
Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane


Abstract
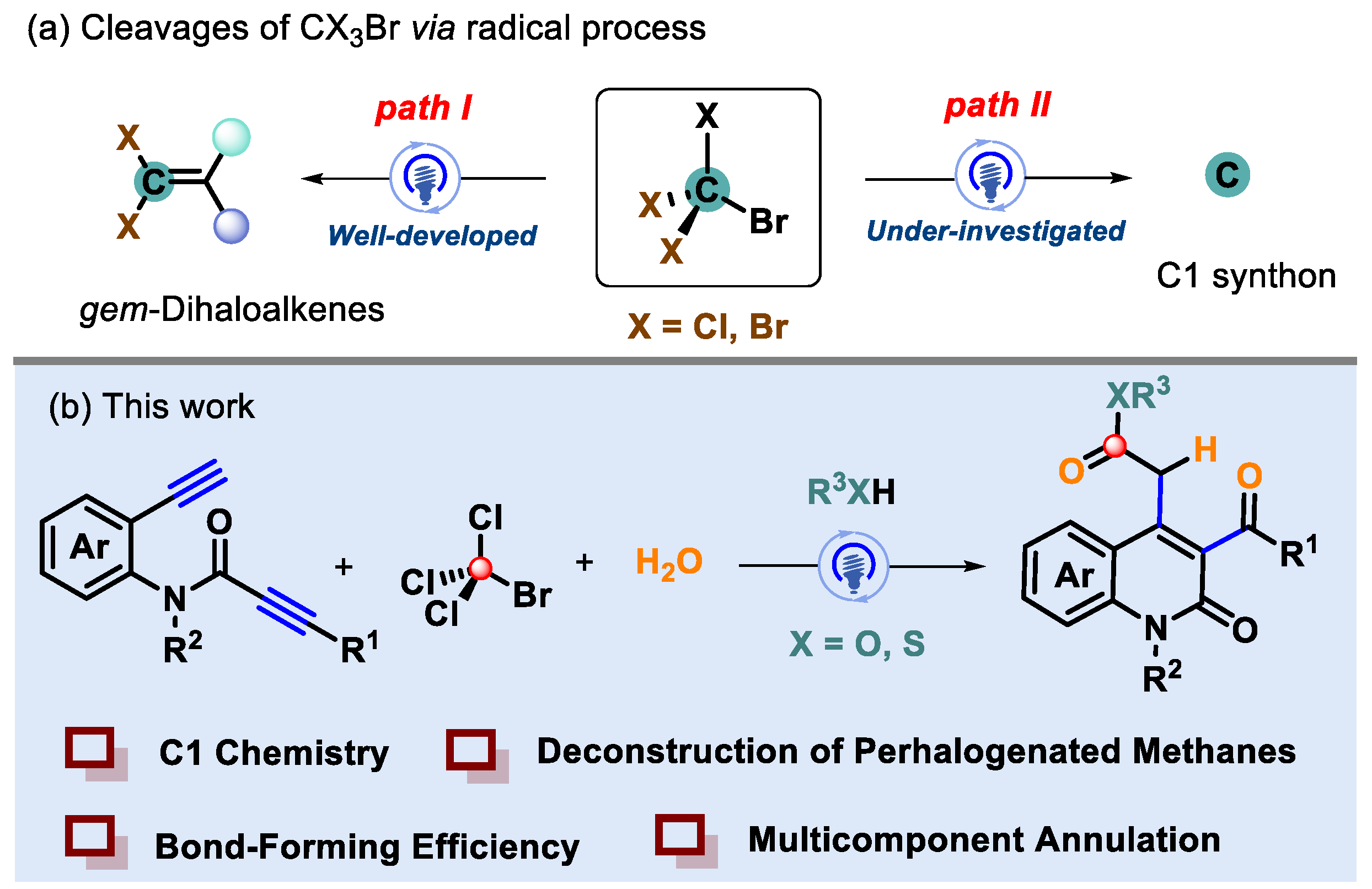
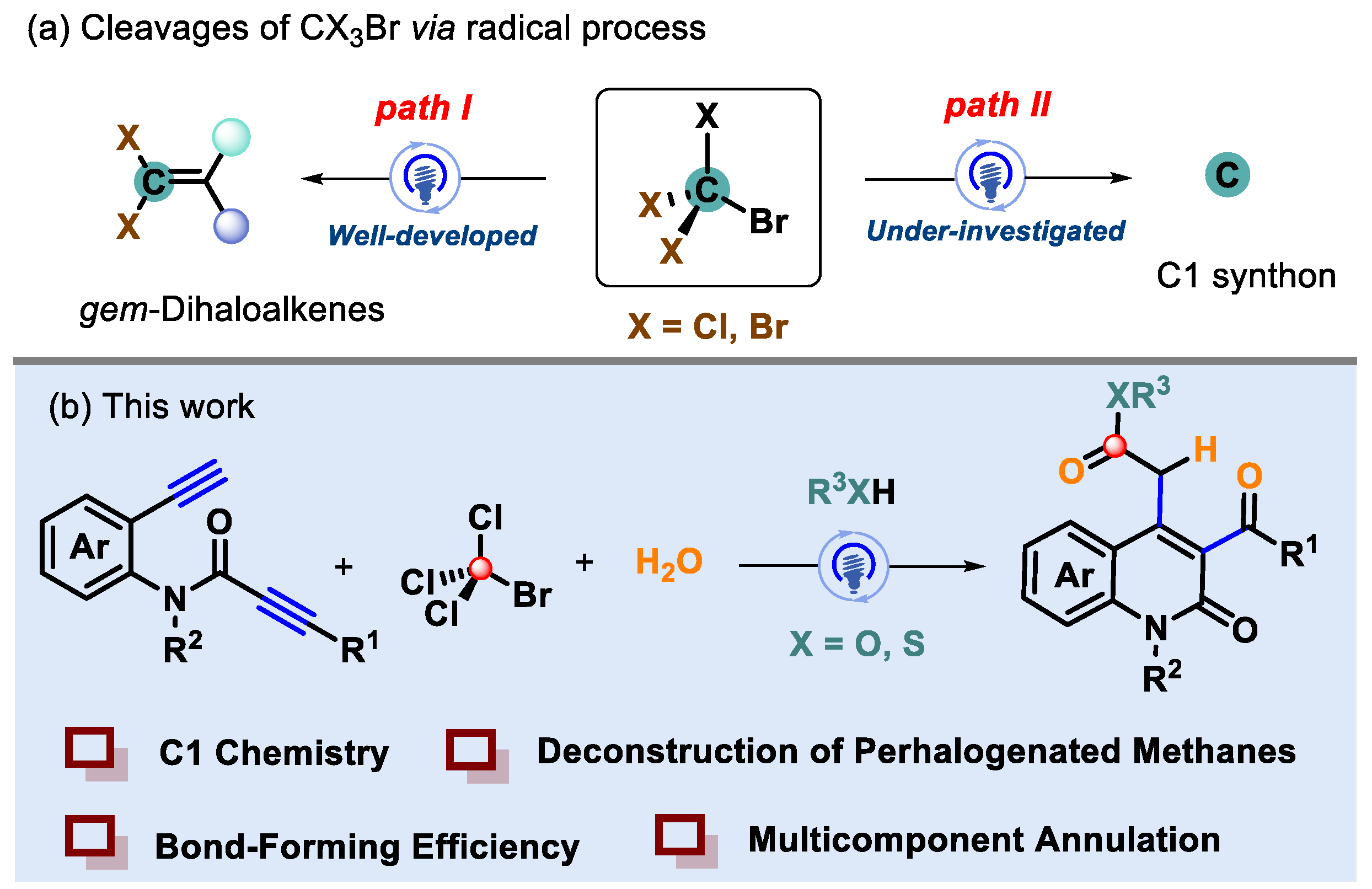
1. Introduction
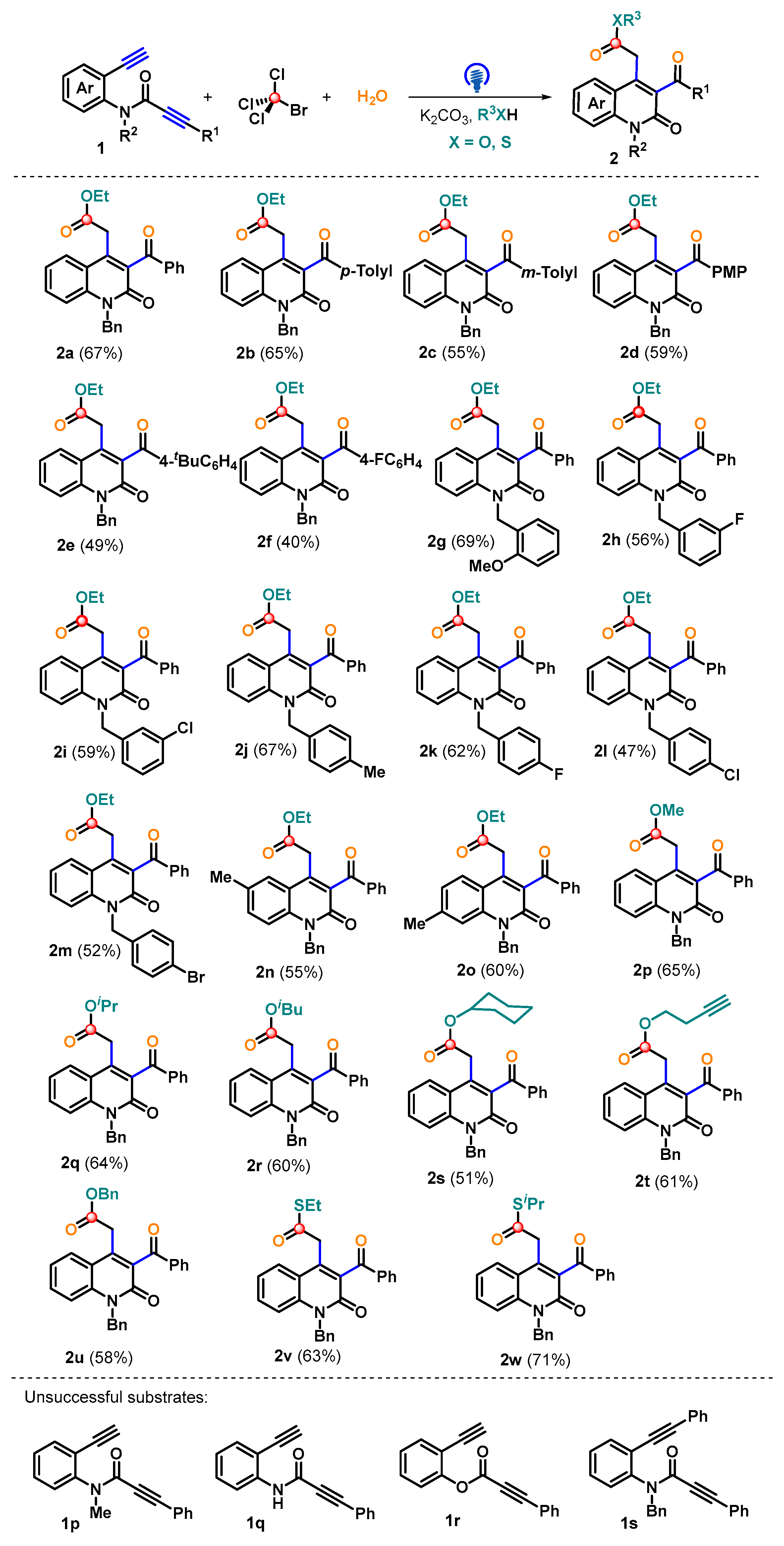
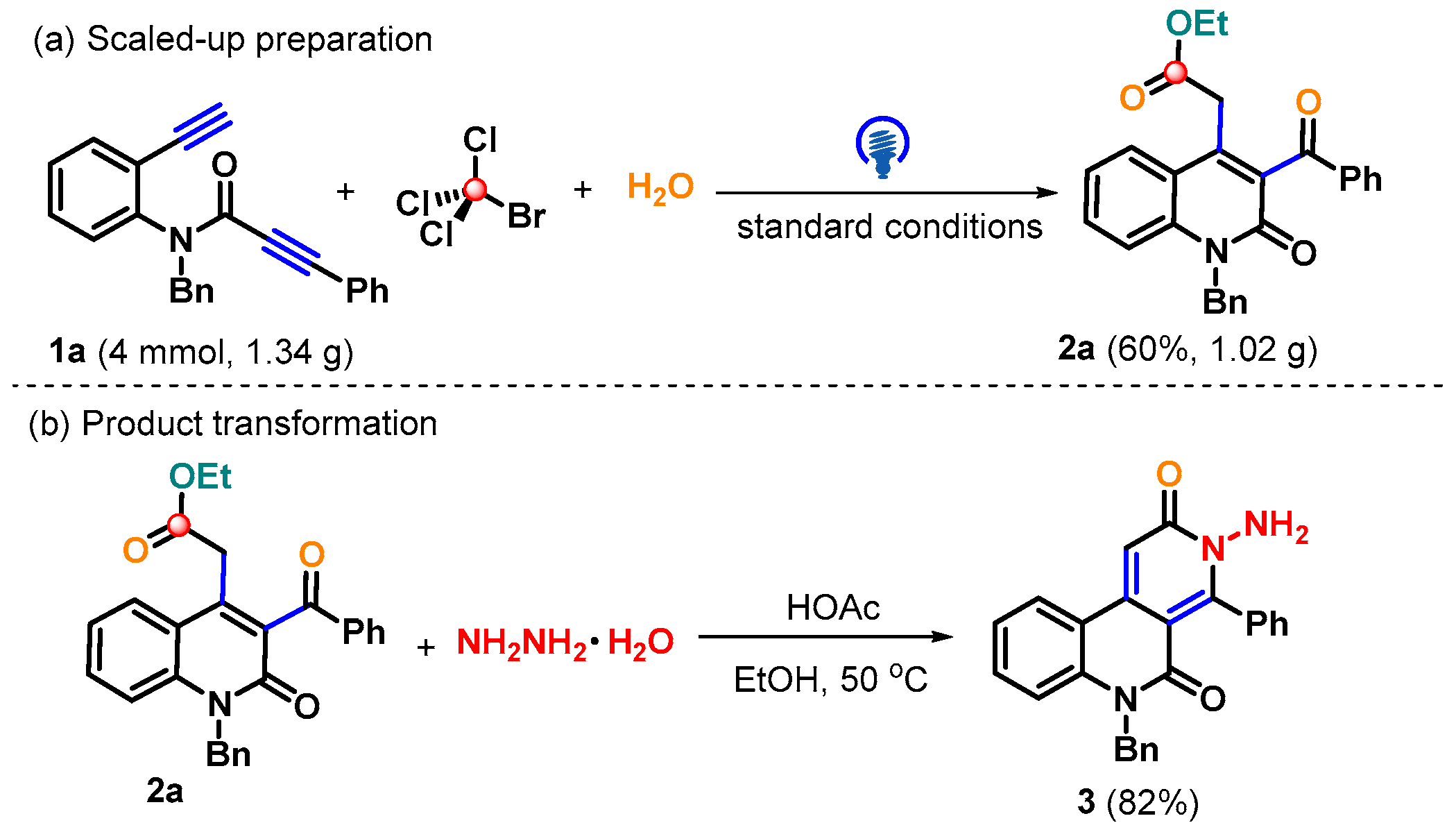
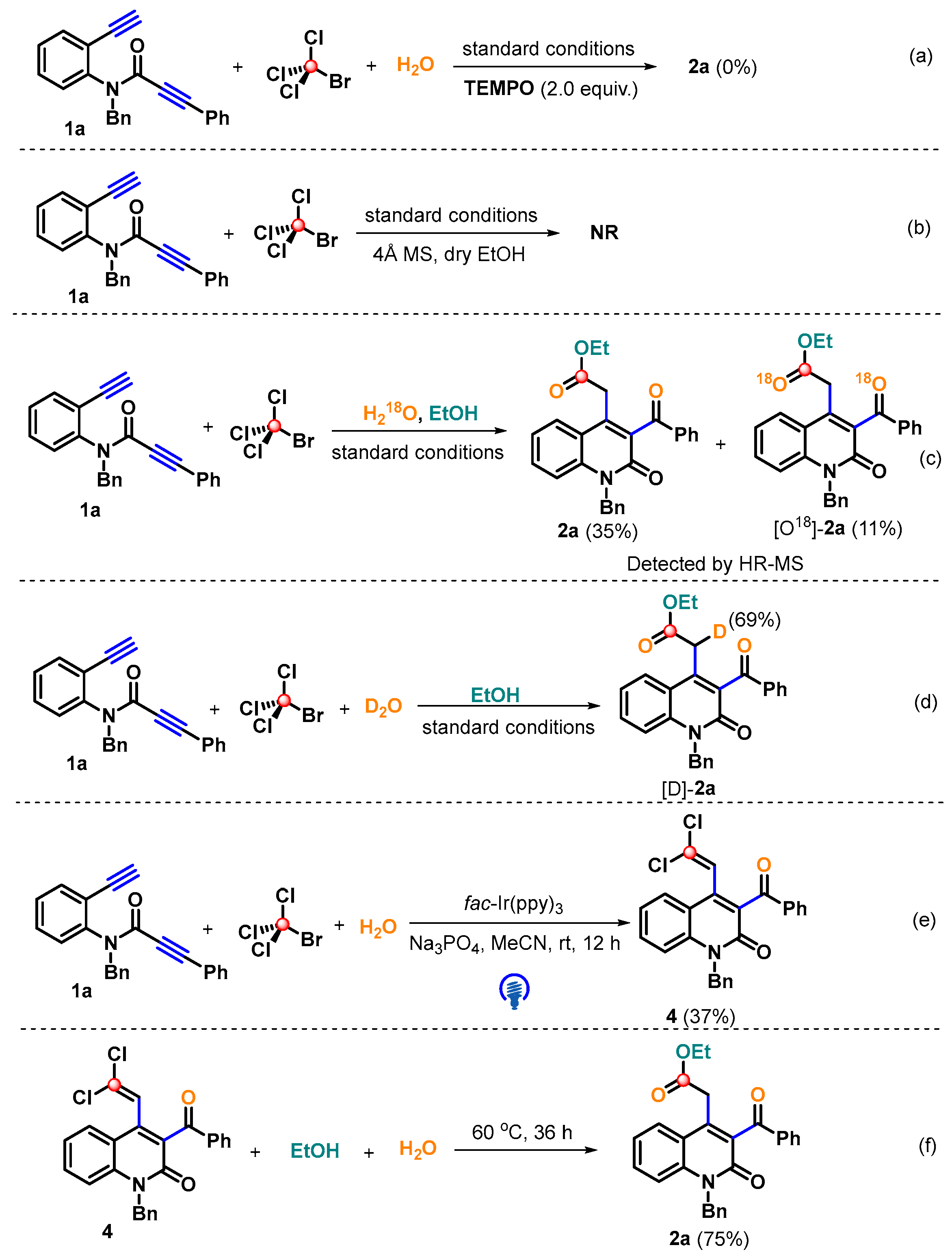
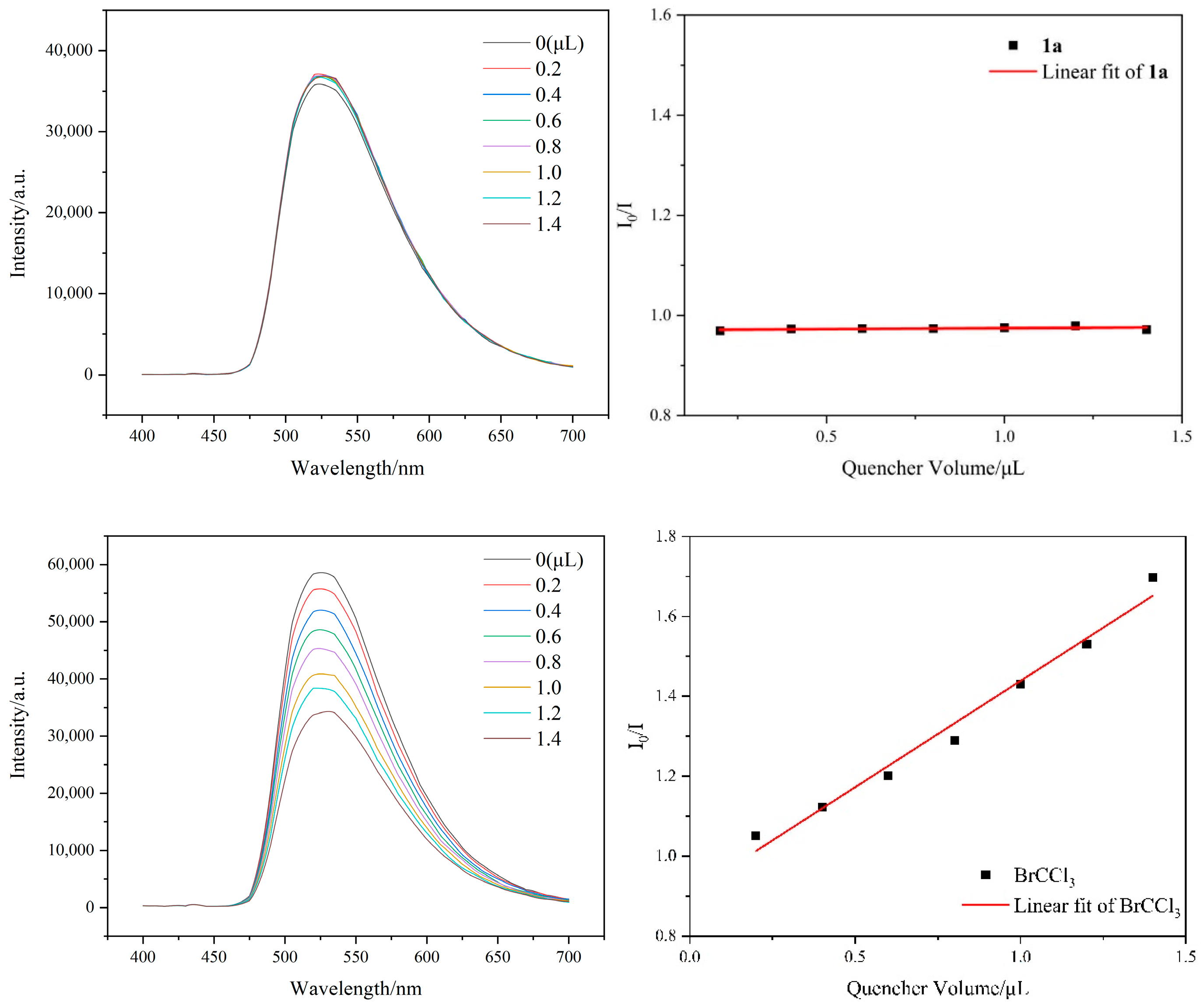
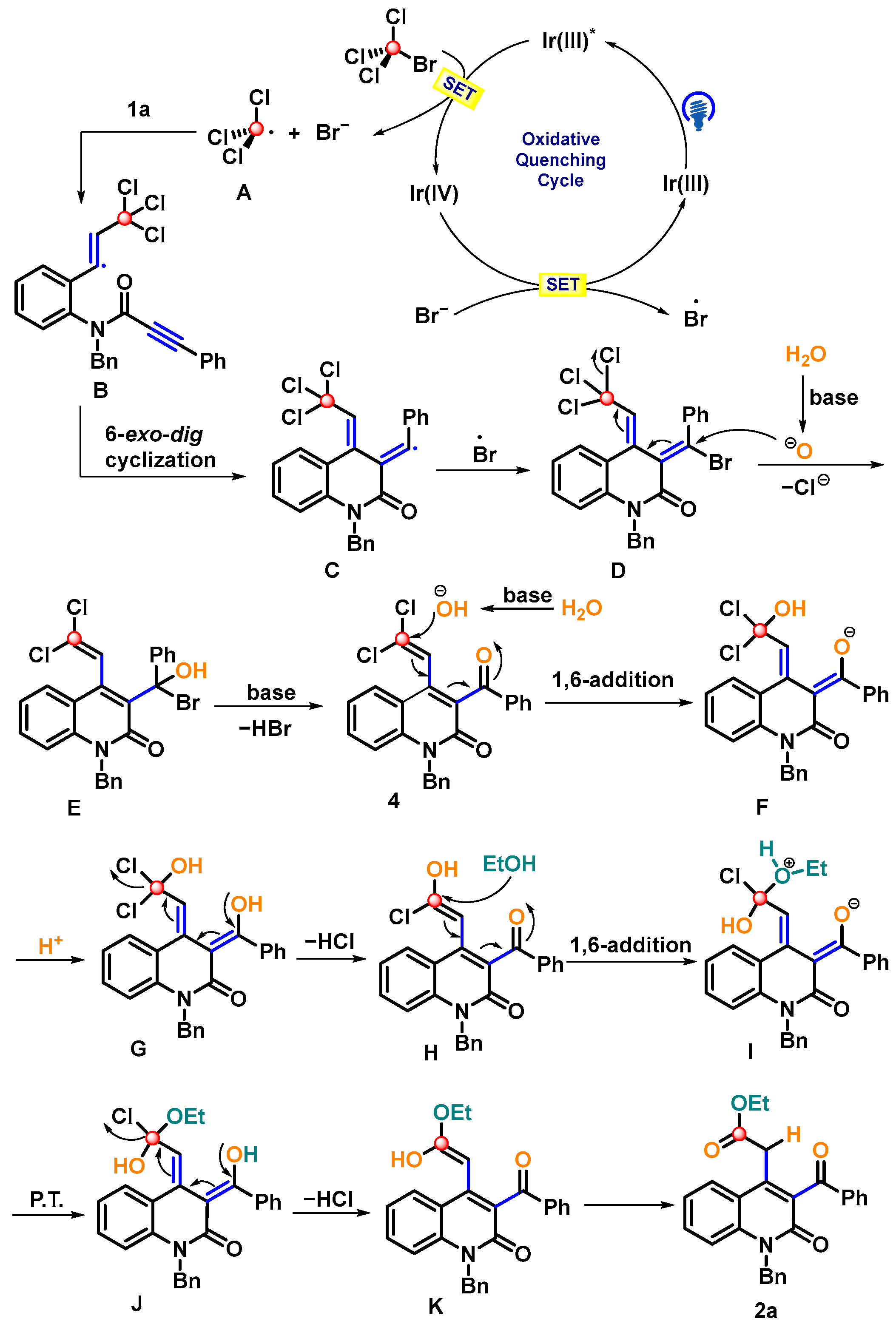
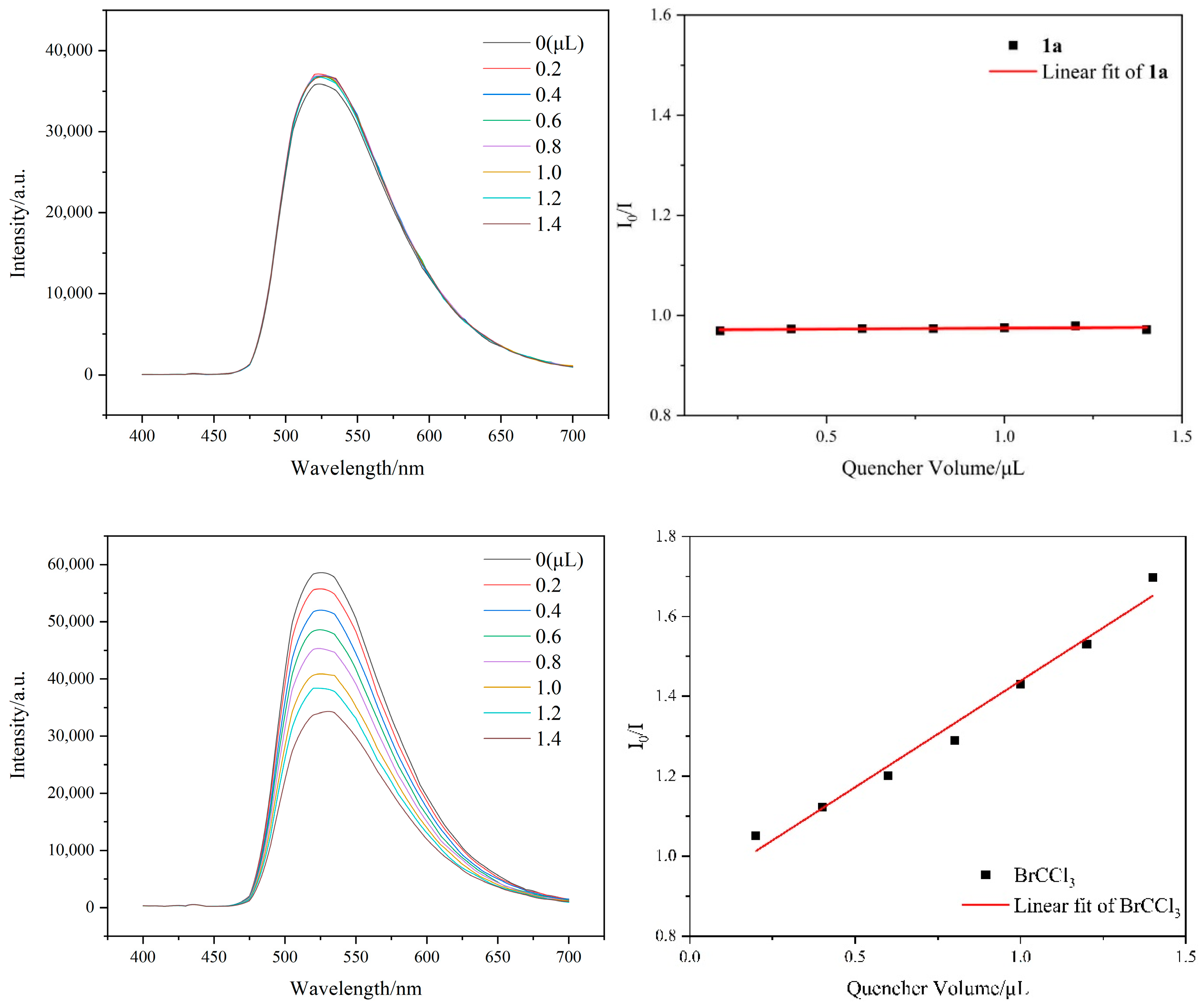
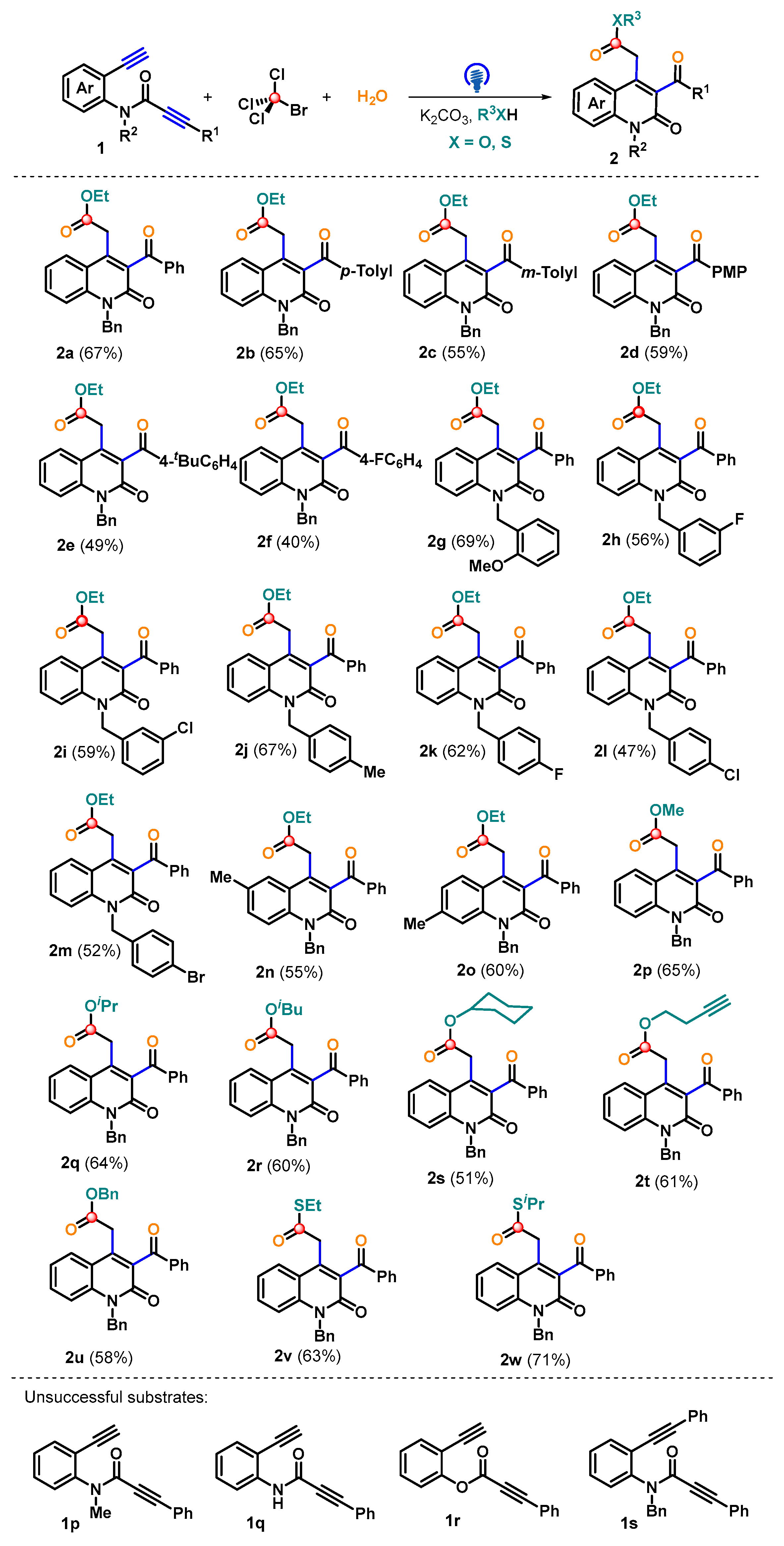
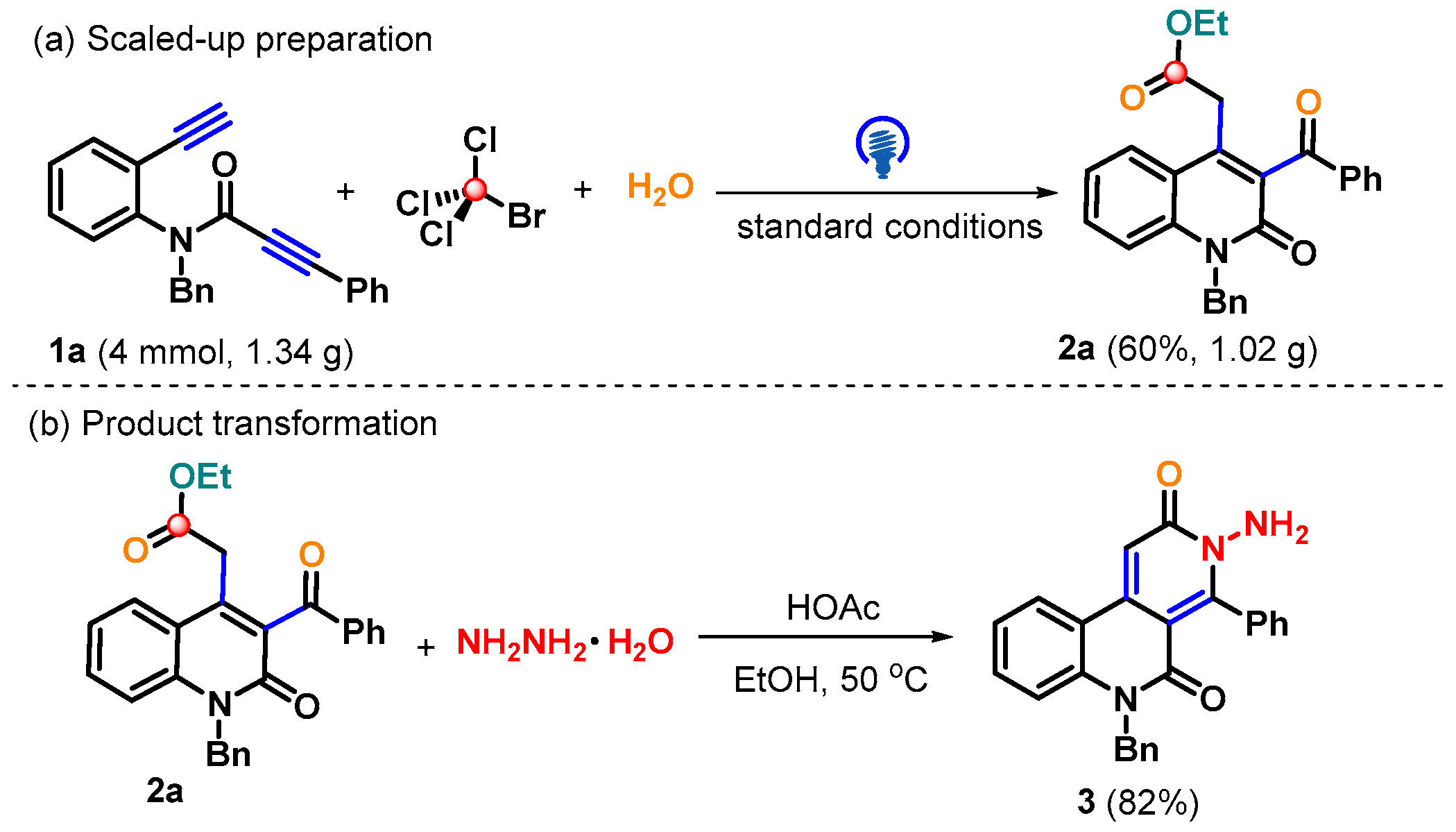
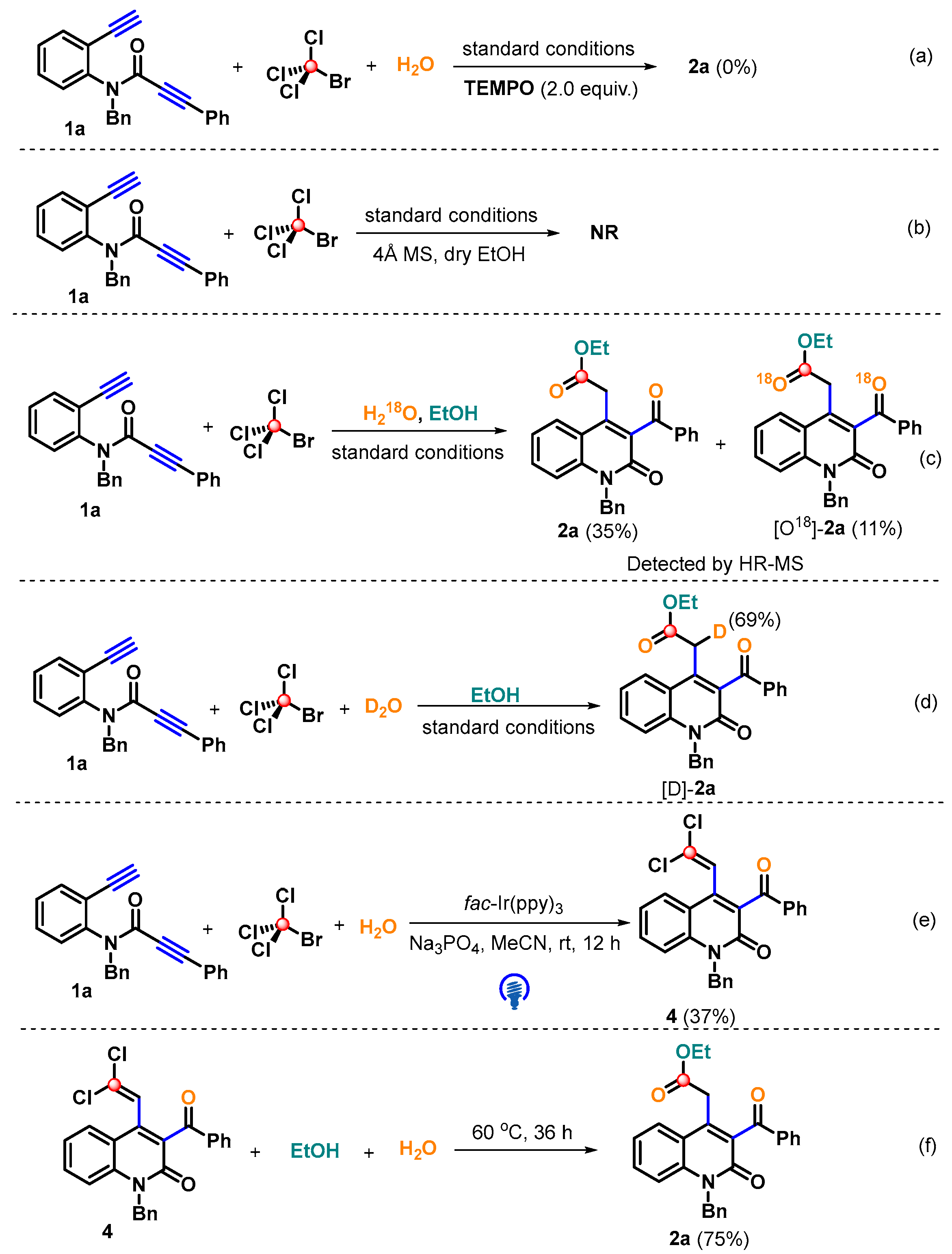
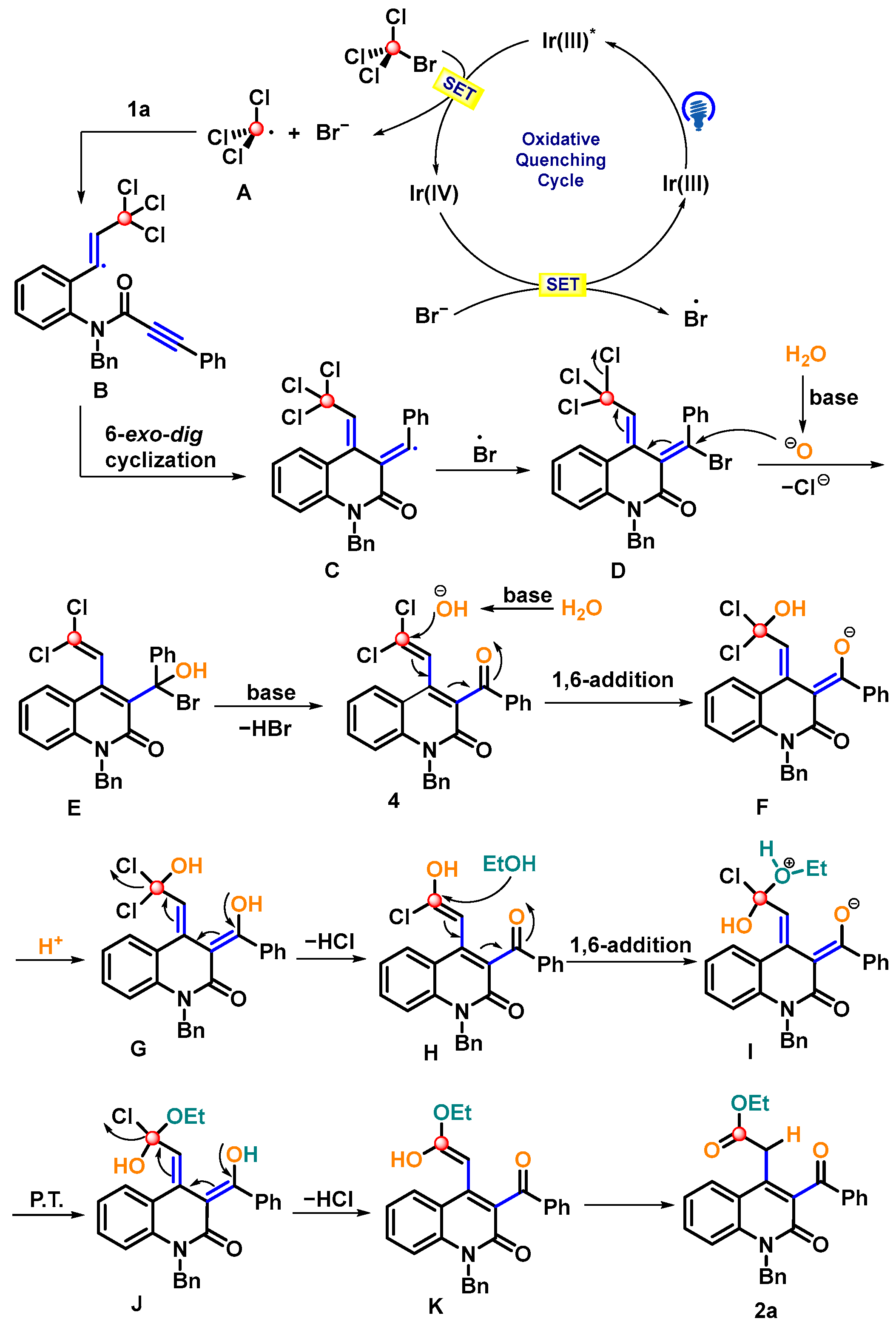
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Huang, K.; Sun, C.-L.; Shi, Z.-J. Transition-metal-catalyzed C–C bond formation through the fixation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 2435–2452. [Google Scholar] [CrossRef]
- Yu, C.; Ma, X.; Song, Q. Palladium-catalyzed cyanation of aryl halides with in situ generated CN− from ClCF2H and NaNH2. Org. Chem. Front. 2020, 7, 2950–2954. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Hu, X. Organic molecules as mediators and catalysts for photocatalytic and electrocatalytic CO2 reduction. Chem. Soc. Rev. 2013, 42, 2253–2261. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933–5947. [Google Scholar] [CrossRef]
