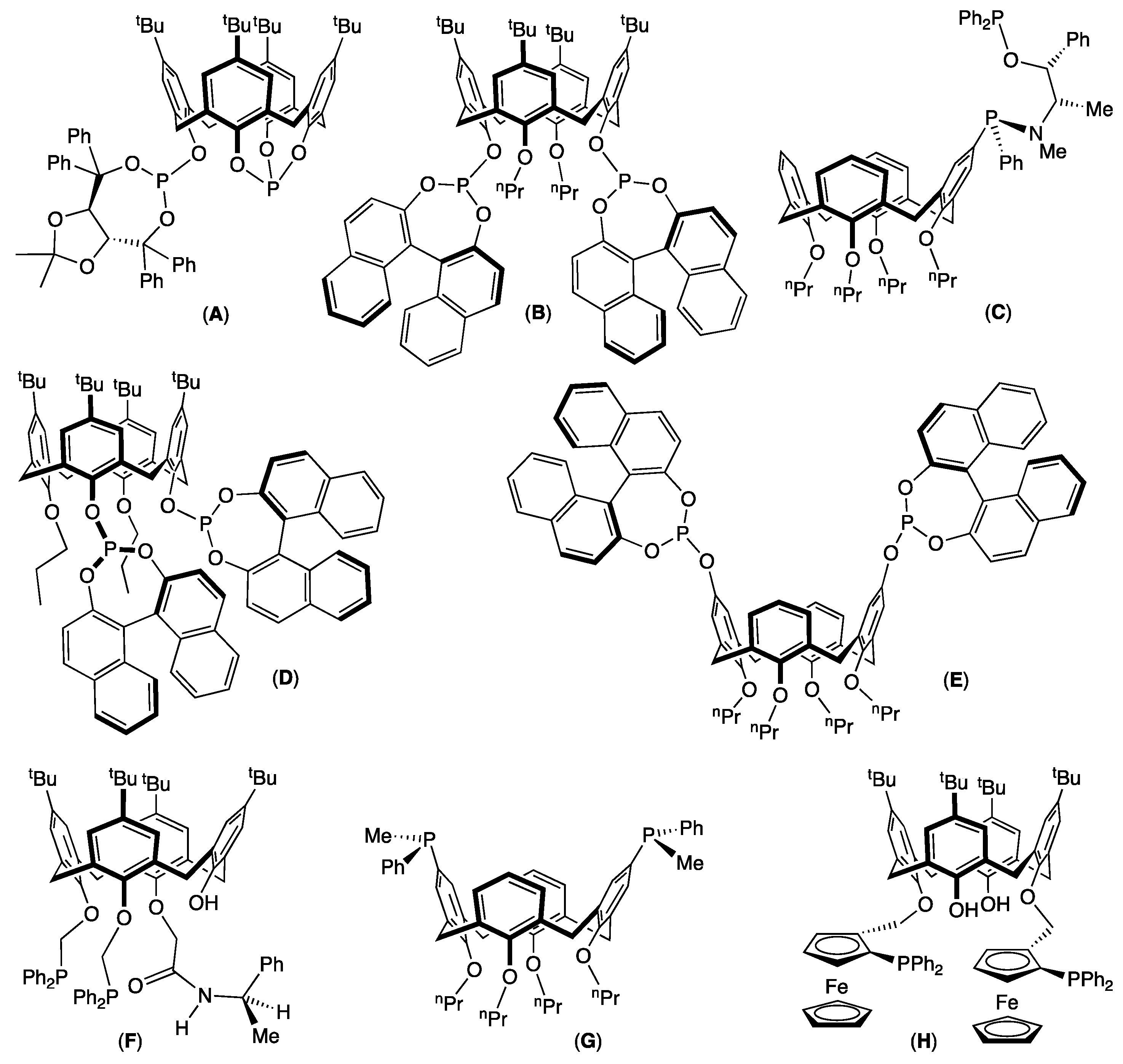
1. Introduction
2. Results and Discussion
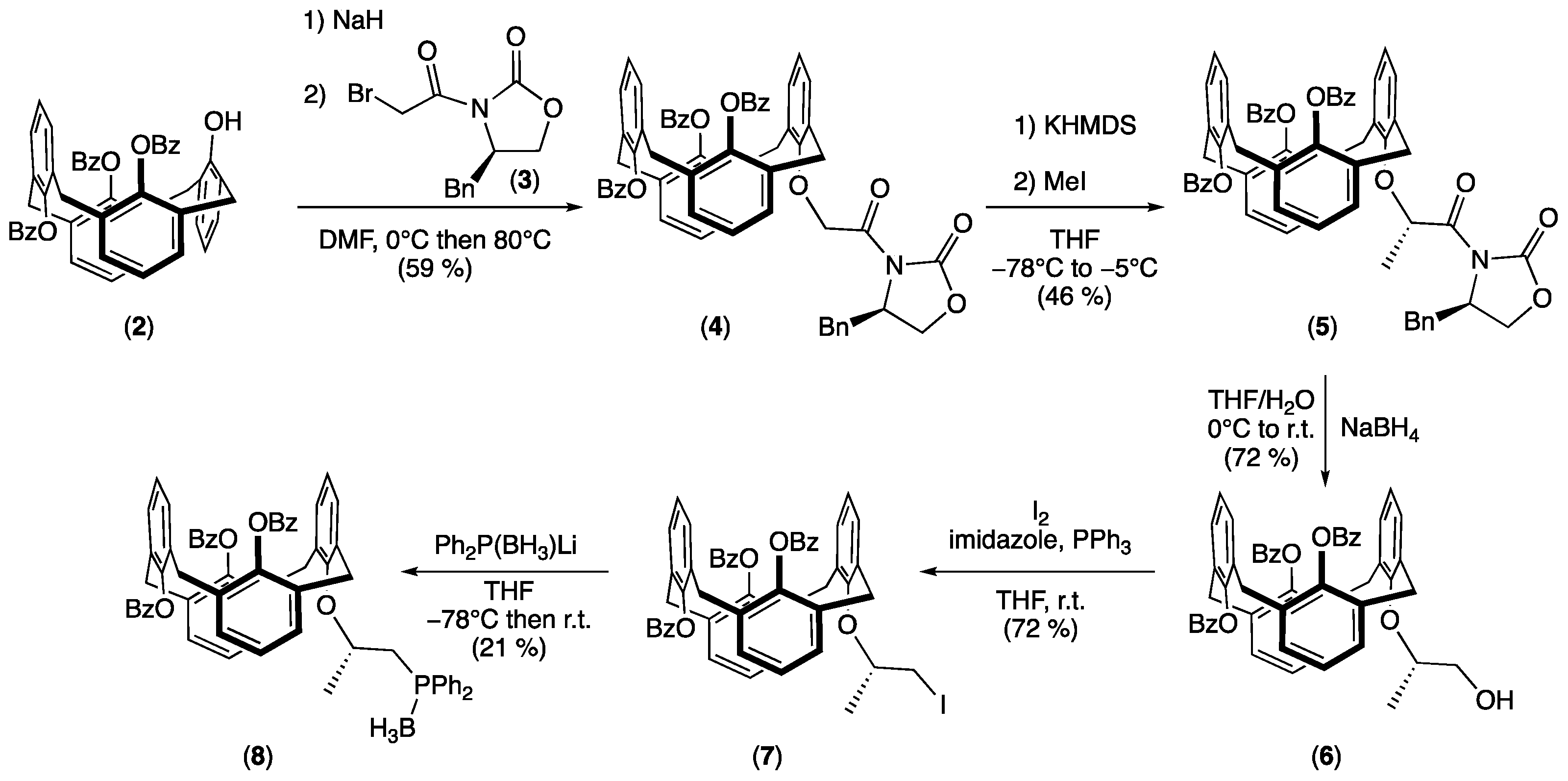
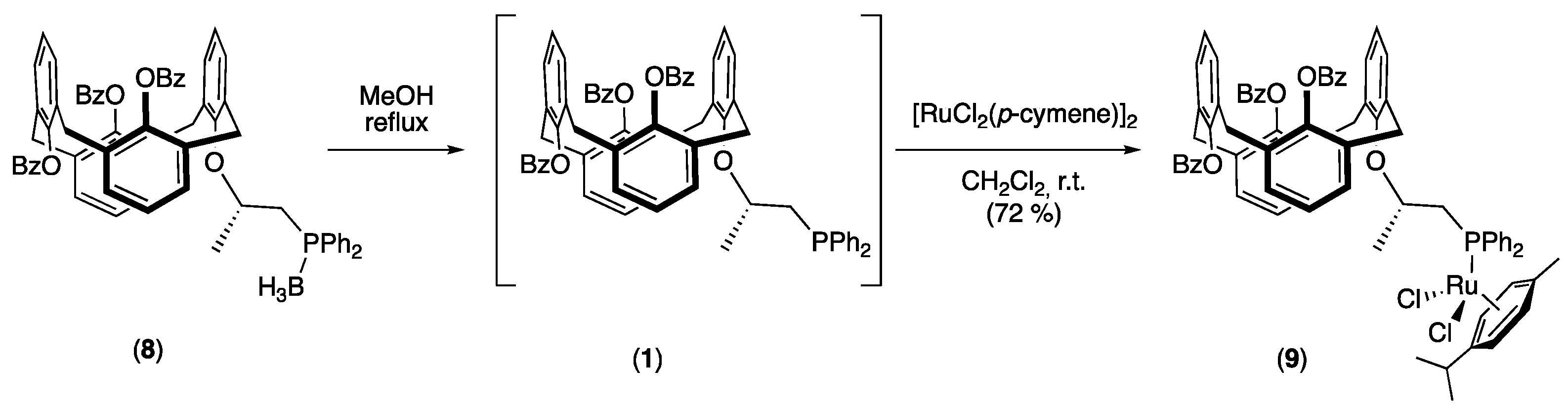
2.1. Synthesis of the Phosphine Borane Adduct
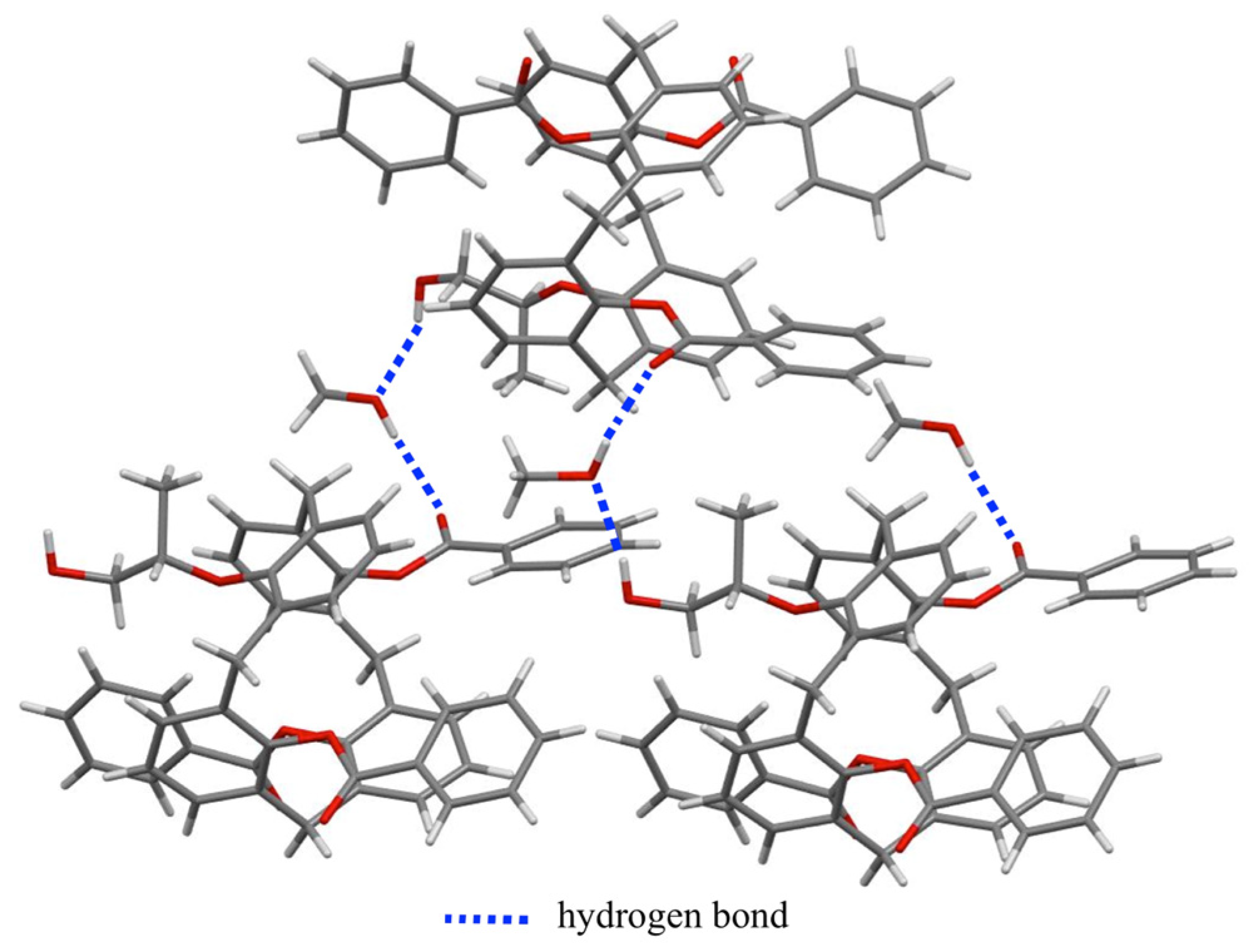
2.2. Single-Crystal X-ray Diffraction Study
2.3. Synthesis of the Ruthenium Complex and Its Catalytic Activity
3. Materials and Methods
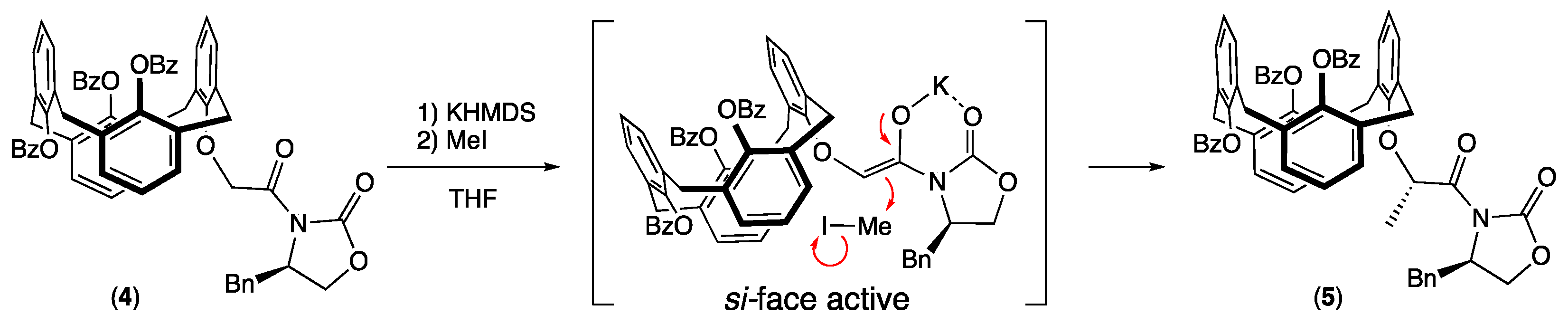
3.1. Synthesis of 25,26,27-Tribenzoyl-28-[2-((R)-4-benzyl-2-oxooxazolidin-3-yl)-2-oxoethoxy] calix[4]arene (1,3-alternate) (4)
3.2. Synthesis of 25,26,27-Tribenzoyl-28-{(S)-1-[((R)-4-benzyl-2-oxooxazolidin-3-yl)- 1-oxopropan-2-yl]oxy}-calix[4]arene) (1,3-alternate) (5)
3.3. Synthesis of 25,26,27-Tribenzoyl-28-[((S)-1-hydroxypropan-2-yl)oxy]-calix[4]arene (1,3-alternate) (6)
3.4. Synthesis of 25,26,27-Tribenzoyl-28-[((S)-1-iodopropan-2-yl)oxy]-calix[4]arene (1,3-alternate) (7)
3.5. Synthesis of {25,26,27-Tribenzoyl-28-[((S)-1-diphenylphosphanyl-propan-2-yl)oxy]-calix[4] arene} borane (1,3-alternate) (8)
3.6. Synthesis of Dichloro-P-{25,26,27-tribenzoyl-28-[((S)-1-diphenylphosphanyl-propan-2-yl) oxy]-calix[4]arene} (p-cymene)ruthenium(II) (1,3-alternate) (9)
3.7. X-ray Crystal Structure Analysis
3.8. General Procedure for Ruthenium-Catalyzed Reduction of Acetophenone
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harvey, P.D. Wide-rim and outer-face functionalizations of calix[4]arene. Coord. Chem. Rev. 2002, 233–234, 289–309. [Google Scholar] [CrossRef]
