
Synthetic Modifications of a Pb2+-Sensor Acridono-Crown Ether for Covalent Attachment and Their Effects on Selectivity


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
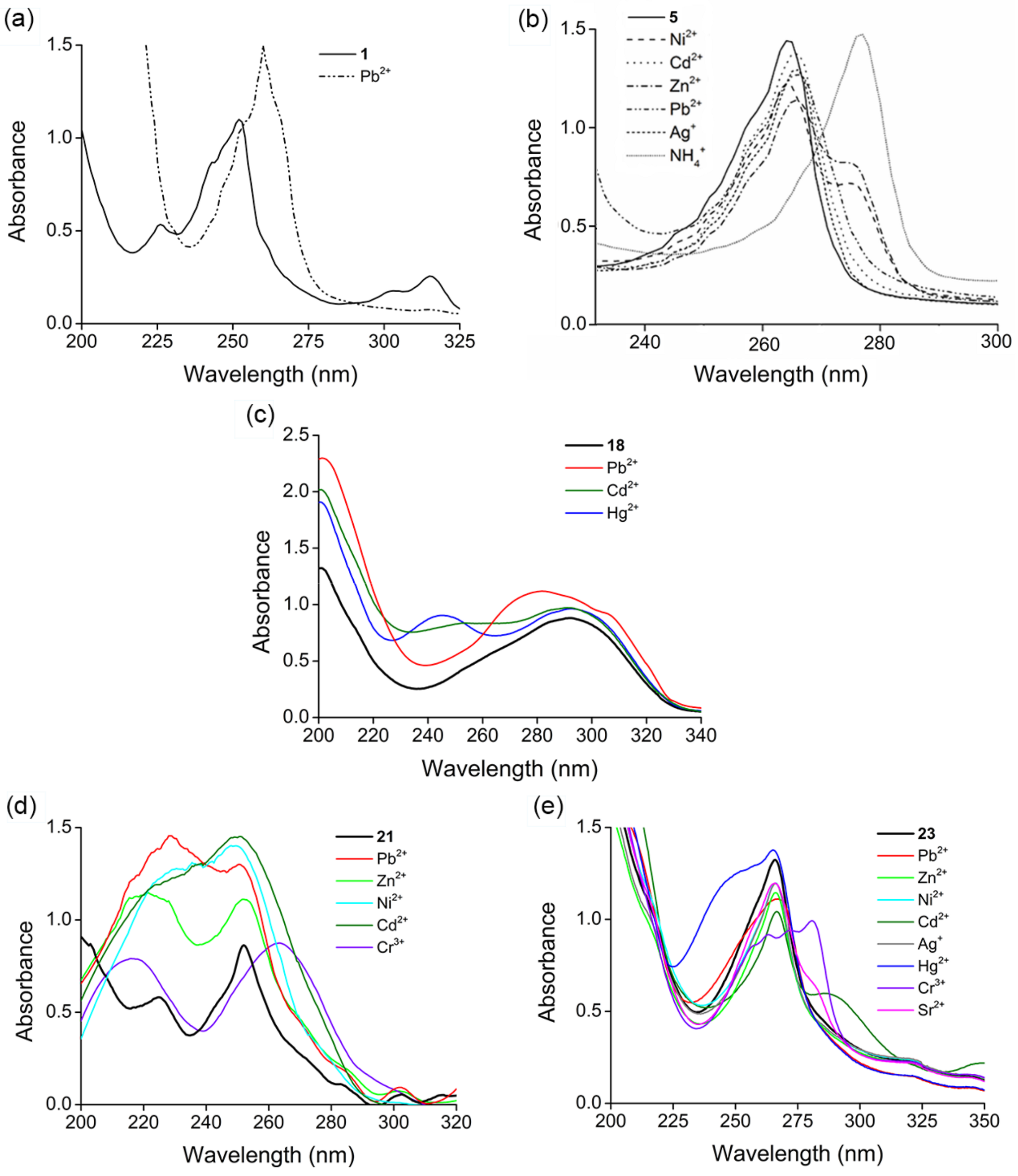
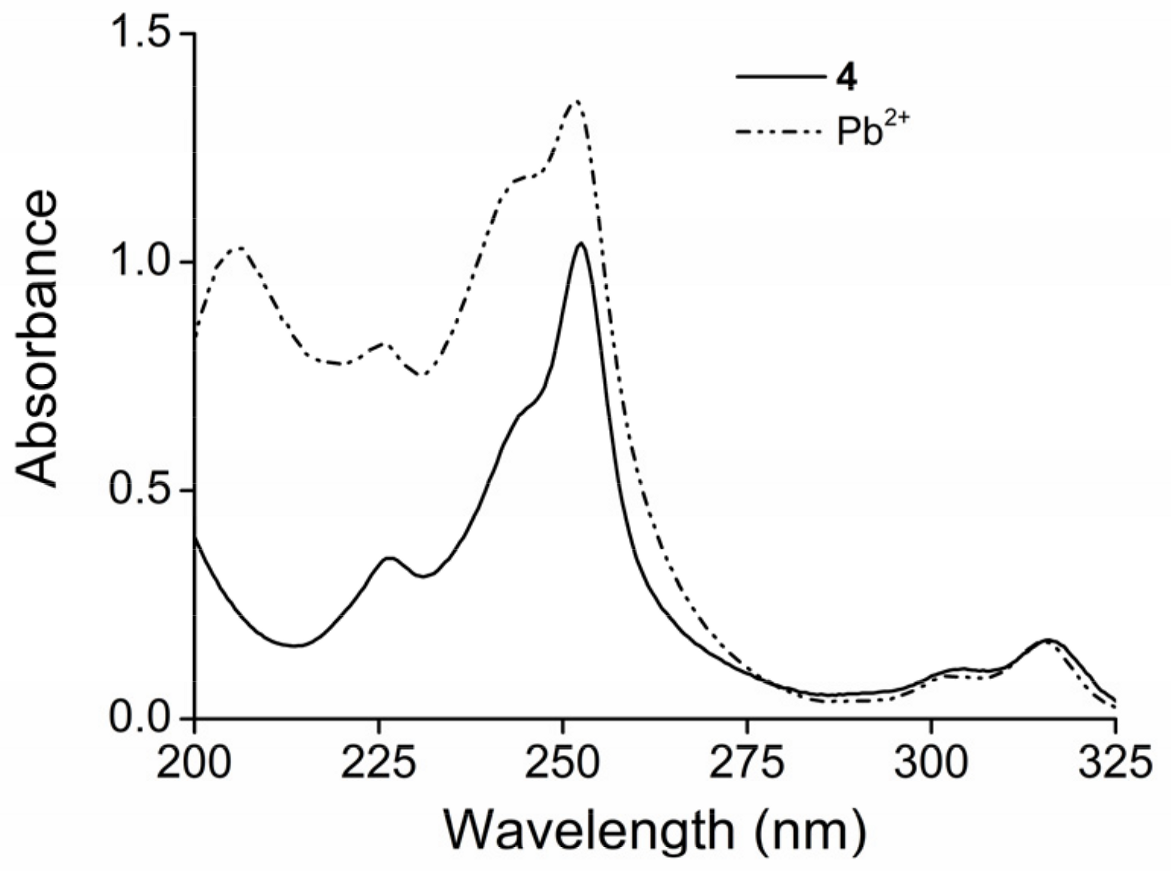
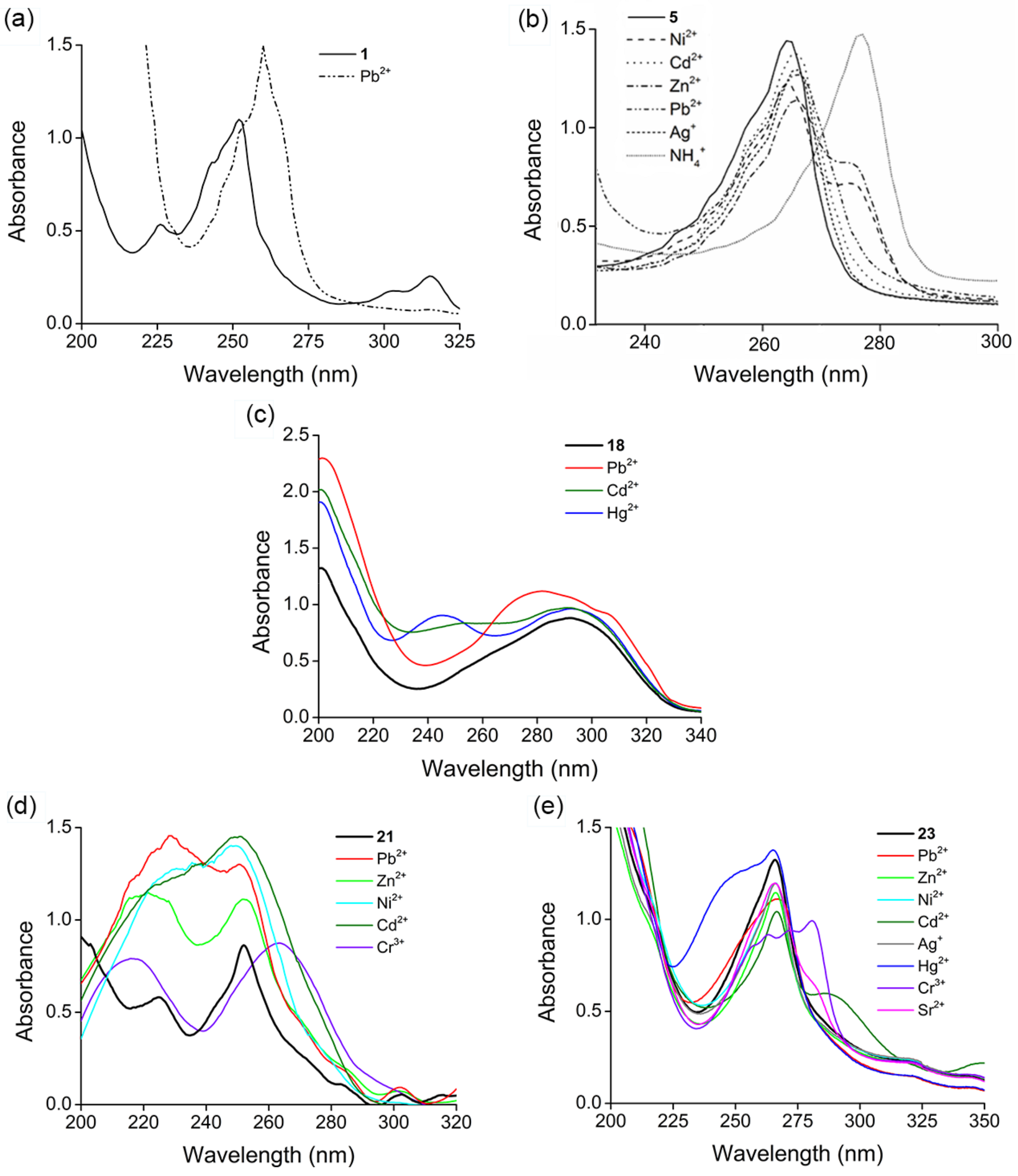
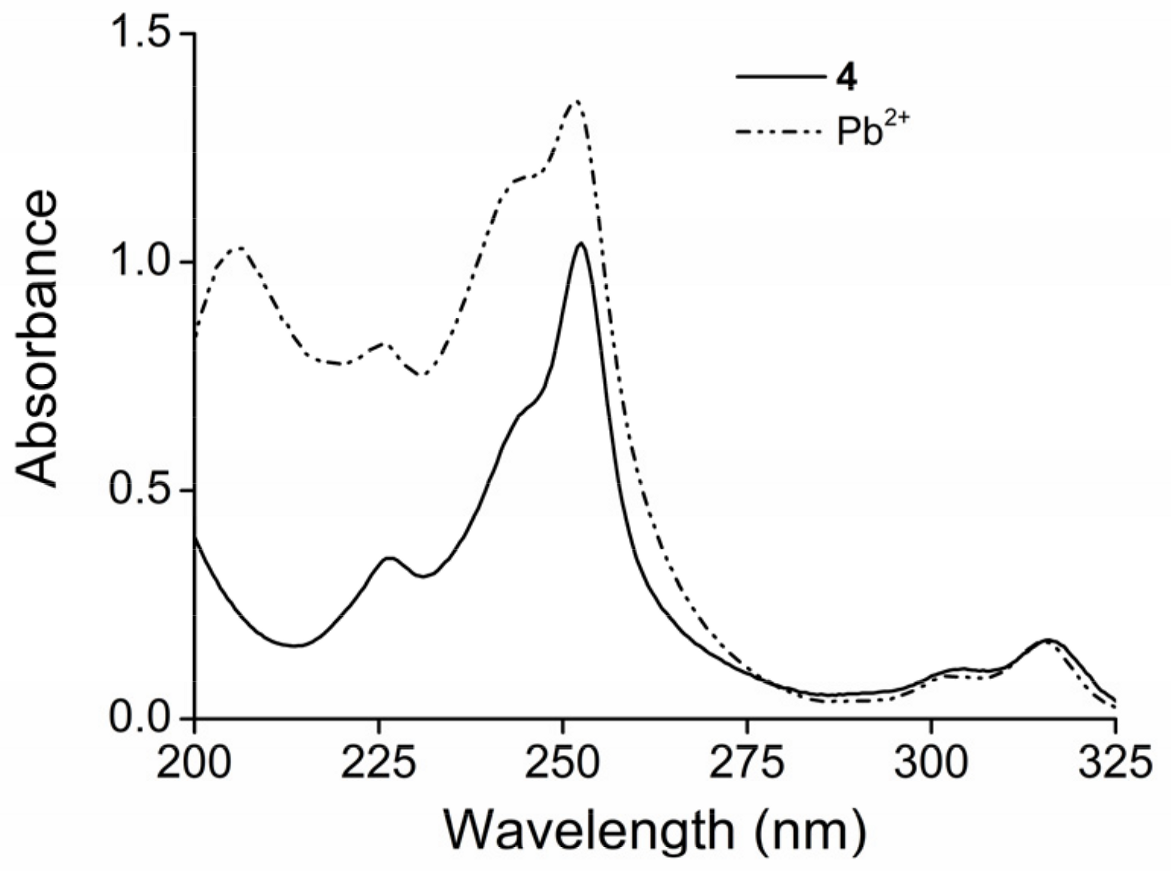
2. Results and Discussion
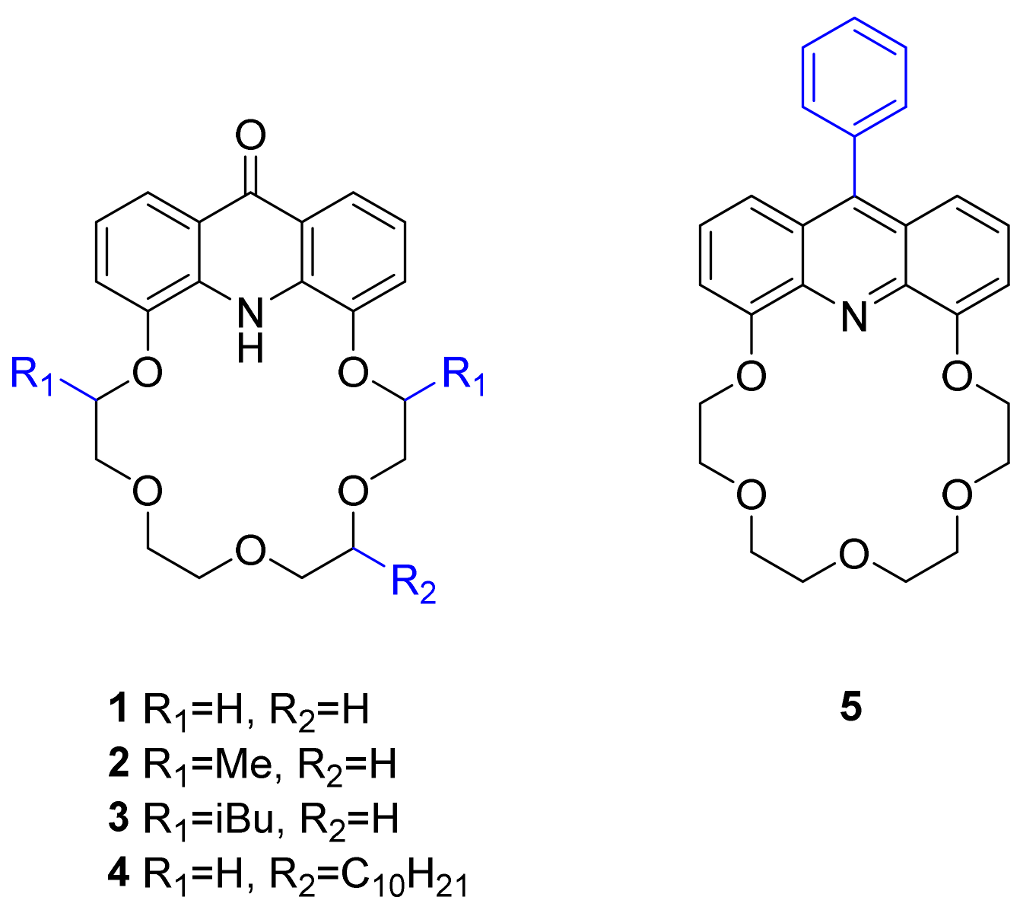
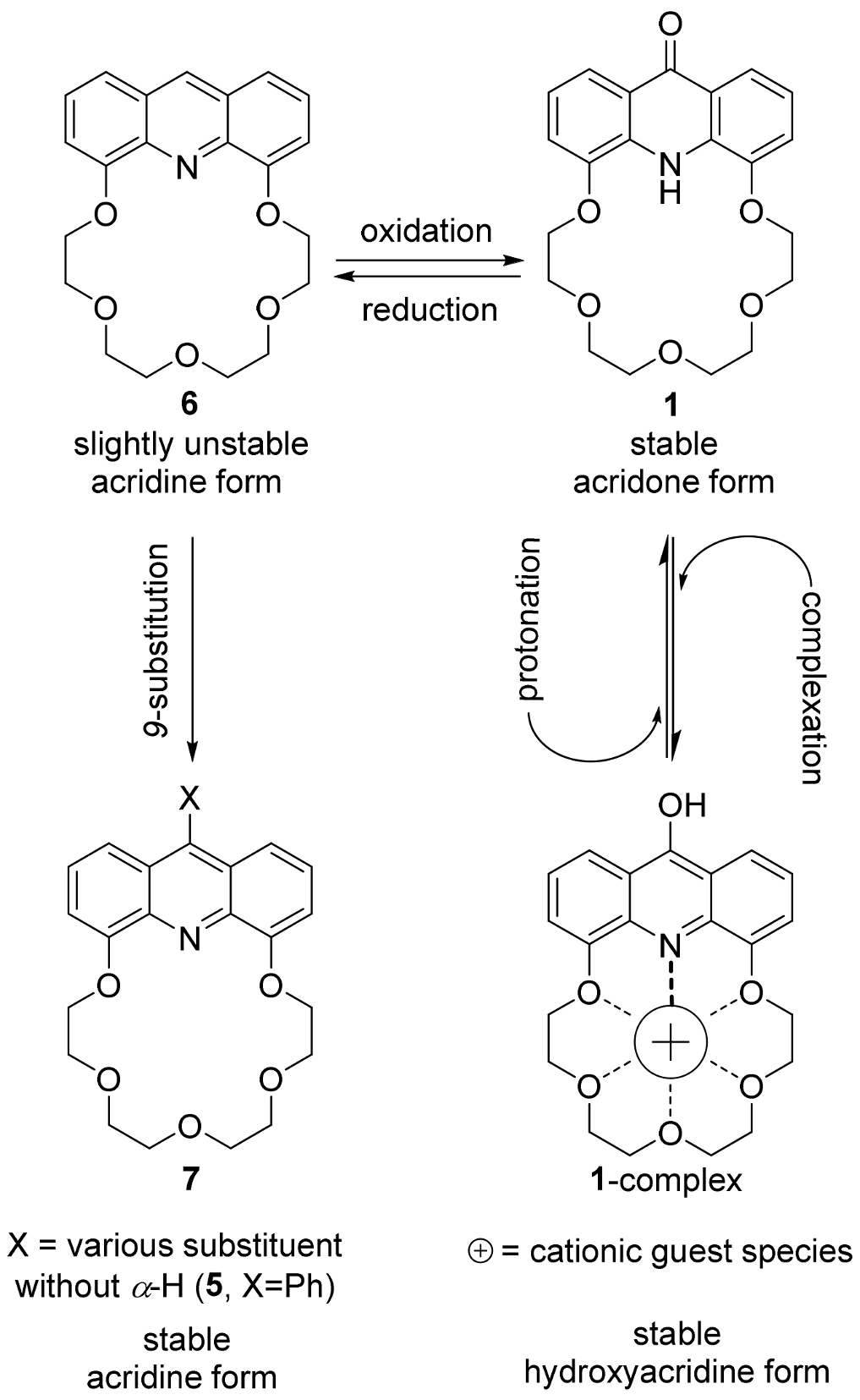
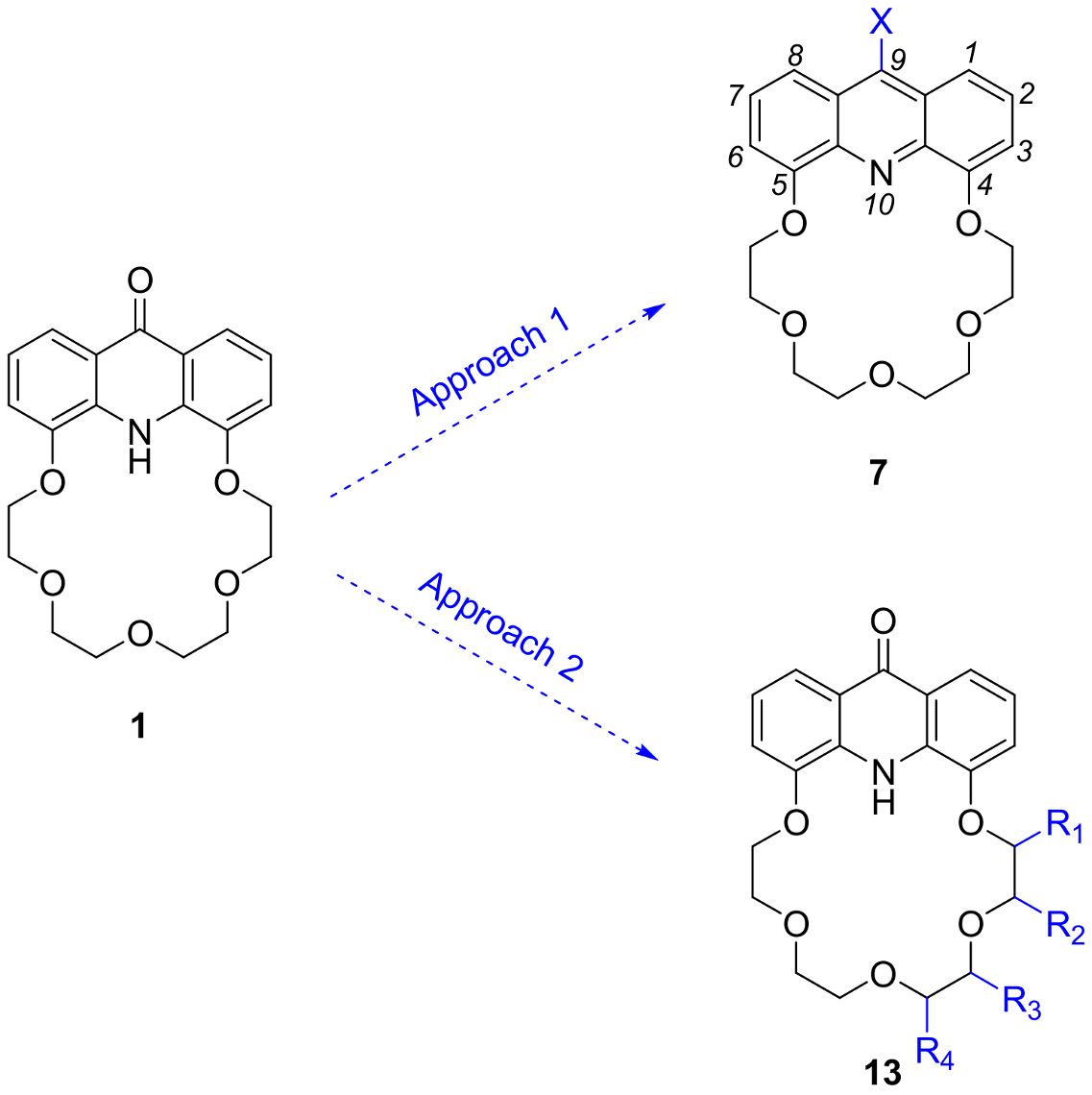