- Ma, X.; Zhou, Y.; Song, Q. Synthesis of β-Aminoenones via Cross-Coupling of In-Situ-Generated Isocyanides with 1,3-Dicarbonyl Compounds. Org. Lett. 2018, 20, 4777–4781. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Mai, S.; Zhou, Y.; Cheng, G.; Song, Q. Dual role of ethyl bromodifluoroacetate in the formation of fluorine-containing heteroaromatic compounds. Chem. Commun. 2018, 54, 8960–8963. [Google Scholar] [CrossRef]
- Ma, X.; Deng, S.; Song, Q. Halodifluoroacetates as formylation reagents for various amines via unprecedented quadruple cleavage. Org. Chem. Front. 2018, 5, 3505–3509. [Google Scholar] [CrossRef]
- Deng, S.; Chen, H.; Ma, X.; Zhou, Y.; Yang, K.; Lan, Y.; Song, Q. S8-Catalyzed triple cleavage of bromodifluoro compounds for the assembly of N-containing heterocycles. Chem. Sci. 2019, 10, 6828–6833. [Google Scholar] [CrossRef]
- Ma, X.; Su, J.; Zhang, X.; Song, Q. Chlorodifluoromethane as a C1 Synthon in the Assembly of N-Containing Compounds. iScience 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Ma, X.; Song, Q. Recent progress on selective deconstructive modes of halodifluoromethyl and trifluoromethylcontaining reagents. Chem. Soc. Rev. 2020, 49, 9197–9219. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, H.I.; Kim, D.Y. Visible light-mediated photocatalytic bromination of 2-arylimidazo[1,2-a]pyridines using CBr4 as bromine source. Synth. Commun. 2020, 50, 197–206. [Google Scholar] [CrossRef]
- Kumar, S.; Shah, T.A.; Punniyamurthy, T. Recent advances in the application of tetrabromomethane in organic synthesis. Org. Chem. Front. 2021, 8, 4288–4314. [Google Scholar] [CrossRef]