- Homden, D.M.; Redshaw, C. The use of calixarenes in metal-based catalysis. Chem. Rev. 2008, 108, 5086–5130. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Samanta, K.; Rao, C.P. Conjugates of calixarenes emerging as molecular entities of nanoscience. Coord. Chem. Rev. 2012, 256, 2096–2125. [Google Scholar] [CrossRef]
- Sémeril, D.; Matt, D. Synthesis and catalytic relevance of P(III) and P(V)-functionalised calixarenes and resorcinarenes. Coord. Chem. Rev. 2014, 279, 58–95. [Google Scholar] [CrossRef]
- Rodik, R.; Cherenok, S.; Kalchenko, O.; Yesypenko, O.; Lipkowski, J.; Kalchenko, V. Functional calixarenes for material and life science. Curr. Org. Chem. 2018, 22, 2196–2218. [Google Scholar] [CrossRef]
- Santoro, O.; Redshaw, C. Metallocalix[n]arenes in catalysis: A 13-year update. Coord. Chem. Rev. 2021, 448, 214173. [Google Scholar] [CrossRef]
- Sachdeva, G.; Vaya, D.; Srivastava, C.M.; Kumar, A.; Rawat, V.; Singh, M.; Verma, M.; Rawat, P.; Rao, G.K. Calix[n]arenes and its derivatives as organocatalysts. Coord. Chem. Rev. 2022, 472, 214791. [Google Scholar] [CrossRef]
- Pan, Y.-C.; Tian, J.-H.; Guo, D.-S. Molecular Recognition with Macrocyclic receptors for application in precision medicine. Acc. Chem. Res. 2023, 56, 3626–3639. [Google Scholar] [CrossRef]
- Yang, X.; Xu, L.; Zhang, A.; Xiao, C. Organophosphorus extractants: A critical choice for actinides/lanthanides separation in nuclear fuel cycle. Chem. Eur. J. 2023, 29, e202300456. [Google Scholar] [CrossRef] [PubMed]
- Mourer, M.; Regnouf-de-Vains, J.-B.; Duval, R.E. Functionalized calixarenes as promising antibacterial drugs to face antimicrobial resistance. Molecules 2023, 28, 6954. [Google Scholar] [CrossRef]
