
Mechanistic Studies on Rhodium-Catalyzed Chemoselective Cycloaddition of Ene-Vinylidenecyclopropanes: Water-Assisted Proton Transfer


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Mechanistic Studies through Experiments
2.2. Two Proposed Reaction Pathways Investigated through DFT Calculations
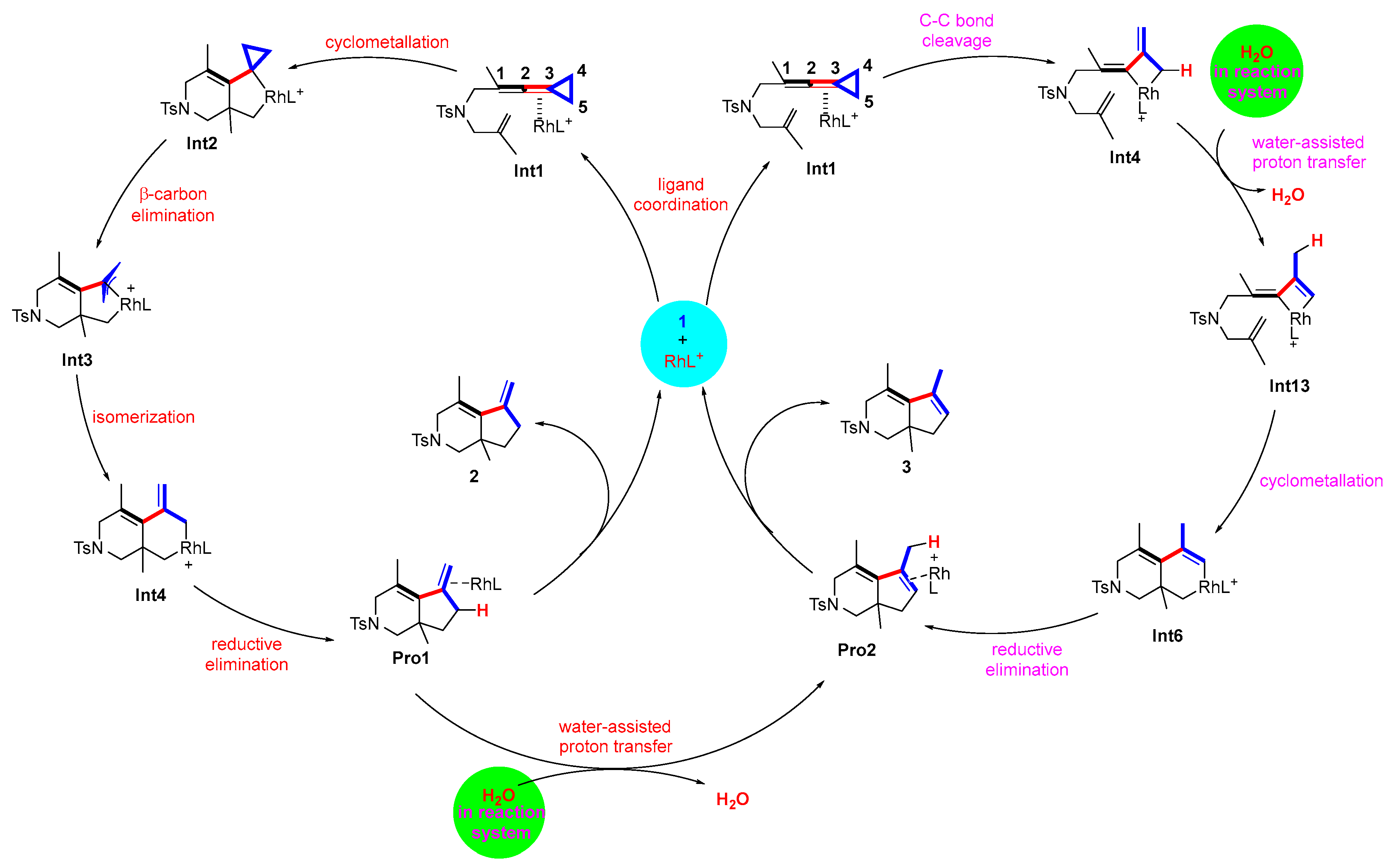
2.3. Theoretical Investigation of the Reaction Pathways Involving Water-Assisted Proton Transfer
3. Materials and Methods
3.1. General Information
3.2. Experimental Procedures for Control Experiments
3.2.1. Experimental Procedures for 1 to 2 with Standard Conditions
3.2.2. Experimental Procedures for 1 to 3 with Standard Conditions
3.2.3. Experimental Procedures for 2 to 3 without Catalysts and Ligands
3.2.4. Experimental Procedures for 2 to 3 with Standard Conditions
3.2.5. Experimental Procedures for 3 to 2 without Catalysts and Ligands
3.2.6. Experimental Procedures for 3 to 2 with Standard Conditions
3.3. Experimental Procedures for Control Experiments Involving Water
3.3.1. Experimental Procedures for 1 to 3 with Standard Conditions and Water
3.3.2. Experimental Procedures for 2 to 3 with Standard Conditions and Water
3.3.3. Experimental Procedures for 3 to 2 with Standard Conditions and Water
3.3.4. Experimental Procedures for 1 to 3 with Standard Conditions and 4Å Molecular Sieves
3.3.5. Experimental Procedures for 2 to 3 with Standard Conditions and 4Å Molecular Sieves
3.3.6. Experimental Procedures for 2 to 3 with HBF4
3.4. Experimental Procedures for Control Experiments to Monitor the Course of Reaction
3.4.1. Experimental Procedures for 1 to 3 with Standard Conditions
3.4.2. Experimental Procedures for 1 to 3 with Standard Conditions and Water
3.4.3. Experimental Procedures for 2 to 3 with Standard Conditions
3.4.4. Experimental Procedures for 2 to 3 with Standard Conditions and Water
3.5. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nakamura, I.; Yamamoto, Y. Transition Metal-Catalyzed Reactions of Methylenecyclopropanes. Adv. Synth. Catal. 2002, 344, 111–129. [Google Scholar] [CrossRef]
- Masarwa, A.; Marek, I. Selectivity in Metal-Catalyzed Carbon—Carbon Bond Cleavage of Alkylidenecyclopropanes. Chem. Eur. J. 2010, 16, 9712–9721. [Google Scholar] [CrossRef]
- Ackermann, L.; Kozhushkov, S.I.; Yufit, D.S. Ruthenium-Catalyzed Hydroarylation of Methylenecyclopropanes through C–H Bond Cleavage: Scope and Mechanism. Chem. Eur. J. 2012, 18, 12068–12077. [Google Scholar] [CrossRef]
- Zhang, D.-H.; Tang, X.-Y.; Shi, M. Gold-Catalyzed Tandem Reactions of Methylenecyclopropanes and Vinylidenecyclopropanes. Acc. Chem. Res. 2014, 47, 913–924. [Google Scholar] [CrossRef]
- Wang, F.; Yu, S.; Li, X. Transition Metal-Catalysed Couplings between Arenes and Strained or Reactive Rings: Combination of C–H Activation and Ring Scission. Chem. Soc. Rev. 2016, 45, 6462–6477. [Google Scholar] [CrossRef]