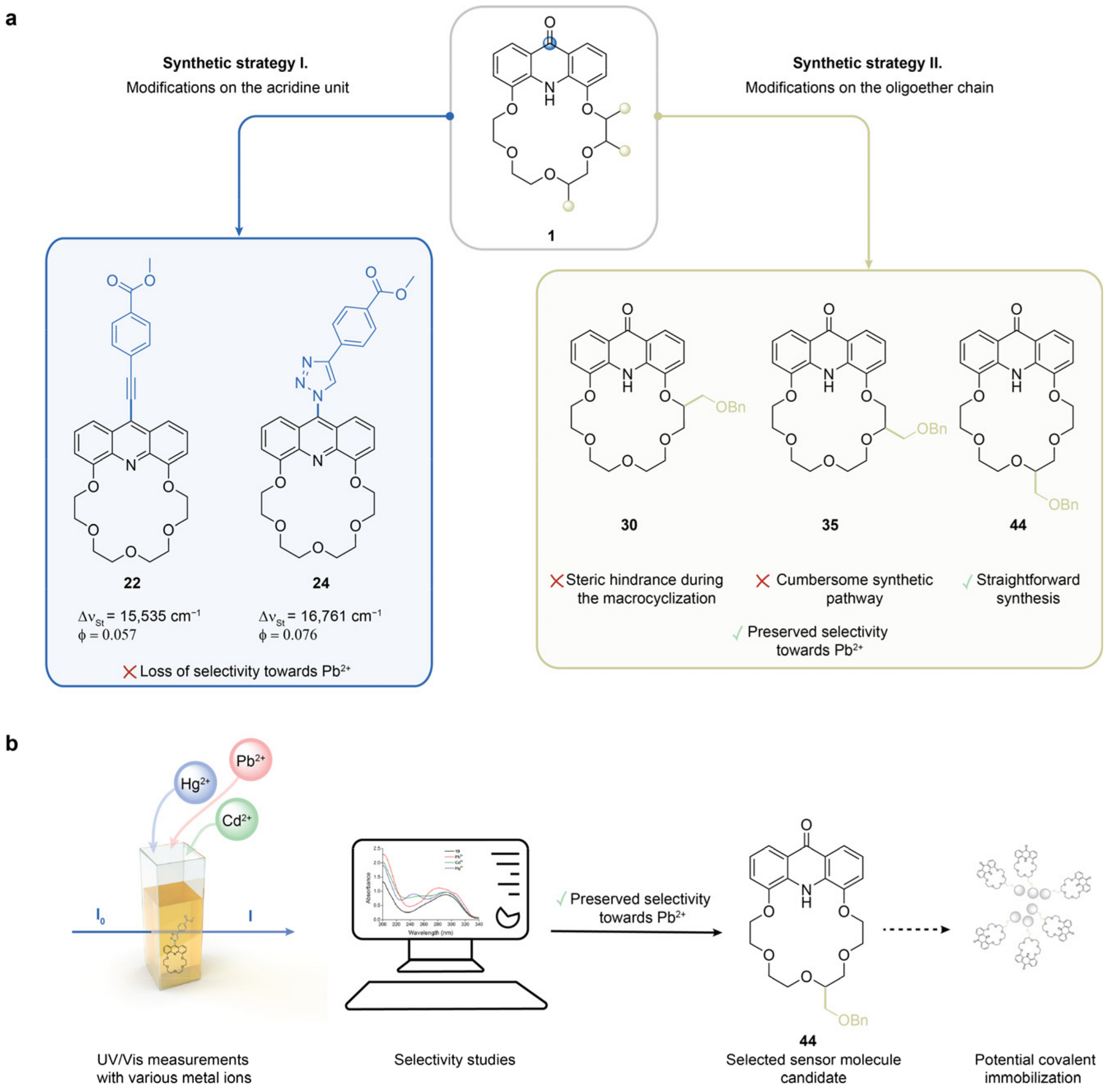
2.1. Structure Design
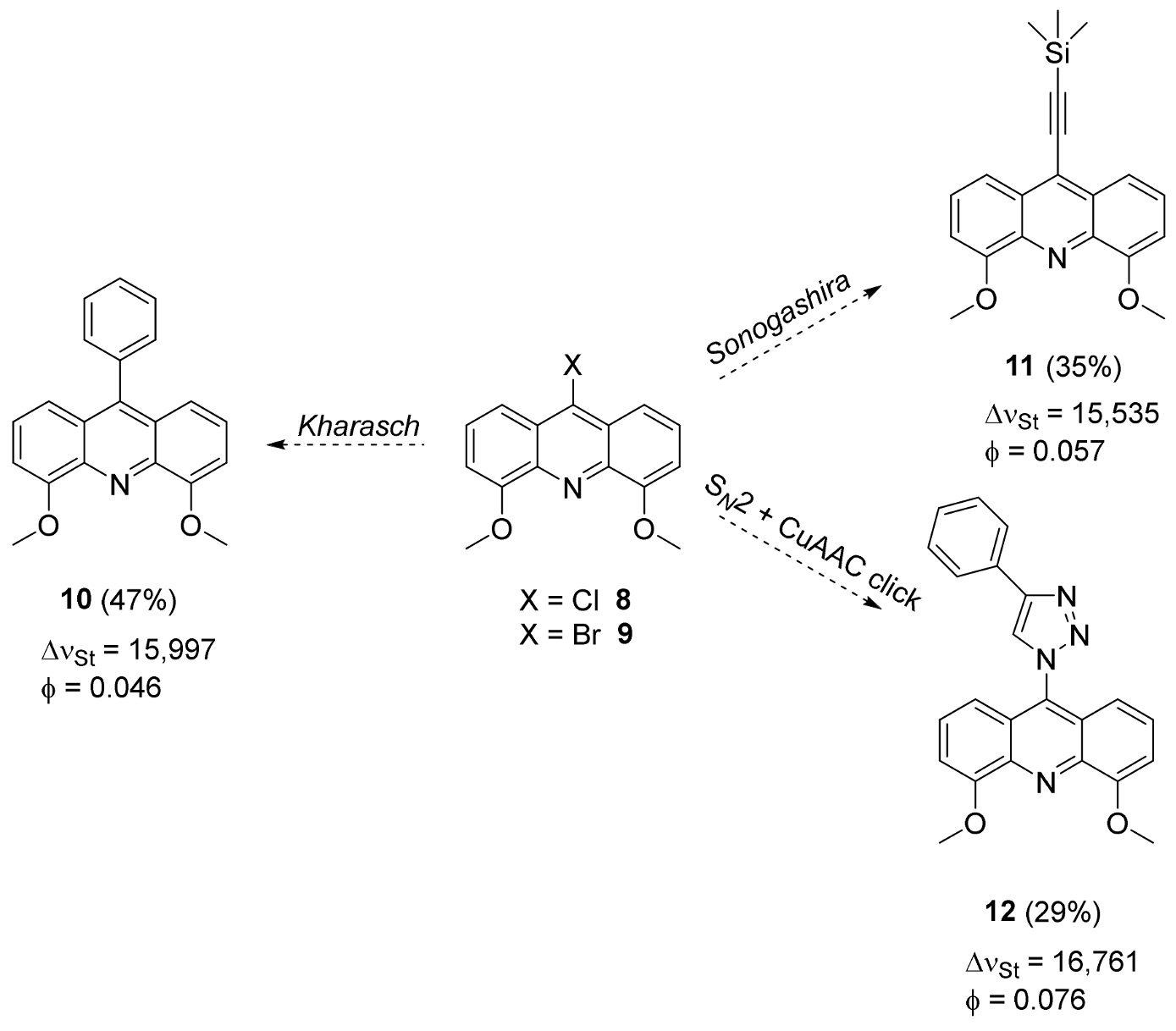
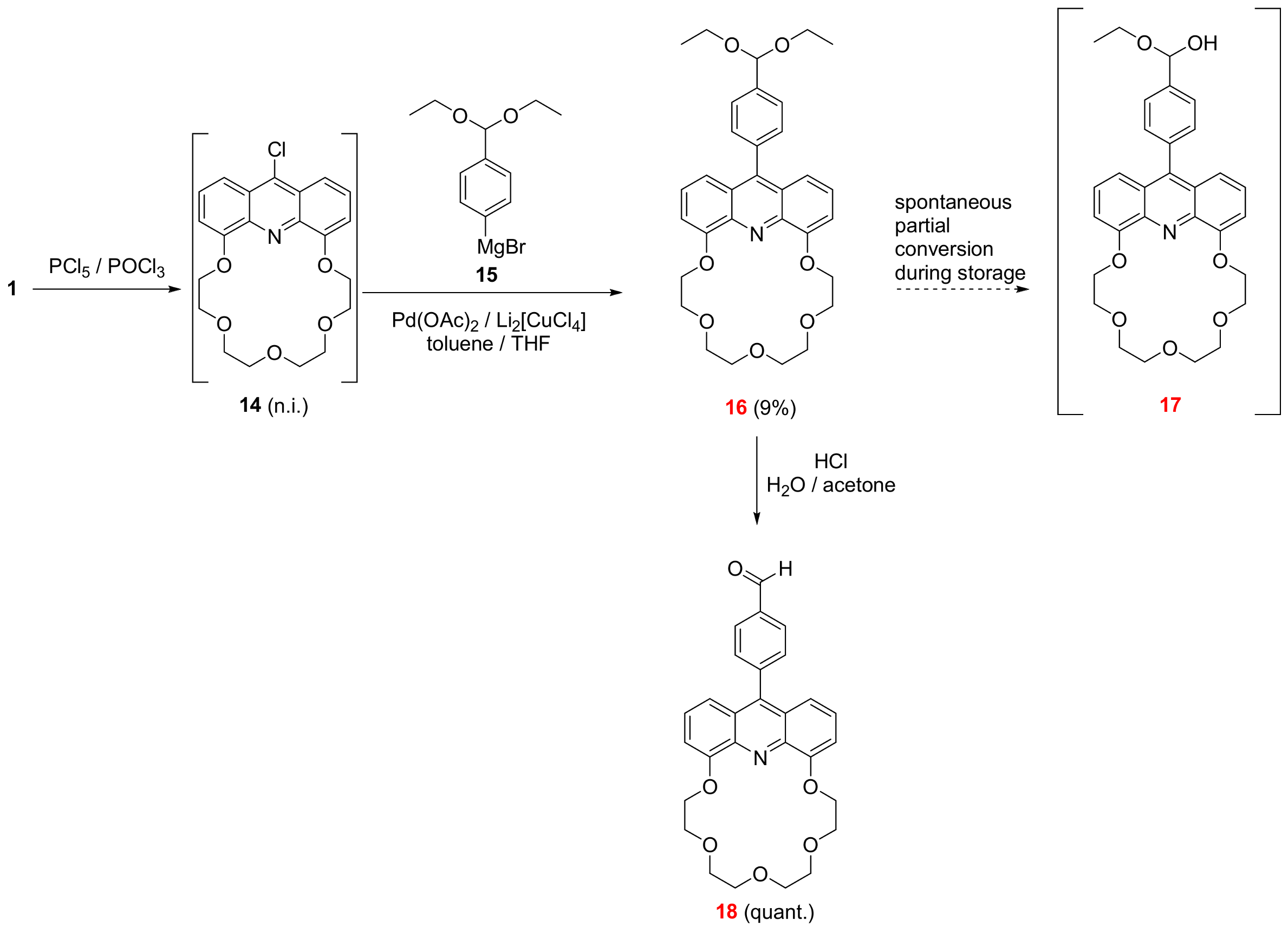
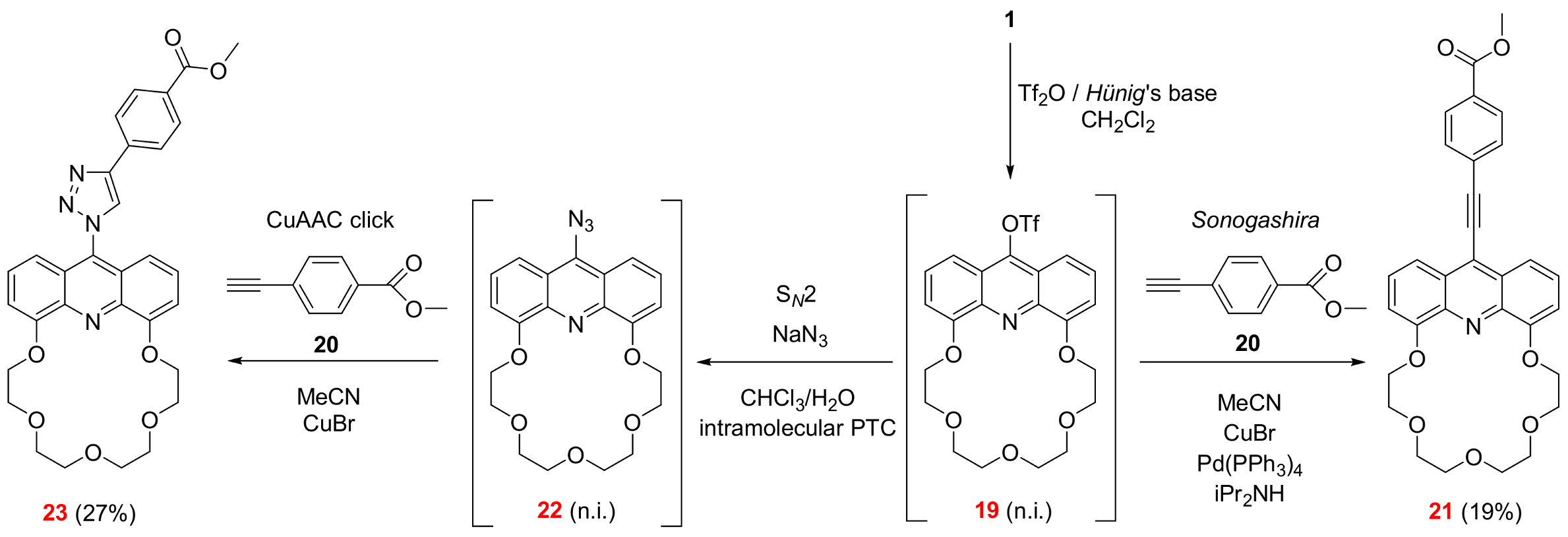
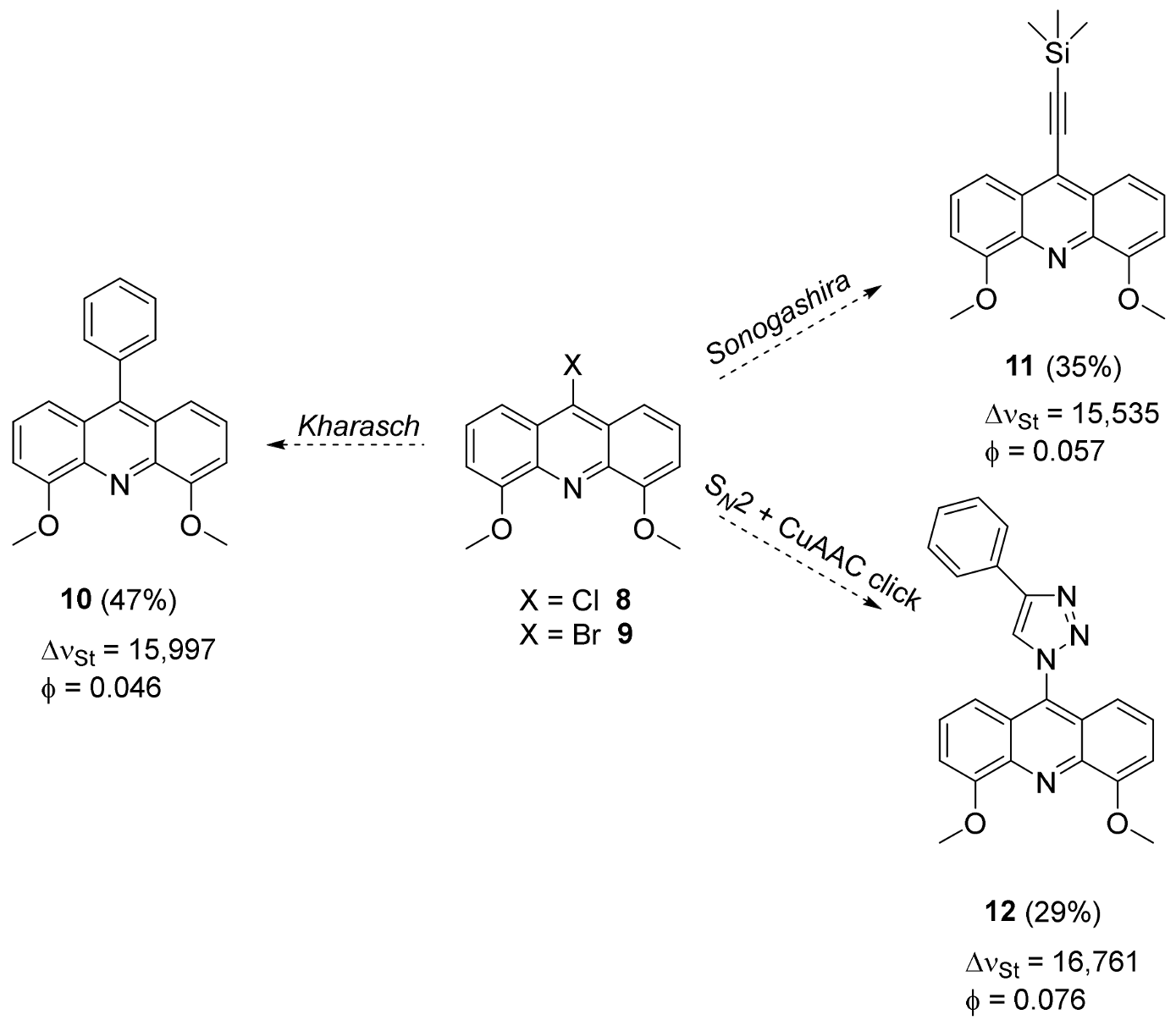
2.2. Functionalization via 9-Substitution of the Acridine Unit (Approach 1)
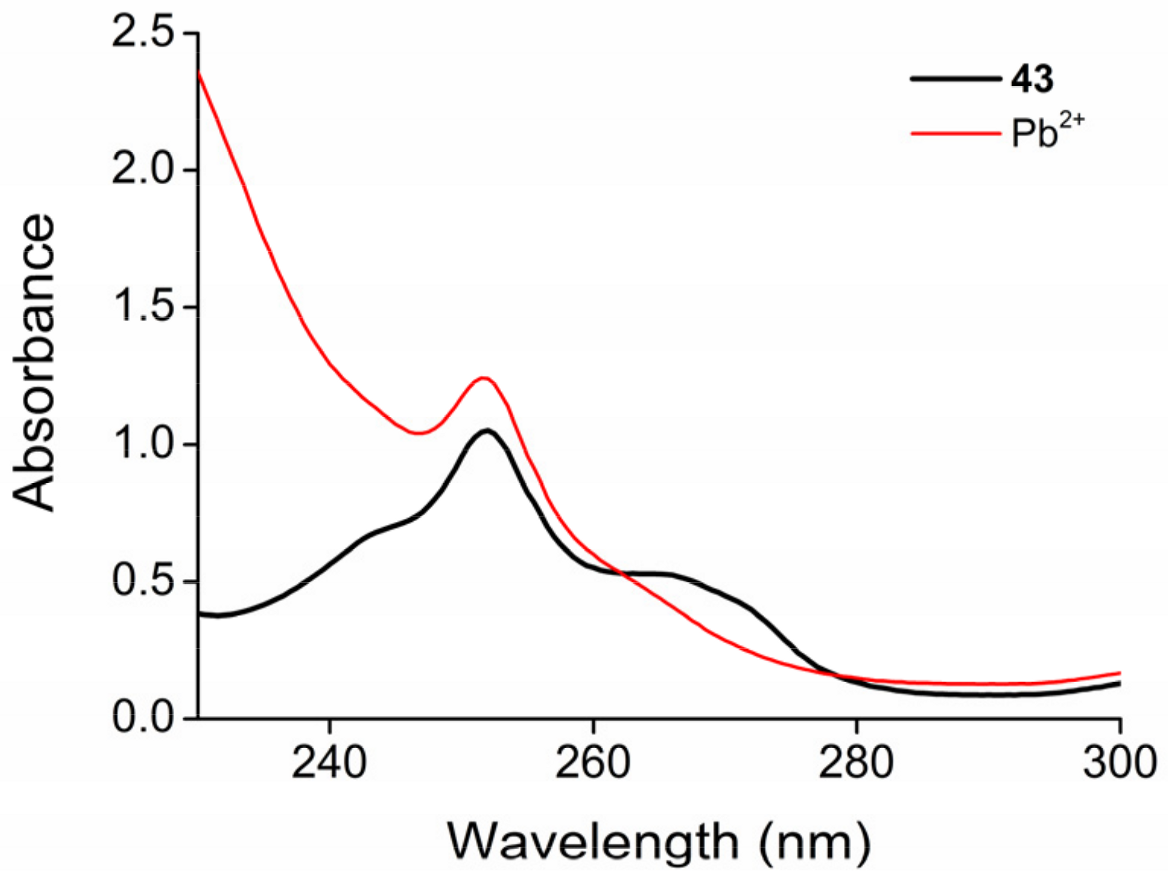
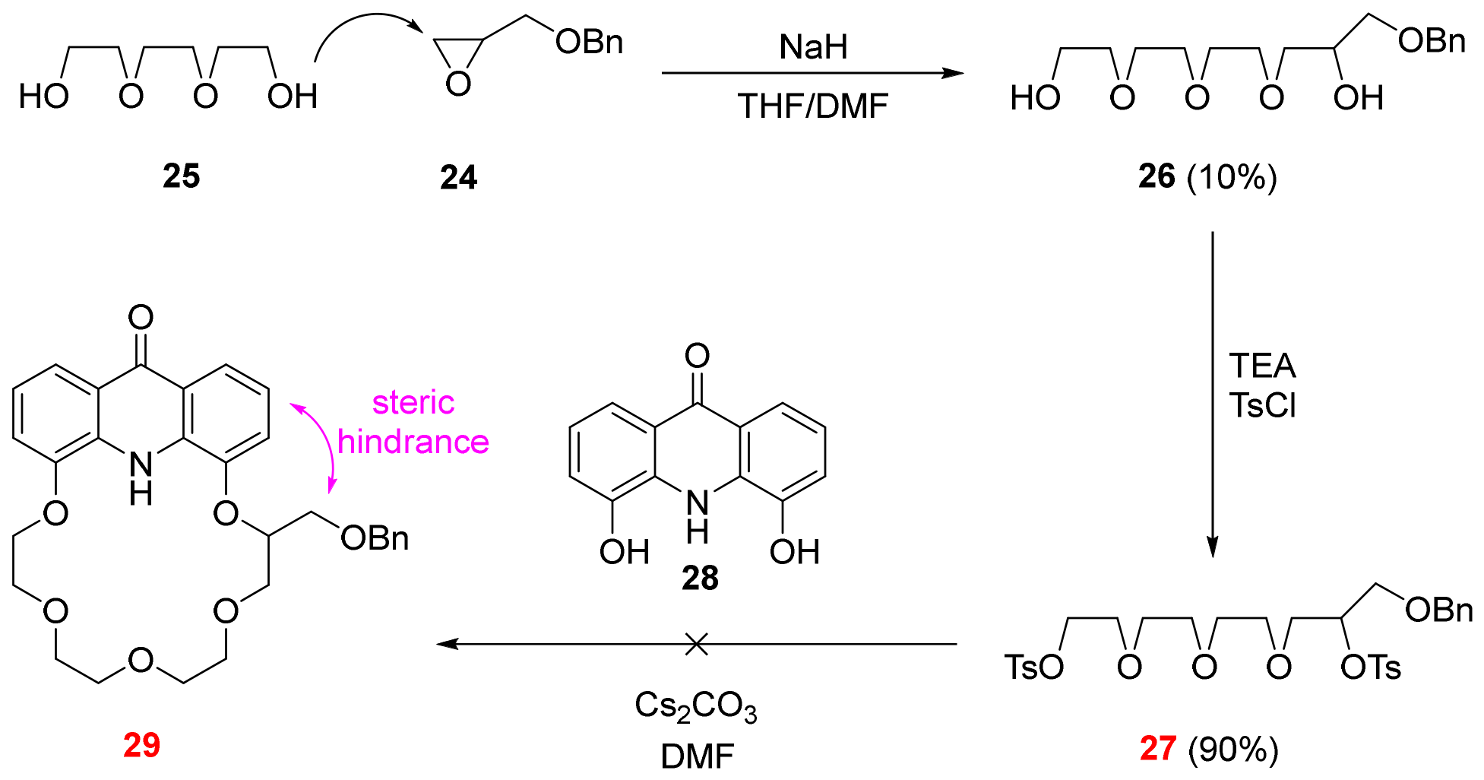
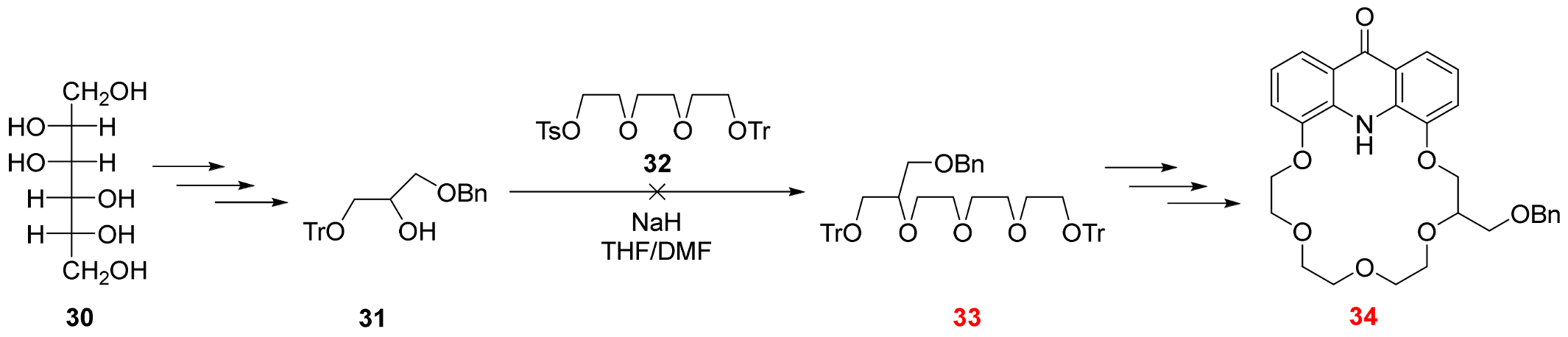
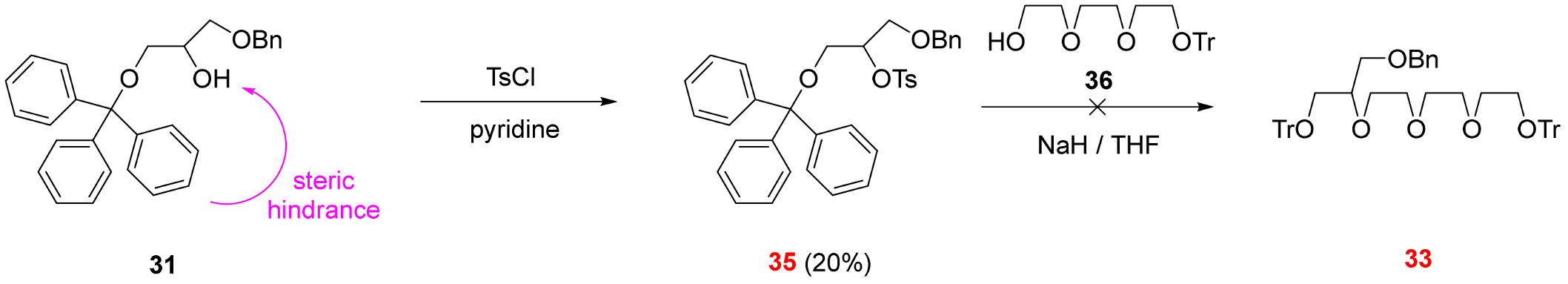