- Sachdeva, G.; Bamal, Y.; Ladan, A.; Tiwari, O.S.; Rawat, V.; Yadav, P.; Verma, V.P. Calixarene-metal complexes in lactide polymerization: The story so far. ACS Omega 2023, 8, 13479–13491. [Google Scholar] [CrossRef]
- Ren, H.; Wang, H.; Wen, W.; Li, S.; Li, N.; Huo, F.; Yin, C. A summary of calixarene-based fluorescent sensors developed during the past five years. Chem. Commun. 2023, 59, 13790–13799. [Google Scholar] [CrossRef]
- Gutsche, C.D. Calixarenes. Acc. Chem. Res. 1983, 16, 161–170. [Google Scholar] [CrossRef]
- Otsuka, H.; Shinkai, S. Stereochemical control of calixarenes useful as rigid and conformationally diversiform platforms for molecular design. Supramol. Sci. 1996, 3, 189–205. [Google Scholar] [CrossRef]
- Marson, A.; Freixa, Z.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. Chiral calix[4]arene-based diphosphites as ligands in the asymmetric hydrogenation of prochiral olefins. Eur. J. Inorg. Chem. 2007, 4587–4591. [Google Scholar] [CrossRef]
- Liu, S.; Sandoval, C.A. Evaluation of calix[4]arene-based chiral diphosphite ligands in Rh-catalyzed asymmetric hydrogenation of simple dehydroamino acid derivatives. J. Mol. Catal. A Chem. 2010, 325, 65–72. [Google Scholar] [CrossRef]
- Khiri, N.; Bertrand, E.; Ondel-Eymin, M.-J.; Rousselin, Y.; Bayardon, J.; Harvey, P.D.; Jugé, S. Enantioselective hydrogenation catalysis aided by a σ-bonded calix[4]arene to a P-chirogenic aminophosphane phosphinite rhodium complex. Organometallics 2010, 29, 3622–3631. [Google Scholar] [CrossRef]