- Dinda, T.K.; Mal, P. Activation of C-Br Bond of CBr4 and CBrCl3 Using 9-Mesityl-10-methylacridinium Perchlorate Photocatalyst. J. Org. Chem. 2023, 88, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; Chen, F.; Li, M.; Bu, Q.; Du, Z.; Liu, J.; Dai, B.; Liu, N. Visible-light-promoted synthesis of gem-dihaloenones. Green Chem. 2023, 25, 1191–1200. [Google Scholar] [CrossRef]
- Wu, D.; Hao, W.-J.; Rao, Q.; Lu, Y.; Tu, S.-J.; Jiang, B. Engaging 1,7-diynes in a photocatalytic Kharasch-type addition/1,5-(SN″)-substitution cascade toward β-gem-dihalovinyl carbonyls. Chem. Commun. 2021, 57, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Y.; Liu, Y.-P.; Liu, X.; Fu, R.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Photocatalytic Chemodivergent Synthesis of α-gem-Dihalovinyl Ketones and Chromen-2-Ones from Monoalkynes. Adv. Synth. Catal. 2022, 364, 2666–2672. [Google Scholar] [CrossRef]
- Rossi-Ashton, J.-A.; Clarke, A.-K.; Unsworth, W.-P.; Taylor, R.-J. Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 2020, 10, 7250–7261. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Xiao, W.-J.; Chen, J.-R. When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [Google Scholar] [CrossRef] [PubMed]
- Pagire, S.-K.; Foell, T.; Reiser, O. Shining visible light on vinyl halides: Expanding the horizons of photocatalysis. Acc. Chem. Res. 2020, 53, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Qin, Y. Indole Alkaloid Synthesis Facilitated by Photoredox Catalytic Radical Cascade Reactions. Acc. Chem. Res. 2019, 52, 1877–1891. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, W.; Gao, X.; Ren, X.; Dong, C.; Wang, C.; Liu, L.; Li, J. Synthesis of [1,2,3]Triazolo-[1,5-a]quinoxalin-4(5H)-ones through Photoredox-Catalyzed [3 + 2] Cyclization Reactions with Hypervalent Iodine(III) Reagents. J. Org. Chem. 2022, 87, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Zhao, W.; Wen, J.; Dong, C.; Hu, C.; Li, J. Photoredox-Catalyzed Cascade sp2 C−H Bond Functionalization to Construct Substituted Acridine with Diarylamine and Hypervalent Iodine(III) Reagents. Org. Lett. 2023, 25, 592–596. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.; Awad, J.M.; Xie, G.; Qiu, W.; Zhang, W. One-pot synthesis of tetrahydro-pyrrolobenzodiazepinones through sequential 1,3-dipolar cycloaddition/N-alkylation(N-acylation)Staudinger/aza-Witting reactions. Green Chem. 2019, 21, 4489–4494. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhang, W. Remote Radical 1,3-,1,4-,1,5-,1,6- and 1,7-Difunctionalization Reaction. Molecules 2023, 28, 3027. [Google Scholar] [CrossRef]
- Wang, C.-S.; Dixneuf, P.H.; Soule, J.-F. Photoredox Catalysis for Building C—C Bonds from C(sp2)—H Bonds. Chem. Rev. 2018, 118, 7532–7585. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Lang, X.; Zhao, J.; Chen, X. Cooperative photoredox catalysis. Chem. Soc. Rev. 2016, 45, 3026–3038. [Google Scholar] [CrossRef]
- Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Photocatalysis in organic and polymer synthesis. Chem. Soc. Rev. 2016, 45, 6165–6212. [Google Scholar] [CrossRef]
- Narayanama, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: Applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar] [CrossRef]
- Rotondo, D.M.A.; McCusker, J.K. The photophysics of photoredox catalysis: A roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. [Google Scholar] [CrossRef]
- Zhu, S.-S.; Zhou, J.-N.; Wu, Q.-L.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Photoinduced double [2 + 2] cycloaddition relay of yne-allenones for highly diastereoselective synthesis of hexacyclic 1-naphthols. Org. Chem. Front. 2020, 7, 2975–2980. [Google Scholar] [CrossRef]
- Zheng, J.-L.; Wu, D.; Lin, N.; Liu, Y.-P.; Wang, L.; Zhu, X.-T.; Hao, W.-J.; Wang, S.-L.; Jiang, B. Kharasch-type photocyclization of 1,7-diynes for the stereospecific synthesis of tetrahydronaphthalen-1-ols. Tetrahedron Lett. 2021, 85, 153485. [Google Scholar] [CrossRef]
- Wang, L.; Shen, Y.-T.; Wang, Y.-X.; Wang, H.-Y.; Hao, W.-J.; Jiang, B. Multicomponent Annulative SO2 Insertion of Heteroatom-Linked 1,7-Diynes for Accessing Tricyclic Sulfones. Adv. Synth. Catal. 2023, 365, 1693–1698. [Google Scholar] [CrossRef]
- Wang, L.; Xu, T.; Rao, Q.; Zhang, T.-S.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Photocatalytic Biheterocyclization of 1,7-Diynes for Accessing Skeletally Diverse Tricyclic 2-Pyranones. Org. Lett. 2021, 23, 7845–7850. [Google Scholar] [CrossRef] [PubMed]