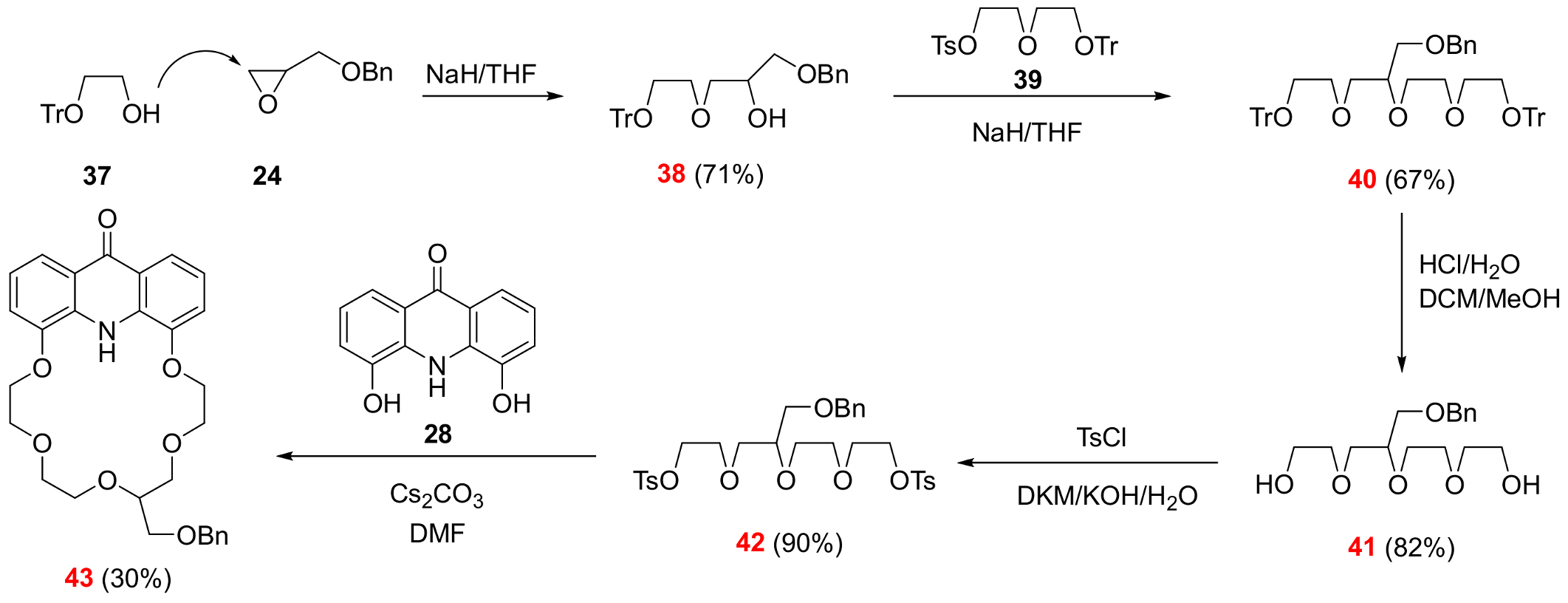
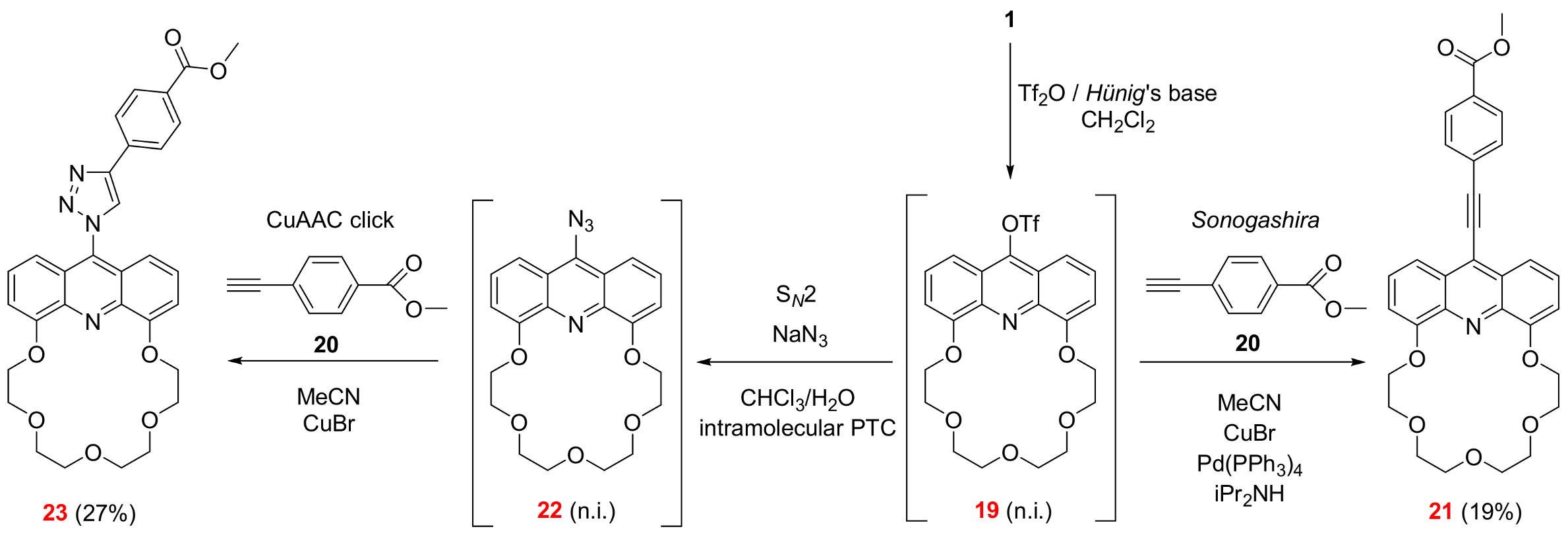
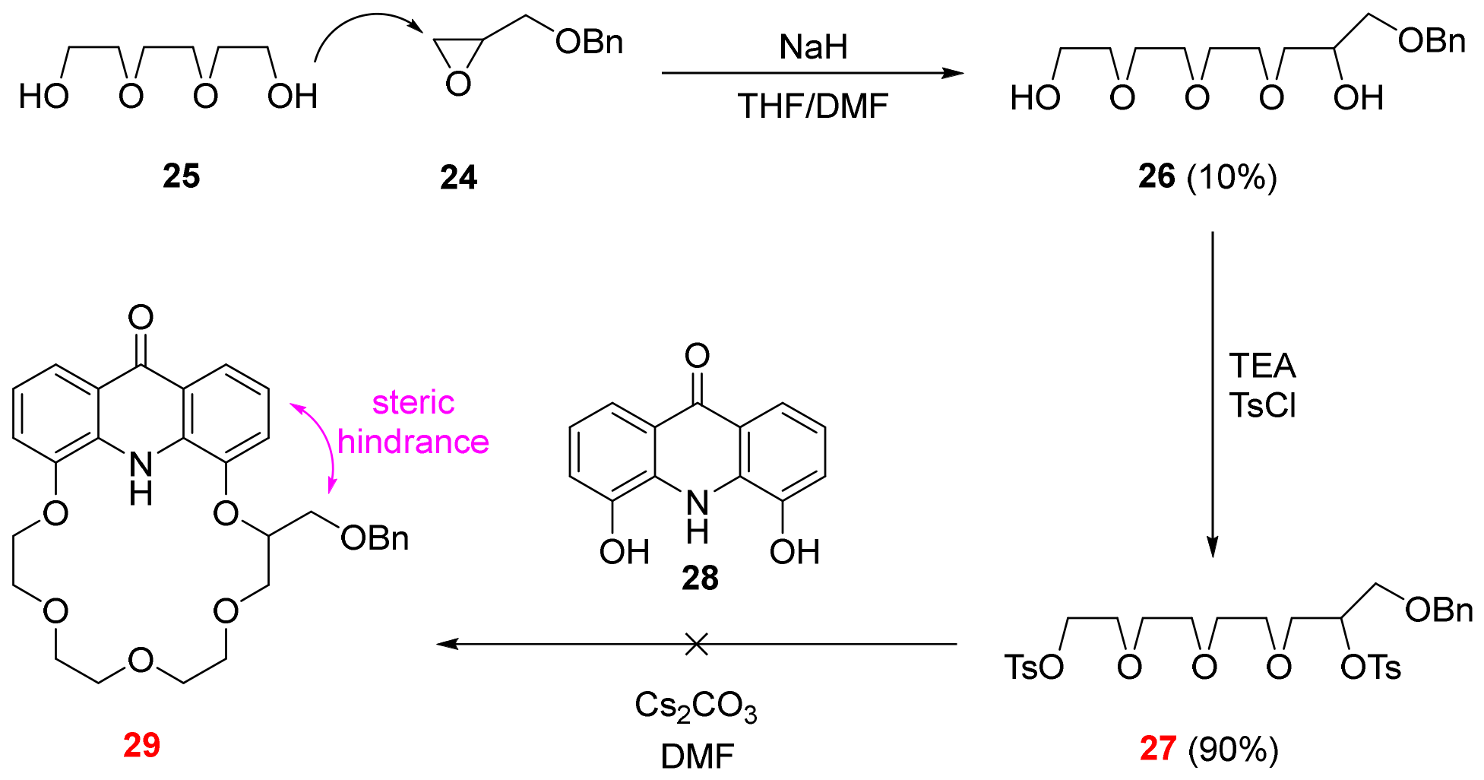
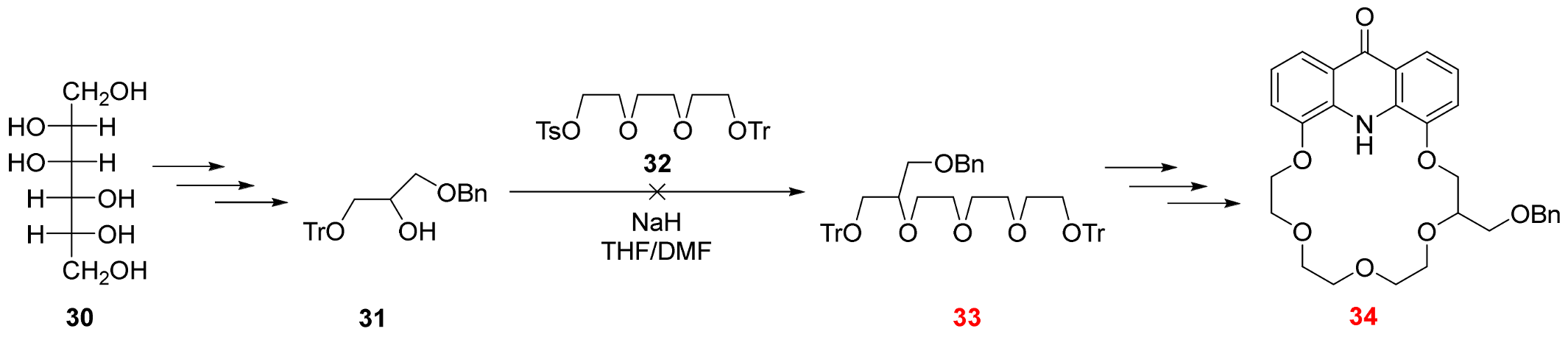
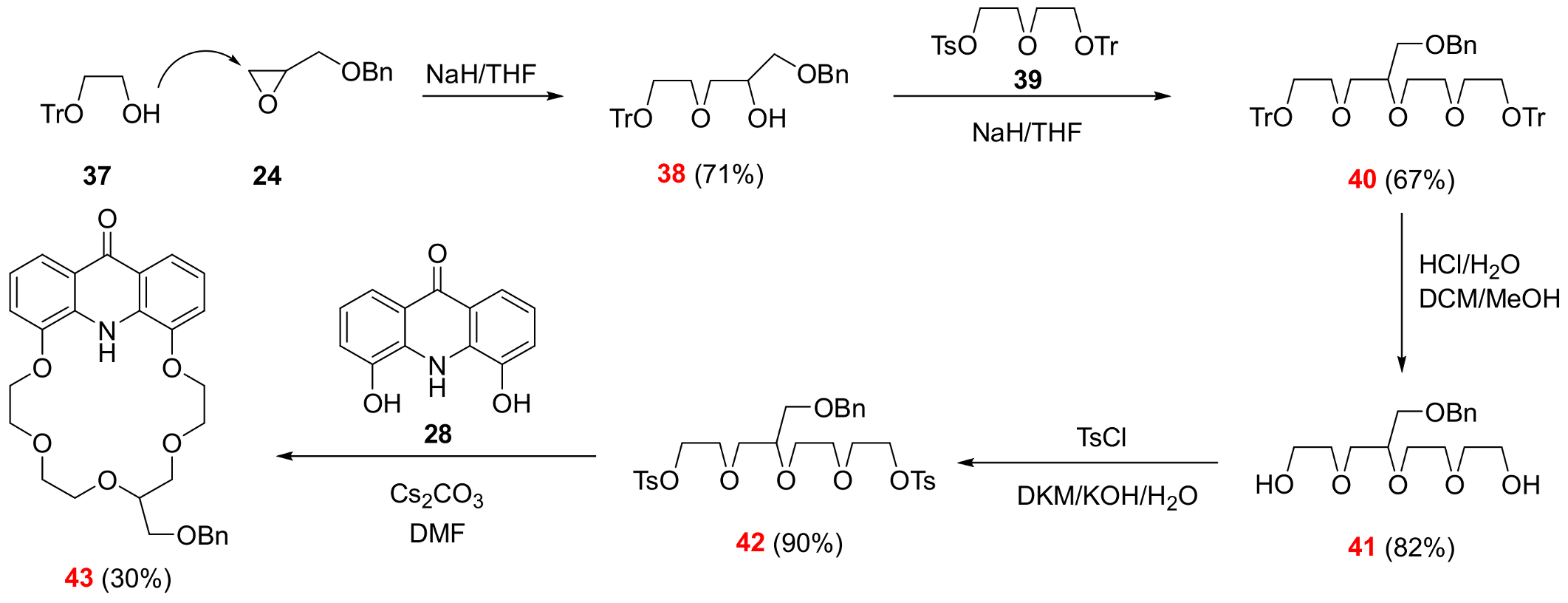
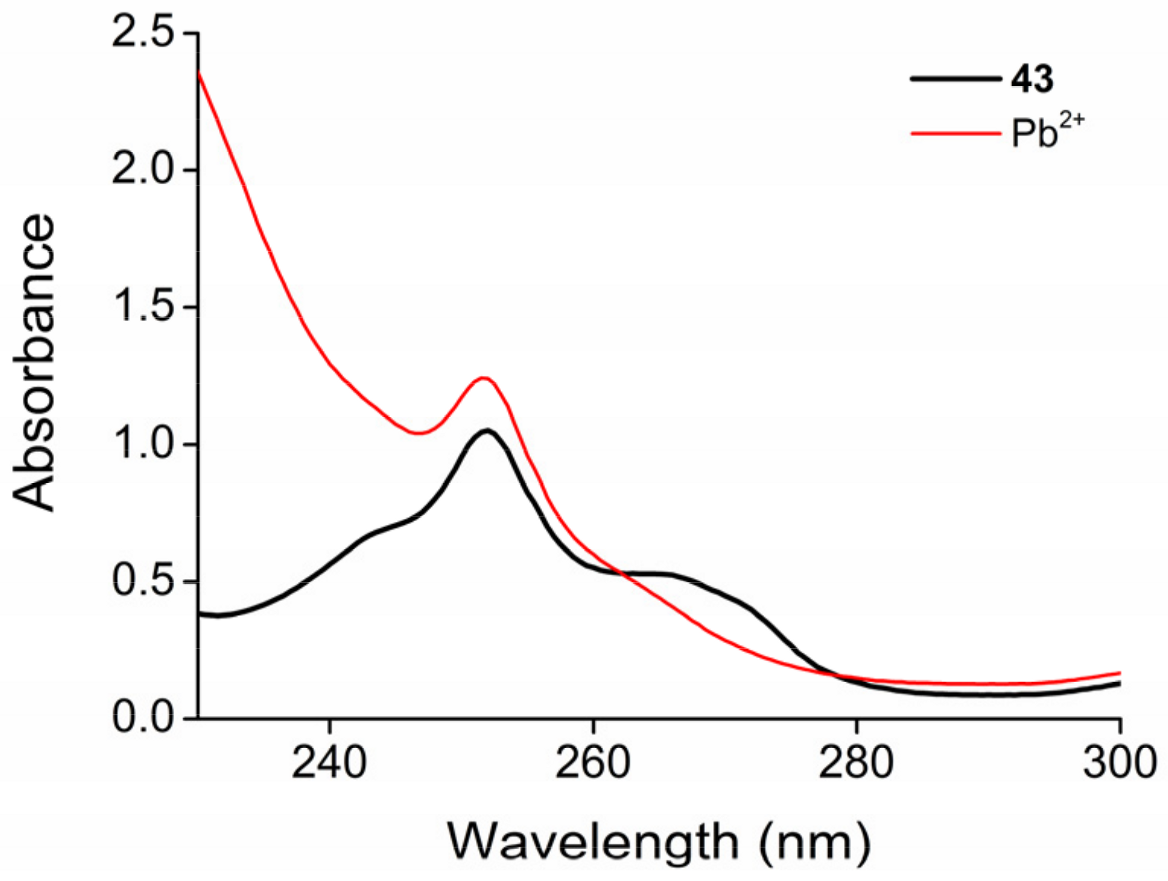
2.3. Functionalization via the Oligoether Unit (Approach 2)
3. Experimental Section
3.1. General
3.2. Synthesis of the New Compounds
3.2.1. 27-[4-(Diethoxymethyl)phenyl]-6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaene (See 16 in Scheme 1)