- Hkiri, S.; Sémeril, D. How do positions of phosphito units on a calix[4]arene platform affect the enantioselectivity of a catalytic reaction? Organics 2022, 3, 470–480. [Google Scholar] [CrossRef]
- Arnott, G.E. Inherently chiral calixarenes: Synthesis and applications. Chem. Eur. J. 2018, 24, 1744–1754. [Google Scholar] [CrossRef]
- Dieleman, C.; Steyer, S.; Jeunesse, C.; Matt, D. Diphosphines based on an inherently chiral calix[4]arene scaffold: Synthesis and use in enantioselective catalysis. J. Chem. Soc. Dalton Trans. 2001, 2508–2517. [Google Scholar] [CrossRef]
- Khiri-Meribout, N.; Bertrand, E.; Bayardon, J.; Eymin, M.-J.; Rousselin, Y.; Cattey, H.; Fortin, D.; Harvey, P.D.; Jugé, S. P-Chirogenic phosphines supported by calix[4]arene: New insight into palladium-catalyzed asymmetric allylic substitution. Organometallics 2013, 32, 2827–2839. [Google Scholar] [CrossRef]
- Karpus, A.; Yesypenko, O.; Boiko, V.; Poli, R.; Daran, J.-C.; Voitenko, Z.; Kalchenko, V.; Manoury, E. Chiral phosphinoferrocenyl-calixarenes. Eur. J. Org. Chem. 2016, 3386–3394. [Google Scholar] [CrossRef]
- Natarajan, N.; Pierrevelcin, M.-C.; Sémeril, D.; Bauder, C.; Matt, D.; Ramesh, R. Chiral calixarene and resorcinarene derivatives. Conical cavities substituted at their up-per rim by two phosphito units and their use as ligands in Rh-catalysed hydroformyla-tion. Catal. Commun. 2019, 118, 70–75. [Google Scholar] [CrossRef]
- Bauder, C.; Sémeril, D. Synthesis of optically pure calix[4]arenes derived from Evans oxazolidinone and/or pyranose. Open J. Chem. 2022, 8, 1–7. [Google Scholar]
