


1H-1,2,3-triazolyl-1,6-naphthyridin-7(6H)-ones as Potential Fluorescent Nucleoside Analogues: Synthesis and Optical Properties

Abstract
1. Introduction
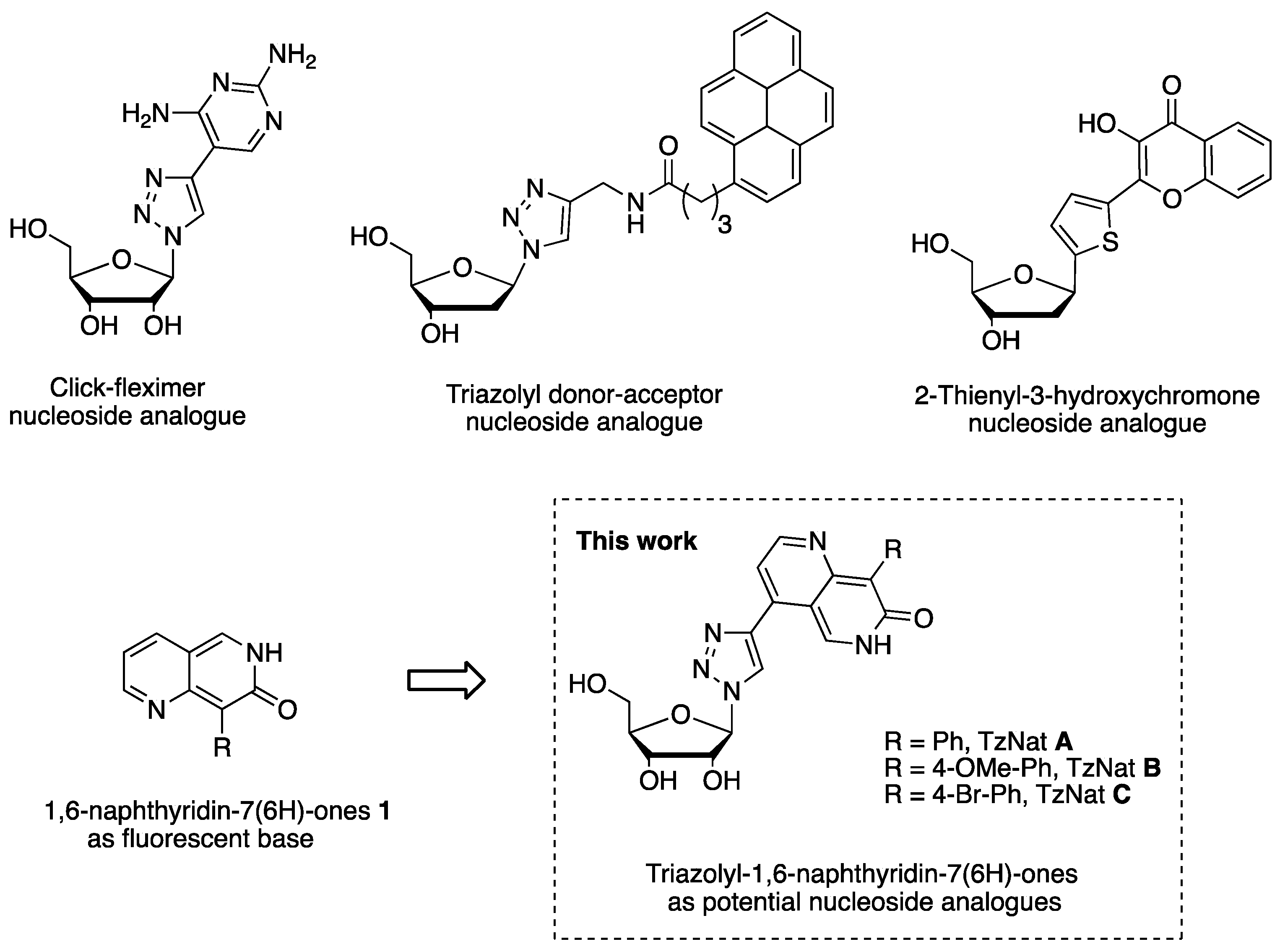
2. Results
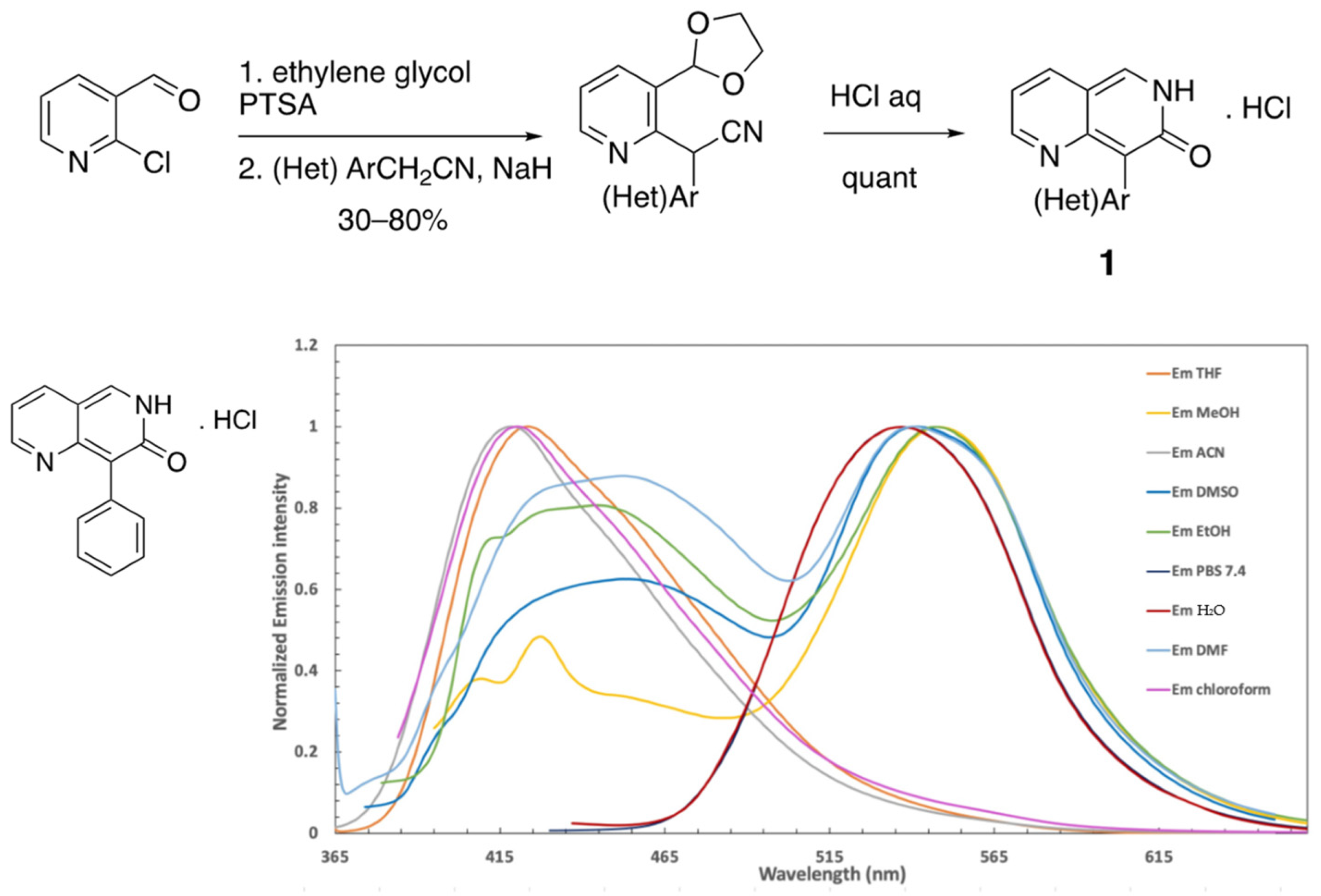
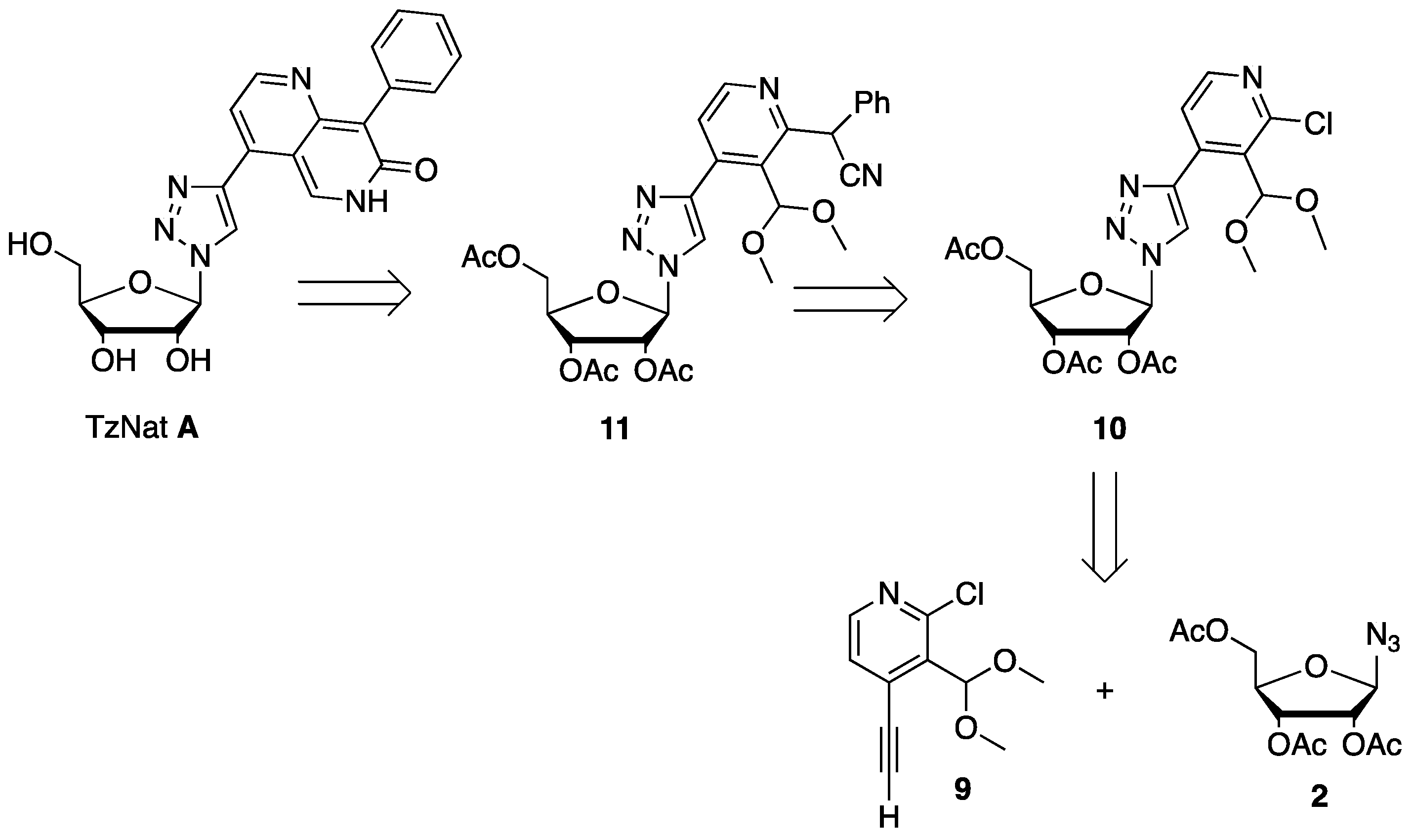
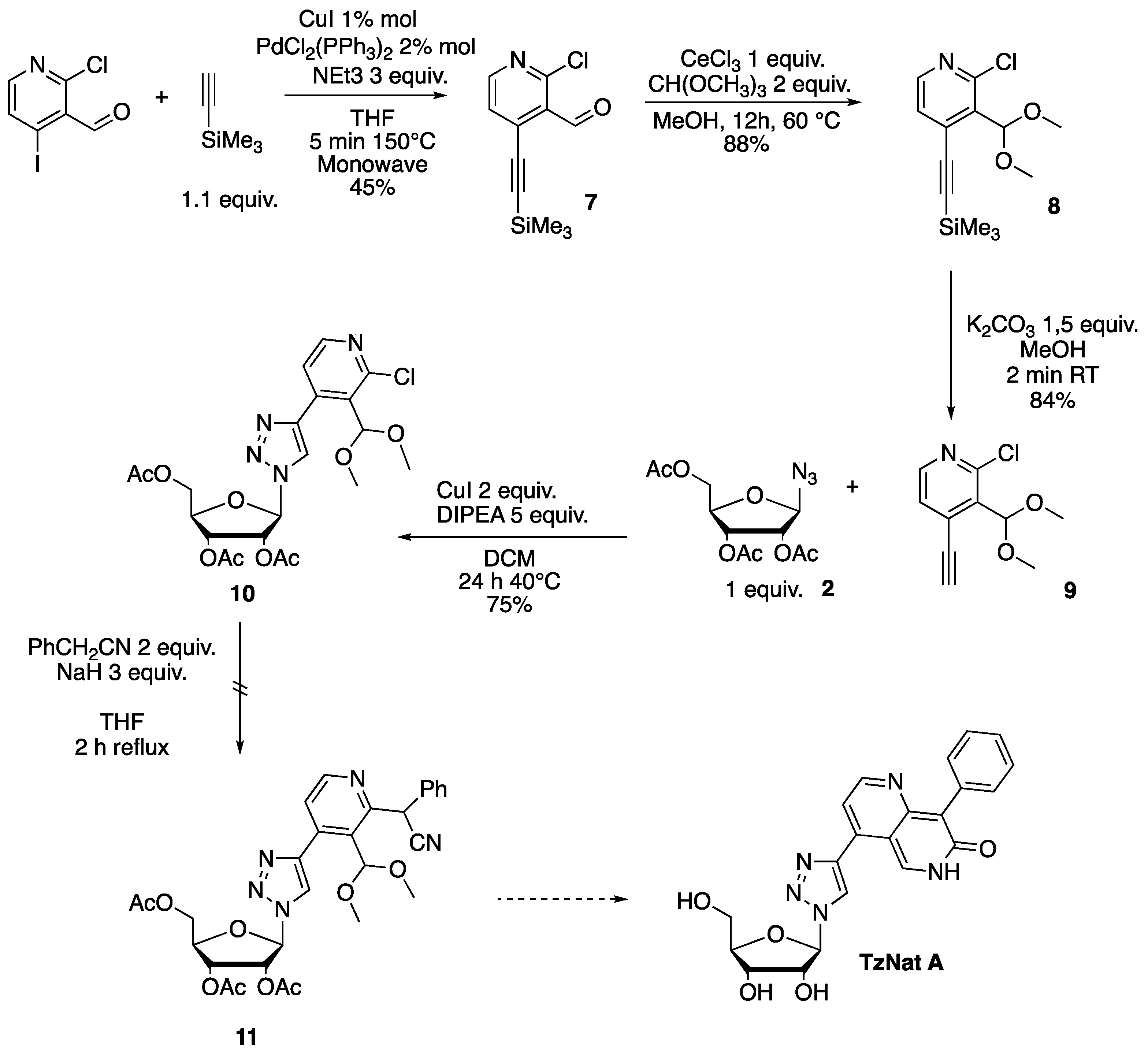
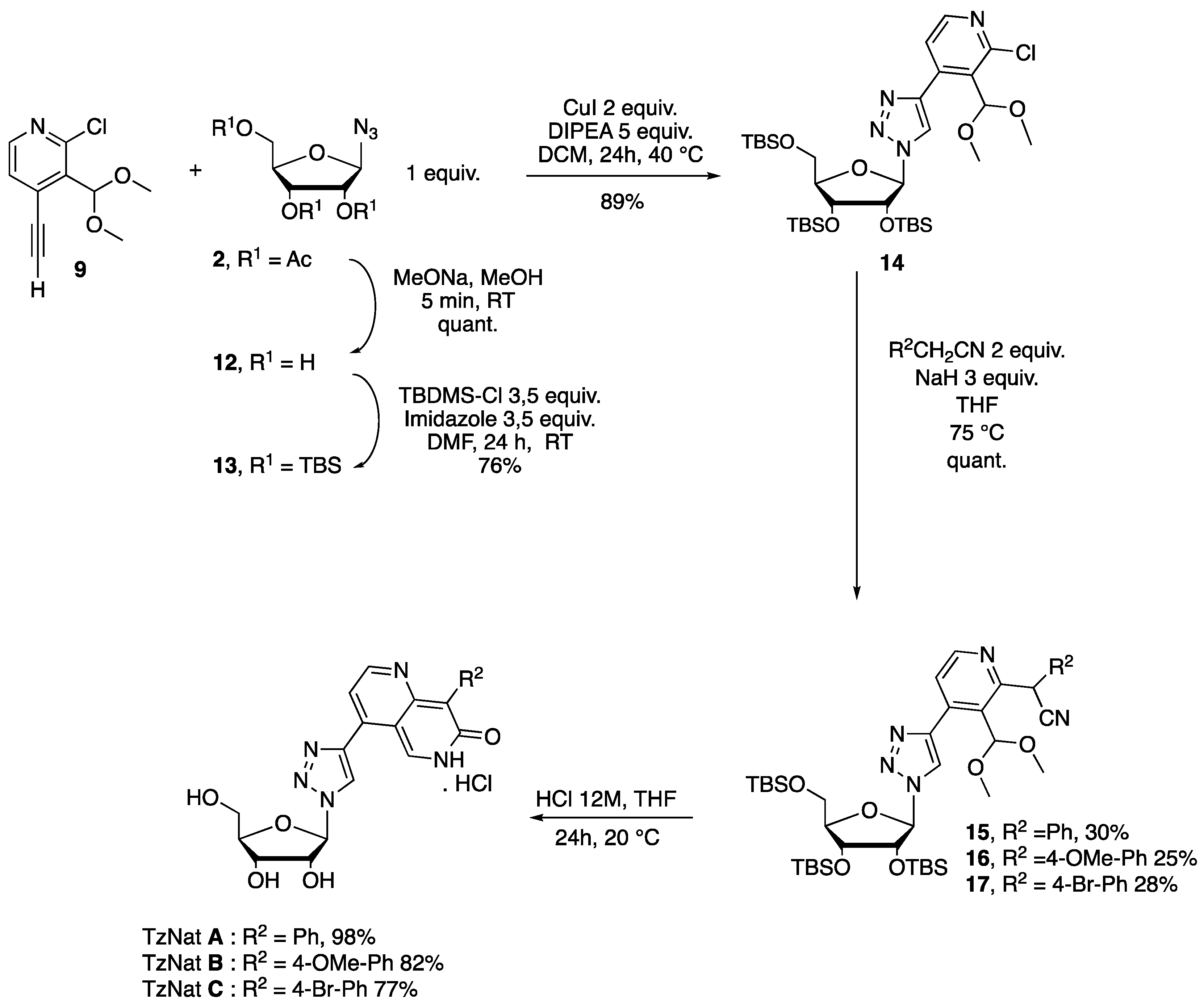
2.1. Fluorescent Nucleosides Synthesis
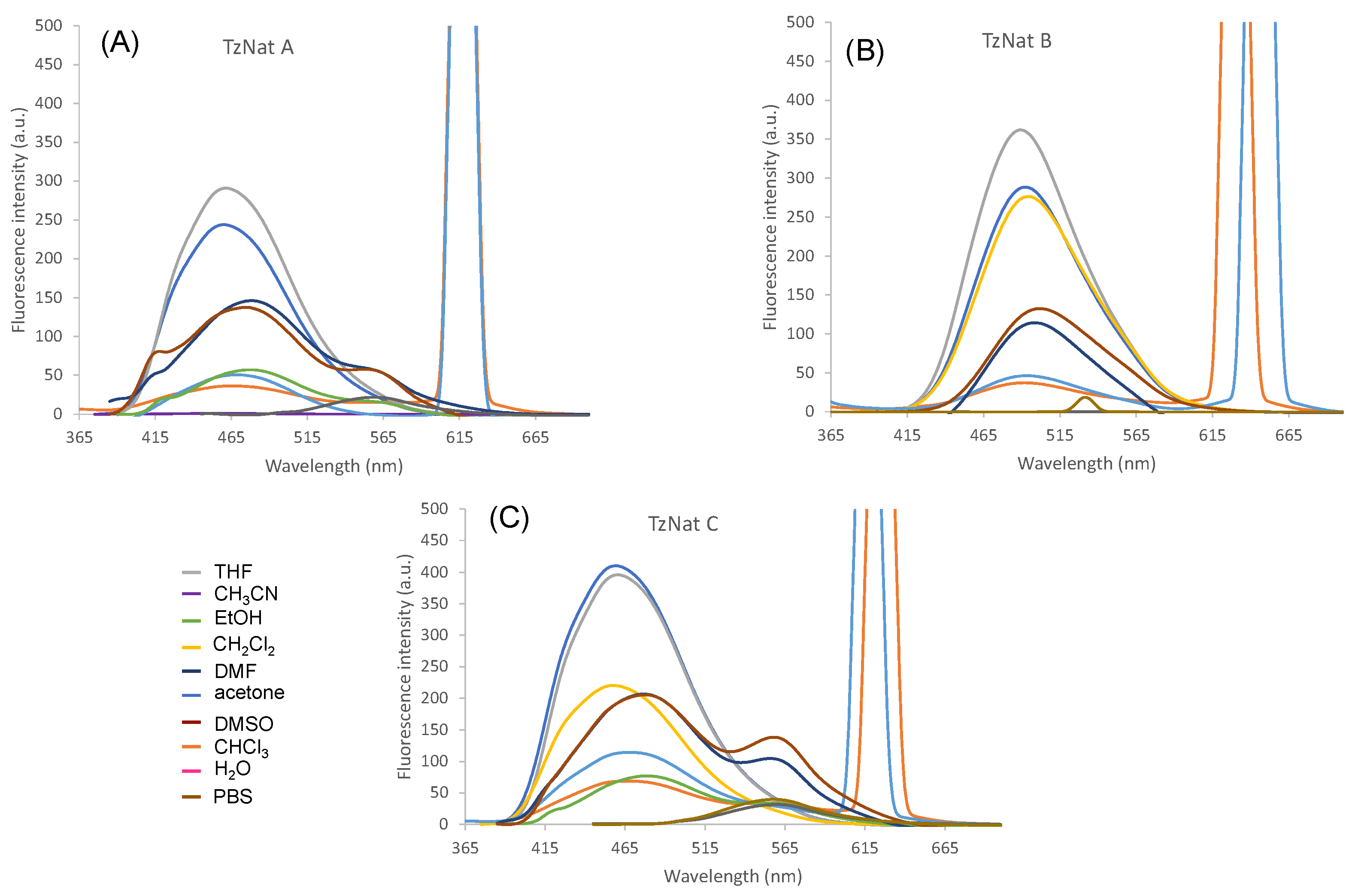
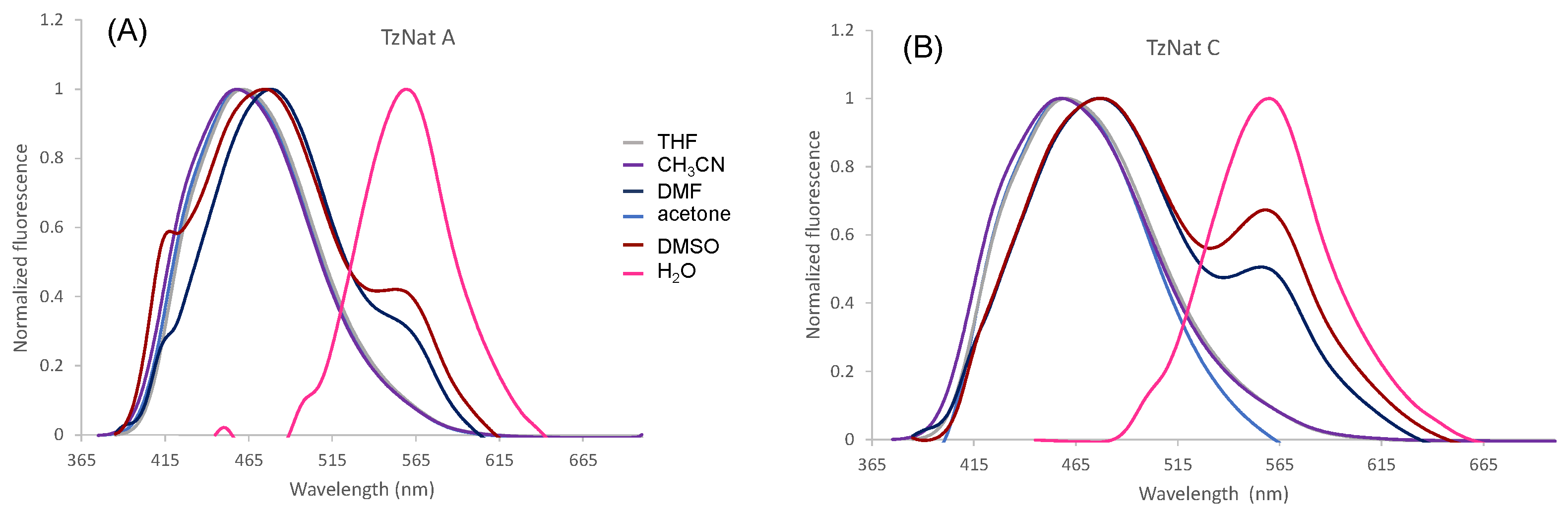
2.2. Optical Properties of TzNat Molecules
3. Materials and Methods
3.1. General Information
3.2. Attempts to Prepare Triazole Conjugate 4 from Acetylated Ribosyl Azide 2 and Compounds 5
3.2.1. Synthesis of 2,3,5-Tri-O-acetyl-β-d-ribofuranosyl Azide 2
3.2.2. Synthesis of (2R,3R,4R,5R)-2-(4-(4-((3-(1,3-dioxolan-2-yl)pyridin-2-yl)(cyano)methyl)phenyl)-1H-1,2,3-triazol-1-yl)-5-(acetoxymethyl)tetrahydrofuran-3,4-diyl Diacetate 6
3.3. Synthesis of the Alkyne Partner 9 for the Click Reaction
3.3.1. Synthesis of 2-chloro-4-((trimethylsilyl)ethynyl)nicotinaldehyde 7
3.3.2. Synthesis of 2-chloro-3-(dimethoxymethyl)-4-((trimethylsilyl)ethynyl)pyridine 8
3.3.3. Synthesis of 2-chloro-3-(dimethoxymethyl)-4-ethynylpyridine 9
3.4. Synthesis of Target 1,2,3 Triazole Nucleoside TzNat A, B, and C