- Fumagalli, G.; Stanton, S.; Bower, J.F. Recent Methodologies That Exploit C–C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev. 2017, 117, 9404–9432. [Google Scholar] [CrossRef]
- Yang, S.; Shi, M. Recent Advances in Transition-Metal-Catalyzed/Mediated Transformations of Vinylidenecyclopropanes. Acc. Chem. Res. 2018, 51, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.A.; De, P.B.; Pradhan, S.; Banerjee, S.; Punniyamurthy, T. Exploiting Strained Rings in Chelation Guided C−H Functionalization: Integration of C−H Activation with Ring Cleavage. Chem. Asian J. 2019, 14, 4520–4533. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, R.; Wei, Y.; Shi, M. Recent Developments in Cyclopropane Cycloaddition Reactions. Trends Chem. 2019, 1, 779–793. [Google Scholar] [CrossRef]
- Pirenne, V.; Muriel, B.; Waser, J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2021, 121, 227–263. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, Y.; O’Brien, M.; Evans, P. Stereocontrolled Preparation of Bicyclic Alkaloid Analogues: An Approach towards the Kinabalurine Skeleton. Tetrahedron 2009, 65, 8259–8268. [Google Scholar] [CrossRef]
- Belhassen, E.; Filippi, J.-J.; Brévard, H.; Joulain, D.; Baldovini, N. Volatile Constituents of Vetiver: A Review: Volatile Constituents of Vetiver. Flavour Fragr. J. 2015, 30, 26–82. [Google Scholar] [CrossRef]
- Lecourt, M.; Antoniotti, S. Chemistry, Sustainability and Naturality of Perfumery Biotech Ingredients. ChemSusChem 2020, 13, 5600–5610. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Li, W.; Wang, X.; Su, G.; Li, M.; Zhang, J.-P.; Jia, H.-L.; Li, Y.-H.; Wang, R.-B.; Gan, M.; et al. Illihenin A: An Antiviral Sesquiterpenoid with a Cage-like Tricyclo[6.2.2.01,5]dodecane Skeleton from Illicium henryi. J. Org. Chem. 2021, 86, 2017–2022. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Li, C.-C. Synthesis of Natural Products Containing Highly Strained Trans-Fused Bicyclo[3.3.0]Octane: Historical Overview and Future Prospects. Chem. Soc. Rev. 2021, 50, 9430–9442. [Google Scholar] [CrossRef]
- Cui, S.; Zhang, Y.; Wu, Q. Rh(III)-Catalyzed C–H Activation/Cycloaddition of Benzamides and Methylenecyclopropanes: Divergence in Ring Formation. Chem. Sci. 2013, 4, 3421–3426. [Google Scholar] [CrossRef]
- Liu, C.-H.; Yu, Z.-X. Rhodium(I)-Catalyzed Bridged [5 + 2] Cycloaddition of Cis-Allene-Vinylcyclopropanes to Synthesize the Bicyclo[4.3.1]Decane Skeleton. Angew. Chem. Int. Ed. 2017, 56, 8667–8671. [Google Scholar] [CrossRef]
- Su, Y.; Inglesby, P.A.; Evans, P.A. Intramolecular Thioether Migration in the Rhodium-Catalyzed Ene-Cycloisomerization of Alkenylidenecyclopropanes by a Metal-Mediated β-Sulfide Elimination. Angew. Chem. Int. Ed. 2018, 57, 673–677. [Google Scholar] [CrossRef]
- Feng, S.; Wang, K.; Ping, Y.; Wang, J. Experimental and Computational Studies on Rh(I)-Catalyzed Reaction of Siloxyvinylcyclopropanes and Diazoesters. J. Am. Chem. Soc. 2020, 142, 21032–21039. [Google Scholar] [CrossRef]