- Aldrich, L.N.; Berry, C.B.; Bates, B.S.; Konkol, L.C.; So, M.; Lindsley, G.W. Towards the total synthesis of Marineosin A: Construction of the macrocyclic pyrrole and an advanced, functionalized spiroaminal model. Eur. J. Org. Chem. 2013, 4215–4218. [Google Scholar] [CrossRef]
- Hoogenboom, J.; Fiers, M.; Immink, R.; Zuilhof, H.; Wennekes, T. Synthesis and evaluation of locostatin-based chemical probes towards PEBP-proteins. Tetrahedron Lett. 2016, 57, 2406–2409. [Google Scholar] [CrossRef]
- Jaime, C.; de Mendoza, J.; Prados, P.; Nieto, P.M.; Sánchez, C. 13C NMR Chemical shifts. A single rule to determine the conformation of calix[4]arenes. J. Org. Chem. 1991, 56, 3372–3376. [Google Scholar] [CrossRef]
- Evans, D.A.; Takacs, J.M.; McGee, L.R.; Ennis, M.D.; Mathre, D.J.; Bartroli, J. Chiral enolate design. Pure Appl. Chem. 1981, 53, 1109–1127. [Google Scholar] [CrossRef]
- Evans, D.A.; Ennis, M.D.; Mathre, D.J. Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of α-substituted carboxylic acid derivatives. J. Am. Chem. Soc. 1982, 104, 1737–1739. [Google Scholar] [CrossRef]
- Smith, T.E.; Richardson, D.P.; Truran, G.A.; Belecki, K.; Onishi, M. Acylation, diastereoselective alkylation, and cleavage of an oxazolidinone chiral auxiliary. A multistep asymmetric synthesis experiment for advanced undergraduates. J. Chem. Ed. 2008, 85, 695–697. [Google Scholar] [CrossRef]
- Prashad, M.; Har, D.; Kim, H.-Y.; Repic, O. A new, economical, practical and racemization-free method for the reductive removal of 2-oxazolidinones from N-acyloxazolidinones with sodium borohydride. Tetrahedron Lett. 1998, 39, 7067–7070. [Google Scholar] [CrossRef]