3.4.1. Synthesis of (2R,3R,4R,5R)-2-(acetoxymethyl)-5-(4-(2-chloro-3-(dimethoxymethyl)pyridin-4-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-3,4-diyl Diacetate 10
3.4.2. Synthesis of β-d-ribofuranosyl Azide 12
3.4.3. Synthesis of 2,3,5-Tri-O-(tertbutyldimethylsilyle)-β-d-ribofuranosyl Azide 13
3.4.4. Synthesis of 4-(1-((2R,3R,4R,5R)-3,4-bis((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl)-1H-1,2,3-triazol-4-yl)-2-chloro-3-(dimethoxymethyl)pyridine 14
3.4.5. Synthesis of 2-(4-(1-((2R,3R,4R,5R)-3,4-bis((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl)-1H-1,2,3-triazol-4-yl)-3-(dimethoxymethyl)pyridin-2-yl)-2-phenylacetonitrile 15
3.4.6. Synthesis of 2-(4-(1-((2R,3R,4R,5R)-3,4-bis((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl)-1H-1,2,3-triazol-4-yl)-3-(dimethoxymethyl)pyridin-2-yl)-2-(4-methoxyphenyl)acetonitrile 16
3.4.7. Synthesis of 2-(4-(1-((2R,3R,4R,5R)-3,4-bis((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl)-1H-1,2,3-triazol-4-yl)-3-(dimethoxymethyl)pyridin-2-yl)-2-(4-bromophenyl)acetonitrile 17
3.4.8. Synthesis of 4-(1-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-1H-1,2,3-triazol-4-yl)-8-phenyl-1,6-naphthyridin-7(6H)-one hydrochloride Salt TzNat A