- Yu, H.; Lu, Q.; Dang, Z.; Fu, Y. Mechanistic Study of the Rhodium-Catalyzed [3 + 2 + 2] Carbocyclization of Alkenylidenecyclopropanes with Alkynes. Chem. Asian J. 2013, 8, 2262–2273. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, T.; Xia, Y. Mechanistic Understanding of the Aryl-Dependent Ring Formations in Rh(III)-Catalyzed C–H Activation/Cycloaddition of Benzamides and Methylenecyclopropanes by DFT Calculations. Organometallics 2015, 34, 3012–3020. [Google Scholar] [CrossRef]
- Wang, T.; Lv, S.; Guo, X.; Li, Z.; Li, J. Rhodium-Catalyzed Ene-Cycloisomerization of Allylic-Sulfide-Tethered Alkylidenecyclopropanes: DFT Analysis of Origins of Regio- and Diastereo-Selectivities. Org. Chem. Front. 2020, 7, 678–688. [Google Scholar] [CrossRef]
- Rui, K.-H.; Yang, S.; Wei, Y.; Shi, M. Rh(I)-Catalyzed Stereoselective Intramolecular Cycloaddition Reactions of Ene-Vinylidenecyclopropanes for the Construction of Fused 6,5-Bicyclic Skeletons with a Quaternary All-Carbon Stereocenter. Org. Chem. Front. 2019, 6, 2506–2513. [Google Scholar] [CrossRef]
- Xia, Y.; Liang, Y.; Chen, Y.; Wang, M.; Jiao, L.; Huang, F.; Liu, S.; Li, Y.; Yu, Z.-X. An Unexpected Role of a Trace Amount of Water in Catalyzing Proton Transfer in Phosphine-Catalyzed (3 + 2) Cycloaddition of Allenoates and Alkenes. J. Am. Chem. Soc. 2007, 129, 3470–3471. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wei, Y.; Shi, M. Thermally-Induced Intramolecular [4+2] Cycloaddition of Allylamino- or Allyloxy-Tethered Alkylidenecyclopropanes. Chem. Asian J. 2021, 16, 2463–2468. [Google Scholar] [CrossRef]
- Qiu, Y.; Fu, C.; Zhang, X.; Ma, S. Studies on [PtCl2]− or [AuCl]− Catalyzed Cyclization of 1-(Indol-2-yl)-2,3-Allenols: The Effects of Water/Steric Hindrance and 1,2-Migration Selectivity. Chem. Eur. J. 2014, 20, 10314–10322. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, J. [DBU-H]+ and H2O as Effective Catalyst Form for 2,3-Dihydropyrido[2,3-d]Pyrimidin-4(1H)-Ones: A DFT Study. J. Comput. Chem. 2015, 36, 1295–1303. [Google Scholar] [CrossRef]
- Li, G.; Wang, B.; Resasco, D.E. Water-Mediated Heterogeneously Catalyzed Reactions. ACS Catal. 2020, 10, 1294–1309. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, S.; Xia, Y.; Li, Y.; Yu, Z.-X. Mechanism, Regioselectivity, and the Kinetics of Phosphine-Catalyzed [3+2] Cycloaddition Reactions of Allenoates and Electron-Deficient Alkenes. Chem. Eur. J. 2008, 14, 4361–4373. [Google Scholar] [CrossRef]
- Cheng, Q.; Yan, W.; Li, T.; Jiao, Y.; Tang, Z. Insights into the Regioselectivity and Diastereoselectivity of the Nazarov Cyclization of 3-Alkenyl-2-Indolylmethanol with Tryptophol. Org. Chem. Front. 2023, 10, 1721–1730. [Google Scholar] [CrossRef]
- Castle, L.; Honeybone, C.A.; Jickells, S.M.; Philo, M.R.; Sharman, M. Practical Aspects of Testing Food Contact Materials for Migration. Food Addit. Contam. 1994, 11, 177–185. [Google Scholar] [CrossRef]
- Decaro, C.; Ruegg, K.; Deagostini, A. Coulometric Karl Fischer Titration with a Diaphragm-Free Cell: Cell Design and Applications. Food Chem. 2006, 96, 431–435. [Google Scholar] [CrossRef]