- Join, B.; Lohier, J.-F.; Delacroix, O.; Gaumont, A.-C. Convenient mild and selective hydrophosphination of functionalized alkenes: Access to P, O and P, S derivatives. Synthesis 2008, 3121–3125. [Google Scholar] [CrossRef]
- Fecher, G.-H.; Kübler, J.; Felser, C. Chirality in the solid state: Chiral crystal structures in chiral and achiral space groups. Materials 2022, 15, 5812. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B 2013, 69, 249–259. [Google Scholar] [CrossRef]
- Van Overschelde, M.; Verveckena, E.; Modha, S.G.; Cogen, S.; Van der Eycken, E. Catalyst-free alcoholysis of phosphane-boranes: A smooth, cheap, and efficient deprotection procedure. Tetrahedron 2009, 65, 6410–6415. [Google Scholar] [CrossRef]
- Kagan, H.B.; Tahar, M.; Fiaud, J.-C. Rabbit gastric lipase in the biocatalytic resolution of 2-hydroxyalkyldiphenylphosphines. Bioorg. Med. Chem. 1994, 2, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.E.; Durran, S.E.; Elsegood, M.R.J.; Smith, M.B.; Staniland, P.M.; Talib, S.; Dale, S.H. Supramolecular chemistry of half-sandwich organometallic building blocks based on RuCl2(p-cymene)Ph2PCH2Y. J. Organomet. Chem. 2006, 691, 4829–4842. [Google Scholar] [CrossRef]
- Grabulosa, A.; Granell, J.; Font-Bardia, M. Cyclometalated ruthenium complexes with P-stereogenic monophosphines containing a polycyclic aromatic substituent. J. Organomet. Chem. 2019, 896, 51–58. [Google Scholar] [CrossRef]