3.2.2. 4-{6,9,12,15,18-Pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl}benzaldehyde (See 18 in Scheme 1)
3.2.3. 6,9,12,15,18-Pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl trifluoromethanesulfonate (See 19 in Scheme 2)
3.2.4. Methyl 4-(2-{6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl}ethynyl)benzoate (See 21 in Scheme 2)
3.2.5. 27-Azido-6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaene (See 22 in Scheme 2)
3.2.6. Methyl 4-(1-{6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23(27),24-heptaen-27-yl}-1H-1,2,3-triazol-4-yl)benzoate (See 23 in Scheme 2)
3.2.7. 4,14-Bis[(4-methylbenzenesulfonyl)oxy]-1-phenyl-2,6,9,12-tetraoxatetradecane (See 27 in Scheme 3)
3.2.8. General Procedure for Macrocyclizations
3.2.9. 1-(Benzyloxy)-3-(triphenylmethoxy)propan-2-yl 4-methylbenzene-1-sulfonate (See 35 in Scheme 5)
3.2.10. ({2-[3-(Benzyloxy)-2-hydroxypropoxy]ethoxy}diphenylmethyl)benzene (See 38 in Scheme 6)
3.2.11. 7-[(Benzyloxy)methyl]-1,1,1,15,15,15-hexaphenyl-2,5,8,11,14-pentaoxapentadecane (See 40 in Scheme 6)
3.2.12. 2-[3-(Benzyloxy)-2-[2-(2-hydroxyethoxy)ethoxy]propoxy]ethan-1-ol (See 41 in Scheme 6)
3.2.13. 1-({2-[3-(Benzyloxy)-2-(2-{2-[(4-methylbenzenesulfonyl)oxy]ethoxy}ethoxy)propoxy]ethoxy}sulfonyl)-4-methylbenzene (See 42 in Scheme 6)
3.2.14. 13-[(Benzyloxy)methyl]-6,9,12,15,18-pentaoxa-25-azatetracyclo[21.3.1.05,26.019,24]heptacosa-1,3,5(26),19,21,23-hexaen-27-one (See 43 in Scheme 6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gokel, G.W.; Leevy, W.M.; Weber, M.E. Crown ethers: Sensors for ions and molecular scaffolds for materials and biological models. Chem. Rev. 2004, 104, 2723–2750. [Google Scholar] [CrossRef] [PubMed]
- Daicel Chiral Technologies|BioPharma & Academic Research. Available online: https://chiraltech.com/ (accessed on 4 January 2024).