- Tan, Y.S.; Chen, S.; Hong, W.M.; Kan, J.M.; Kwek, E.S.H.; Lim, S.Y.; Lim, Z.H.; Tessensohn, M.E.; Zhang, Y.; Webster, R.D. The Role of Low Levels of Water in the Electrochemical Oxidation of α-Tocopherol (Vitamin E) and Other Phenols in Acetonitrile. Phys. Chem. Chem. Phys. 2011, 13, 12745–12754. [Google Scholar] [CrossRef]
- Zhao, D.; Zhu, B.; Li, L.; Liu, X.; Wen, L.; Song, Y.; Shen, H.; Li, M.; Li, X.; Wu, D. A Review of Methods for Measuring Oil Moisture. Measurement 2023, 217, 113119–113131. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; Version 16; Gaussian Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Boese, A.D.; Martin, J.M.L. Development of Density Functionals for Thermochemical Kinetics. J. Chem. Phys. 2004, 121, 3405–3416. [Google Scholar] [CrossRef] [PubMed]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liao, W.; Wang, Y.; Jiao, L.; Yu, Z.-X. Mechanism and Stereochemistry of Rhodium-Catalyzed [5 + 2 + 1] Cycloaddition of Ene–Vinylcyclopropanes and Carbon Monoxide Revealed by Visual Kinetic Analysis and Quantum Chemical Calculations. J. Am. Chem. Soc. 2022, 144, 2624–2636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-Y.; Zhang, P.; Li, B.-W.; Liu, K.; Li, J.; Yu, Z.-X. Dual Activation Strategy to Achieve C–C Cleavage of Cyclobutanes: Development and Mechanism of Rh and Zn Cocatalyzed [4 + 2] Cycloaddition of Yne-Vinylcyclobutanones. J. Am. Chem. Soc. 2022, 144, 21457–21469. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Legault, C.Y. CYLView, 1.0b; Université de Sherbrooke: Montreal, QC, Canada, 2009; Available online: http://www.cylview.org (accessed on 19 January 2024).
- Wang, Q.; Tang, X.-Y.; Shi, M. Metal-Free Cross-Coupling of Arylboronic Acids and Derivatives with DAST-Type Reagents for Direct Access to Diverse Aromatic Sulfinamides and Sulfonamides. Angew. Chem. Int. Ed. 2016, 55, 10811–10815. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.; Shi, M.; Wei, Y. Mechanistic Studies on Rhodium-Catalyzed Chemoselective Cycloaddition of Ene-Vinylidenecyclopropanes: Water-Assisted Proton Transfer. Molecules 2024, 29, 1085. https://doi.org/10.3390/molecules29051085
Yu Z, Shi M, Wei Y. Mechanistic Studies on Rhodium-Catalyzed Chemoselective Cycloaddition of Ene-Vinylidenecyclopropanes: Water-Assisted Proton Transfer. Molecules. 2024; 29(5):1085. https://doi.org/10.3390/molecules29051085
Chicago/Turabian StyleYu, Ziqi, Min Shi, and Yin Wei. 2024. "Mechanistic Studies on Rhodium-Catalyzed Chemoselective Cycloaddition of Ene-Vinylidenecyclopropanes: Water-Assisted Proton Transfer" Molecules 29, no. 5: 1085. https://doi.org/10.3390/molecules29051085
APA StyleYu, Z., Shi, M., & Wei, Y. (2024). Mechanistic Studies on Rhodium-Catalyzed Chemoselective Cycloaddition of Ene-Vinylidenecyclopropanes: Water-Assisted Proton Transfer. Molecules, 29(5), 1085. https://doi.org/10.3390/molecules29051085