- Hkiri, S.; Gourlaouen, C.; Touil, S.; Samarat, A.; Sémeril, D. 1,3,4-Oxadiazole-functionalized α-amino-phosphonates as ligands for the ruthenium-catalyzed reduction of ketones. New J. Chem. 2021, 45, 11327–11335. [Google Scholar] [CrossRef]
- Mannu, A.; Grabulosa, A.; Baldino, S. Transfer hydrogenation from 2-propanol to acetophenone catalyzed by [RuCl2(η6-arene)P] (P = monophosphine) and [Rh(PP)2]X (PP = diphosphine, X = Cl−, BF4−) complexes. Catalysts 2020, 10, 162. [Google Scholar] [CrossRef]
- Casnati, A.; Fochi, M.; Minari, P.; Pochini, A.; Reggiani, M.; Ungaro, R.; Reinhoudt, D.N. Upper-rim urea-derivatized calix[4]arenes as neutral receptors for monocarboxylate anions. Gazz. Chim. Ital. 1996, 126, 99–105. [Google Scholar]
- Evans, D.A.; Weber, A.E. Synthesis of the cyclic hexapeptide Echinocandin D. New approaches to the asymmetric synthesis of β-hydroxy α-amino acids. J. Am. Chem. Soc. 1987, 109, 7151–7157. [Google Scholar] [CrossRef]
- M86-EXX229V1 APEX3 User Manual; BRUKER AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Strcut. Chem. 2015, 71, 3–8. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
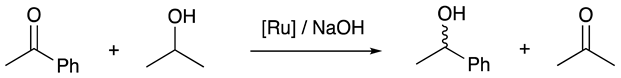 | |||
|---|---|---|---|
| Entry | Temperature (°C) | Conversion (%) | ee (%) |
| 1 | 60 | 60 | 12 |
| 2 | 80 | 68 | 14 |
| 3 | 100 | 67 | 4 |
| CCDC depository | 2313707 | color/shape | colorless/block | ||
| chemical formula | C53H46O9 | formula weight (g mol−1) | 826.90 | ||
| crystal system | orthorhombic | space group | P212121 | ||
| unit cell parameters | a (Å) | 12.790(3) | unit cell parameters | α (°) | 90 |
| b (Å) | 12.890(3) | β (°) | 90 | ||