3.4.9. Synthesis of 4-(1-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-1H-1,2,3-triazol-4-yl)-8-(4-methoxyphenyl)-1,6-naphthyridin-7(6H)-one hydrochloride salt TzNat B
3.4.10. 8-(4-bromophenyl)-4-(1-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-1H-1,2,3-triazol-4-yl)-1,6-naphthyridin-7(6H)-one hydrochloride Salt TzNat C
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tinoco, I. Nucleic Acid Structures, Energetics, and Dynamics. J. Phys. Chem. 1996, 100, 13311–13322. [Google Scholar] [CrossRef]
- Bottaro, S.; Di Palma, F.; Bussi, G. The role of nucleobase interactions in RNA structure and dynamics. Nucleic Acids Res. 2014, 42, 13306–13314. [Google Scholar] [CrossRef]
- Minchin, S.; Lodge, J. Understanding biochemistry: Structure and function of nucleic acids. Essays Biochem. 2019, 63, 433–456. [Google Scholar] [CrossRef]
- Chu, C.K.; Baker, D.C. Nucleosides and Nucleotides as Antitumor and Antiviral Agents; Plenum Press: New York, NY, USA, 1993. [Google Scholar]
- Thomson, J.M.; Lamont, I.L. Nucleoside Analogues as Antibacterial Agents. Front. Microbiol. 2019, 20, 952. [Google Scholar] [CrossRef]