- IBC Advanced Technologies–Molecular Recognition Technology™. Available online: https://ibcmrt.com/ (accessed on 4 January 2024).
- Cerón-Camacho, R.; Cisneros-Dévora, R.; Soto-Castruita, E.; Ramírez-Pérez, J.F.; Martínez-Magadán, J.M.; Oviedo-Roa, R.; Zamudio-Rivera, L.S. Greener process to prepare and scale-up zwitterionic macro cyclic dihydroxy-aza-crown ether and their corresponding supramolecular pairs to enhanced oil recovery application. J. Clean. Prod. 2023, 420, 138446. [Google Scholar] [CrossRef]
- Nicoli, F.; Baroncini, M.; Silvi, S.; Groppi, J.; Credi, A. Direct synthetic routes to functionalised crown ethers. Org. Chem. Front. 2021, 8, 5531–5549. [Google Scholar] [CrossRef] [PubMed]
- Çiçek, B.; Yıldız, A. Synthesis, metal ion complexation and computational studies of thio oxocrown ethers. Molecules 2011, 16, 8670–8683. [Google Scholar] [CrossRef]
- Tsuge, K.; Lim, L.W.; Takeuchi, T. Separation of inorganic anions using an 18-crown-6-ether-modified organic polymer monolithic stationary phase in capillary ion chromatography. Anal. Sci. 2021, 37, 845–850. [Google Scholar] [CrossRef]
- Hu, C.; Li, Z.; Hu, Z.; Li, Q.; Fu, Y.; Chen, Z. Synthesis of multifunctional crown ether covalent organic nanospheres as stationary phase for capillary electrochromatography. J. Chromatogr. A 2022, 1677, 463323. [Google Scholar] [CrossRef]
- Duenas-Ramirez, P.; Bertagnolli, C.; Weiss, R.; Bizeau, J.; Jierry, L.; Choquet, P.; Zaloszyc, A.; Bégin-Colin, S.; Mertz, D. Grafting of crown ether and cryptand macrocycles on large pore stellate mesoporous silica for sodium cation extraction. Molecules 2023, 28, 4622. [Google Scholar] [CrossRef] [PubMed]
- Buriac, O.; Ciopec, M.; Duţeanu, N.; Negrea, A.; Negrea, P.; Grozav, I. Platinum(IV) recovery from waste solutions by adsorption onto dibenzo-30-crown-10 ether immobilized on Amberlite XAD7 resin–factorial design analysis. Molecules 2020, 25, 3692. [Google Scholar] [CrossRef]
- Varga, B.; Ujj, D.; Mátravölgyi, B.; Szolnoki, B.; Koczka, B.; Rapi, Z. Reusable glucose-based crown ethers anchored to PVC. Molecules 2023, 28, 7905. [Google Scholar] [CrossRef]
- Mikhelson, K.N.; Peshkova, M.A. Advances and trends in ionophore-based chemical sensors. Russ. Chem. Rev. 2015, 84, 555. [Google Scholar] [CrossRef]
- Lerchi, M.; Bakker, E.; Rusterholz, B.; Simon, W. Lead-selective bulk optodes based on neutral ionophores with subnanomolar detection limits. Anal. Chem. 1992, 64, 1534–1540. [Google Scholar] [CrossRef]
- Mudipalli, A. Lead hepatotoxicity & potential health effects. Indian J. Med. Res. 2007, 126, 518–527. [Google Scholar]
- Flora, S.J.; Flora, G.; Saxena, G. Environmental occurrence, health effects and management of lead poisoning. In Lead; Casas, J.S., Sordo, J., Eds.; Elsevier Science BV: Amsterdam, The Netherlands, 2006; pp. 158–228. [Google Scholar] [CrossRef]
- Needleman, H. Lead poisoning. Annu. Rev. Med. 2004, 55, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Needleman, H.L.; Bellinger, D. The health effects of low level exposure to lead. Annu. Rev. Public. Health 1991, 12, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Huszthy, P.; Köntös, Z.; Vermes, B.; Pintér, Á. Synthesis of novel fluorescent acridono-and thioacridono-18-crown-6 ligands. Tetrahedron 2001, 57, 4967–4975. [Google Scholar] [CrossRef]
- Szalay, L.; Farkas, V.; Vass, E.; Hollósi, M.; Móczár, I.; Pintér, Á.; Huszthy, P. Synthesis and selective lead(II) binding of achiral and enantiomerically pure chiral acridono-18-crown-6 ether type ligands. Tetrahedron Asymm. 2004, 15, 1487–1493. [Google Scholar] [CrossRef]
- Németh, T.; Tóth, T.; Balogh, G.T.; Huszthy, P. Synthesis and fluorescence spectroscopic studies of novel 9-phenylacridino-18-crown-6 ether type sensor molecules. Period. Polytech. Chem. Eng. 2017, 61, 249–257. [Google Scholar] [CrossRef]
- Golcs, Á.; Horváth, V.; Huszthy, P.; Tóth, T. Fast potentiometric analysis of lead in aqueous medium under competitive conditions using an acridono-crown ether neutral ionophore. Sensors 2018, 18, 1407. [Google Scholar] [CrossRef]
- Bühlmann, P.; Pretsch, E.; Bakker, E. Carrier-based ion-selective electrodes and bulk optodes. 2. Ionophores for potentiometric and optical sensors. Chem. Rev. 1998, 98, 1593–1688. [Google Scholar] [CrossRef]
- Kwon, J.Y.; Jang, Y.J.; Lee, Y.J.; Kim, K.M.; Seo, M.S.; Nam, W.; Yoon, J. A highly selective fluorescent chemosensor for Pb2+. J. Am. Chem. Soc. 2005, 127, 10107–10111. [Google Scholar] [CrossRef]
- Wilson, D.; de los Ángeles Arada, M.; Alegret, S.; del Valle, M. Lead(II) ion selective electrodes with PVC membranes based on two bis-thioureas as ionophores: 1,3-Bis(N′-benzoylthioureido)benzene and 1,3-bis(N′-furoylthioureido)benzene. J. Hazard. Mater. 2010, 181, 140–146. [Google Scholar] [CrossRef]
- Golcs, Á.; Ádám, B.Á.; Vezse, P.; Huszthy, P.; Tóth, T. Synthesis and spectrophotometric studies of 9-substituted-4,5-dimethoxyacridine multifunctionalizable fluorescent dyes and their macrocyclic derivatives. Eur. J. Org. Chem. 2021, 2021, 2485–2497. [Google Scholar] [CrossRef]
- Meng, J.; Pang, M.; Niu, Y.; Zhou, Y.; Wu, B.; Bian, J.; Zhang, J.; Meng, Q. Method for Preparing Diaryl-Substituted Naphthopyran Photochromic Compound. Patent Number CN104557842, 29 April 2015. [Google Scholar]
- Ikeda, I.; Yamamura, S.; Nakatsuji, Y.; Okahara, M. Synthesis of substituted crown ethers from oligoethylene glycols. J. Org. Chem. 1980, 45, 5355–5358. [Google Scholar] [CrossRef]
- Amma, J.P.; Stille, J.K. Chiral 1,2-diphosphine ligands. Synthesis and application to rhodium-catalyzed asymmetric hydrogenations. J. Org. Chem. 1982, 47, 468–473. [Google Scholar] [CrossRef]
- Bouton, J.; Van Hecke, K.; Van Calenbergh, S. Efficient diastereoselective synthesis of a new class of azanucleosides: 2′-Homoazanucleosides. Tetrahedron 2017, 73, 4307–4316. [Google Scholar] [CrossRef] [PubMed]
- Bolchi, C.; Pallavicini, M.; Fumagalli, L.; Moroni, B.; Rusconi, C.; Valoti, E. Univocal syntheses of 2-and 3-hydroxymethyl-2,3-dihydro[1,4]dioxino[2,3-b]pyridine enantiomers. Tetrahedron Asymm. 2005, 16, 3380–3384. [Google Scholar] [CrossRef]
- Zhang, Q.; Ren, H.; Baker, G.L. Synthesis of a library of propargylated and PEGylated α-hydroxy acids toward “Clickable” polylactides via hydrolysis of cyanohydrin derivatives. J. Org. Chem. 2014, 79, 9546–9555. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Sarti, G.; Casnati, A.; Carrettoni, B.; Manet, I.; Schuurman, R.; Guardigli, M.; Sabbatini, N.; Ungaro, R. 2,2′-Bipyridine lariat calixcrowns: A new class of encapsulating ligands forming highly luminescent Eu3+ and Tb3+ complexes. Chem. Eur. J. 2000, 6, 1026–1034. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents: Physical Properties and Methods of Purification, 4th ed.; Wiley-Interscience: New York, NY, USA, 1986; pp. 1344–1400. ISBN 978-047108467-9. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vezse, P.; Gede, M.; Golcs, Á.; Huszthy, P.; Tóth, T. Synthetic Modifications of a Pb2+-Sensor Acridono-Crown Ether for Covalent Attachment and Their Effects on Selectivity. Molecules 2024, 29, 1121. https://doi.org/10.3390/molecules29051121
Vezse P, Gede M, Golcs Á, Huszthy P, Tóth T. Synthetic Modifications of a Pb2+-Sensor Acridono-Crown Ether for Covalent Attachment and Their Effects on Selectivity. Molecules. 2024; 29(5):1121. https://doi.org/10.3390/molecules29051121
Chicago/Turabian StyleVezse, Panna, Martin Gede, Ádám Golcs, Péter Huszthy, and Tünde Tóth. 2024. "Synthetic Modifications of a Pb2+-Sensor Acridono-Crown Ether for Covalent Attachment and Their Effects on Selectivity" Molecules 29, no. 5: 1121. https://doi.org/10.3390/molecules29051121
APA StyleVezse, P., Gede, M., Golcs, Á., Huszthy, P., & Tóth, T. (2024). Synthetic Modifications of a Pb2+-Sensor Acridono-Crown Ether for Covalent Attachment and Their Effects on Selectivity. Molecules, 29(5), 1121. https://doi.org/10.3390/molecules29051121