- Nikitas, N.-F.; Voutyritsa, E.; Gkizis, P.-L.; Kokotos, C.-G. Metal-free Photochemical Atom Transfer Radical Addition (ATRA) of BrCCl3 to Alkenes. Eur. J. Org. Chem. 2021, 2021, 96–101. [Google Scholar] [CrossRef]
- Nguyen, D.; Tucker, J.-W.; Konieczynska, M.-D.; Stephenson, C.-R. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. [Google Scholar] [CrossRef] [PubMed]
- Voutyritsa, E.; Triandafillidi, L.; Tzouras, N.-V.; Nikitas, N.-F.; Pefkianakis, E.-K.; Vougioukalakis, G.-C.; Kokotos, C.-G. Photocatalytic Atom Transfer Radical Addition to Olefins Utilizing Novel Photocatalysts. Molecules 2019, 24, 1644. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.-Z.; Wang, S.-C.; Song, K.-X.; Hao, W.-J.; Jiang, B. Visible-Light-Driven Photocatalytic Kharasch-Type Addition of 1,6-Enynes. Chin. J. Org. Chem. 2021, 41, 4815–4824. [Google Scholar] [CrossRef]
- Ji, X.-S.; Fu, R.; Wang, S.-L.; Hao, W.-J.; Jiang, B. Visible Light Driven Phot ocatalytic Kharasch Reaction of Phenol/Arylamine Linked 1,6 Enynes with Perhalogenated Methane. Chin. J. Org. Chem. 2022, 42, 4282–4291. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhang, S.; Tang, Y.; Yan, S.; Li, G. Copper-Catalyzed Annulation-Trifluoromethyl Functionalization of Enynones. Org. Lett. 2023, 25, 2509–2514. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Li, G.; Hao, W.