- Jordheim, L.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug. Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Cox, R.S., III; Hirao, I. Unnatural base pair systems for sensing and diagnostic applications. Expert. Rev. Mol. Diagn. 2011, 3, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.T.; Kool, E.T. Redesigning the architecture of the base pair: Toward biochemical and biological function of new genetic sets. Chem. Biol. 2009, 16, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chan, K.M.; Kool, E.T. Fluorescent nucleobases as tools for studying DNA and RNA. Nat. Chem. 2017, 11, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.Y.; Dziuba, D.; Benhida, R.; Demchenko, A.P.; Burger, A. Probing of Nucleic Acid Structures, Dynamics, and Interactions with Environment-Sensitive Fluorescent Labels. Front. Chem. 2020, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.D.; Teppang, K.L.; Lee, R.W.; Lokensgard, M.E.; Purse, B.W. Fluorescence turn-on sensing of DNA duplex formation by a tricyclic cytidine analogue. J. Am. Chem. Soc. 2017, 139, 1372–1375. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.P.; Mata, G.; Luedtke, N.W. Fluorescent base analogue reveals T-HgII-T base pairs have high kinetic stabilities that perturb DNA metabolism. J. Am. Chem. Soc. 2016, 138, 14733–14739. [Google Scholar] [CrossRef] [PubMed]