| c (Å) | 25.477(7) | γ (°) | 90 | ||
| volume (Å3) | 4200.4(17) | Z | 4 | ||
| D (g cm−3) | 1.308 | μ (mm−1) | 0.717 | ||
| Tmin, Tmax | 0.5999, 0.7528 | F(000) | 1744 | ||
| crystal size (mm) | 0.220 × 0.140 × 0.090 | index ranges | −15 ≤ h ≤ 15 | ||
| θ range for data collection (°) | 3.469 ≤ θ ≤ 67.329 | −13 ≤ k ≤ 15 | |||
| reflections collected | 19,727 | −27 ≤ l ≤ 30 | |||
| independent/observed | 6600/3288 | Rint | 0.0737 | ||
| data/restraints/parameters | 6600/483/563 | goodness-of-fit on F2 | 1.045 | ||
| final R indices (I > 2.0 σ(I)) | R1 = 0.0968, wR2 = 0.2723 | R indices (all data) | R1 = 0.2383, wR2 = 0.3385 | ||
| Δρmax, Δρmin (eÅ−3) | 0.434, −0.687 | Flack parameter | 0.03(13) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauder, C.; Sémeril, D. Optically Pure Calixarenyl Phosphine via Stereospecific Alkylation on Evans’ Oxazolidinone Moiety. Molecules 2024, 29, 1156. https://doi.org/10.3390/molecules29051156
Bauder C, Sémeril D. Optically Pure Calixarenyl Phosphine via Stereospecific Alkylation on Evans’ Oxazolidinone Moiety. Molecules. 2024; 29(5):1156. https://doi.org/10.3390/molecules29051156
Chicago/Turabian StyleBauder, Claude, and David Sémeril. 2024. "Optically Pure Calixarenyl Phosphine via Stereospecific Alkylation on Evans’ Oxazolidinone Moiety" Molecules 29, no. 5: 1156. https://doi.org/10.3390/molecules29051156
APA StyleBauder, C., & Sémeril, D. (2024). Optically Pure Calixarenyl Phosphine via Stereospecific Alkylation on Evans’ Oxazolidinone Moiety. Molecules, 29(5), 1156. https://doi.org/10.3390/molecules29051156