-J.; Jiang, B. Catalytic Benzannulation Reactions of Enynones for Accessing Heterocycle-Incorporating Diarylmethanes. Synlett 2023, 34, 243–248. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhang, S.; Yuan, Q.; Li, G.; Yan, S. Catalytic Radical-Triggered Annulation/Iododifluoromethylation of Enynones for the Stereospecific Synthesis of 1-Indenones. J. Org. Chem. 2023, 88, 8532–8541. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, D.; Wang, J.-Y.; Yan, S.; Li, G. Four-layer folding framework: Design, GAP synthesis, and aggregation-induced emission. Front. Chem. 2023, 11, 1259609. [Google Scholar] [CrossRef] [PubMed]
- Tyson, E.L.; Ament, M.S.; Yoon, T.P. Transition Metal Photoredox Catalysis of Radical Thiol-Ene Reactions. J. Org. Chem. 2013, 78, 2046–2050. [Google Scholar] [CrossRef]
- Keylor, M.H.; Park, J.E.; Wallentin, C.-J.; Stephenson, C.R.J. Photocatalytic initiation of thioleene reactions: Synthesis of thiomorpholin-3-ones. Tetrahedron 2014, 70, 4264–4269. [Google Scholar] [CrossRef]
- Bacauanu, V.; Cardinal, S.; Yamauchi, M.; Kondo, M.; Fernandez, D.F.; Remy, R.; MacMillan, D.W.C. Metallaphotoredox Difluoromethylation of Aryl Bromides. Angew. Chem. Int. Ed. 2018, 57, 12543–12548. [Google Scholar] [CrossRef]
- Wang, S.-W.; Yu, J.; Zhou, Q.-Y.; Chen, S.-Y.; Xu, Z.-H.; Tang, S. Visible-Light-Induced Atom Transfer Radical Addition and Cyclization of Perfluoroalkyl Halides with 1,n-Enynes. ACS Sustain. Chem. Eng. 2019, 7, 10154–10162. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