- Klöcker, N.; Weissenboeck, F.P.; Rentmeister, A. Covalent labeling of nucleic acids. Chem. Soc. Rev. 2020, 49, 8749–8773. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, D. Environmentally sensitive fluorescent nucleoside analogues as probes for nucleic acid—protein interactions: Molecular design and biosensing applications. Methods Appl. Fluoresc. 2022, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Secrist, J.A., III; Barrio, J.R.; Leonard, N.J.A. Fluorescent modification of adenosine triphosphate with activity in enzyme systems:1,N 6-ethenoadenosine triphosphate. Science 1972, 175, 646–647. [Google Scholar] [CrossRef]
- Coleman, R.S.; Madaras, M.L. Synthesis of a novel coumarin C-riboside as a photophysical probe of oligonucleotide dynamics. J. Org. Chem. 1998, 63, 5700–5703. [Google Scholar] [CrossRef]
- Morales-Rojas, H.; Kool, E.T. A porphyrin C-nucleoside incorporated into DNA. Org. Lett. 2002, 4, 4377–4380. [Google Scholar] [CrossRef]
- Okamoto, A.; Tainaka, K.; Fujiwara, Y. Nile Red nucleoside: Design of a solvatofluorochromic nucleoside as an indicator of micropolarity around DNA. J. Org. Chem. 2006, 71, 3592–3598. [Google Scholar] [CrossRef]
- Gao, J.; Strassler, C.; Tahmassebi, D.; Kool, E.T. Libraries of composite polyfluors built from fluorescent deoxyribosides. J. Am. Chem. Soc. 2002, 124, 11590–11591. [Google Scholar] [CrossRef]
- Hirao, I.; Kimoto, M.; Yamashige, R. Natural versus artificial creation of base pairs in DNA: Origin of nucleobases from the perspectives of unnatural base pair studies. Acc. Chem. Res. 2012, 45, 2055–2065. [Google Scholar] [CrossRef]
- Andreeva, O.V.; Garifullin, B.F.; Zarubaev, V.V.; Slita, A.V.; Yesaulkova, I.L.; Saifina, L.F.; Shulaeva, M.M.; Belenok, M.G.; Semenov, V.E.; Kataev, V.E. Synthesis of 1,2,3-triazolyl nucleoside analogues and their antiviral activity. Mol. Divers. 2021, 25, 473–490. [Google Scholar] [CrossRef]
- Alvarez, R.; Velázquez, S.; San-Félix, A.; Aquaro, S.; De Clercq, E.; Perno, C.F.; Karlsson, A.; Balzarini, J.; Camarasa, M.J. 1,2,3-Triazole-[2′,5′-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]-3′-spiro-5″-(4″-amino-1″,2″-oxathiole 2″,2″-dioxide) (TSAO) analogues: Synthesis and anti-HIV-1 activity. J. Med. Chem. 1994, 37, 4185–4194. [Google Scholar] [CrossRef]
- Chittepu, P.; Sirivolu, V.R.; Seela, F. Nucleosides and oligonucleotides containing 1,2,3-triazole residues with nucleobase tethers: Synthesis via the azide-alkyne ‘click’ reaction. Bioorg. Med. Chem. 2008, 16, 8427–8439. [Google Scholar] [CrossRef] [PubMed]