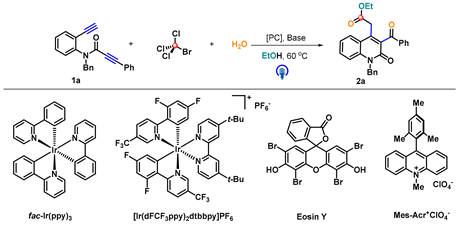 | |||
| Entry | [PC] | Base | Yield (%) b |
| 1 | fac-Ir(ppy)3 | - | 48 |
| 2 | fac-Ir(ppy)3 | K2CO3 | 67 |
| 3 c | fac-Ir(ppy)3 | K2CO3 | 35 |
| 4 d | fac-Ir(ppy)3 | K2CO3 | 52 |
| 5 | - | K2CO3 | NR |
| 6 | [Ir(dFCF3ppy)2dtbbpy]PF6 | K2CO3 | 47 |
| 7 | Eosin Y | K2CO3 | 28 |
| 8 | Mes-Acr+ClO4− | K2CO3 | 16 |
| 9 | fac-Ir(ppy)3 | Na2CO3 | 28 |
| 10 | fac-Ir(ppy)3 | Cs2CO3 | 27 |
| 11 | fac-Ir(ppy)3 | MeONa | 45 |
| 12 | fac-Ir(ppy)3 | Et3N | 55 |
| 13 | fac-Ir(ppy)3 | DMAP | 59 |
| 14 e | fac-Ir(ppy)3 | K2CO3 | 54 |
| 15 f | fac-Ir(ppy)3 | K2CO3 | 39 |
| 16 g | fac-Ir(ppy)3 | K2CO3 | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Bao, Y.; Yan, S.; Wang, J.; Zhang, Y.; Li, G. Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane. Molecules 2024, 29, 782. https://doi.org/10.3390/molecules29040782
Chen D, Bao Y, Yan S, Wang J, Zhang Y, Li G. Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane. Molecules. 2024; 29(4):782. https://doi.org/10.3390/molecules29040782
Chicago/Turabian StyleChen, Daixiang, Yu Bao, Shenghu Yan, Jiayin Wang, Yue Zhang, and Guigen Li. 2024. "Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane" Molecules 29, no. 4: 782. https://doi.org/10.3390/molecules29040782
APA StyleChen, D., Bao, Y., Yan, S., Wang, J., Zhang, Y., & Li, G. (2024). Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane. Molecules, 29(4), 782. https://doi.org/10.3390/molecules29040782