- Ghoteimi, R.; Braka, A.; Rodriguez, C.; Cros-Perrial, E.; Tai Nguyen, V.; Uttaro, J.P.; Mathé, C.; Chaloin, L.; Ménétrier-Caux, C.; Jordheim, L.P.; et al. 4-Substituted-1,2,3-triazolo nucleotide analogues as CD73 inhibitors, their synthesis, in vitro screening, kinetic and in silico studies. Bioorg. Chem. 2021, 107, 104577. [Google Scholar] [CrossRef] [PubMed]
- St. Amant, A.H.; Bean, L.A.; Guthrie, J.P.; Hudson, R.H. Click fleximers: A modular approach to purine base-expanded ribonucleoside analogues. Org. Biomol. Chem. 2012, 10, 6521–6525. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Zhang, L.; Hagos, A.; Quirk, S. “Fleximers”. Design and synthesis of a new class of novel shape-modified nucleosides (1). J. Org. Chem. 2002, 67, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Chudinov, M.V. Nucleoside Analogs with Fleximer Nucleobase. Chem. Heterocycl. Comp. 2020, 56, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K. Flexibility-Not just for yoga anymore! Antivir. Chem. Chemother. 2018, 26, 2040206618756788. [Google Scholar] [CrossRef] [PubMed]
- Bag, S.S.; Talukdar, S.; Das, S.K.; Pradhan, M.K.; Mukherjee, S. Donor/acceptor chromophores-decorated triazolyl unnatural nucleosides: Synthesis, photophysical properties and study of interaction with BSA. Org. Biomol. Chem. 2016, 22, 5088–5108. [Google Scholar] [CrossRef]
- Dziuba, D.; Postupalenko, V.Y.; Spadafora, M.; Klymchenko, A.S.; Guérineau, V.; Mély, Y.; Benhida, R.; Burger, A. A universal nucleoside with strong two-band switchable fluorescence and sensitivity to the environment for investigating DNA interactions. J. Am. Chem. Soc. 2012, 134, 10209–10213. [Google Scholar] [CrossRef]
- Litvinov, V.P. Advances in the chemistry of naphthyridines. Adv. Heterocycl. Chem. 2006, 91, 189–300. [Google Scholar]
- Darakshan; Parvin, T. Domino reaction for the synthesis of pyrazole/isoxazole fused naphthyridine derivatives Involving indole ring opening and double ring formation. J. Org. Chem. 2023, 88, 6847–6856. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, Z.; Chen, F.; Wang, X.; Gou, S. Discovery of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine derivatives as highly selective CK2 inhibitors with potent cancer cell stemness inhibition. J. Med. Chem. 2021, 64, 5082–5098. [Google Scholar] [CrossRef]
- Sun, Q.; Ren, R.; Wu, P.-P.; Zhuo, L.-S.; Dong, H.; Peng, H.-T.; Cao, Y.-F.; Luo, X.-G.; She, N.-F. A 2, 7-naphthyridine-based fluorescent turn-on probe for detection of biothiols in vitro and in vivo. Dyes Pigm. 2020, 182, 108702. [Google Scholar] [CrossRef]
- Che, C.-M.; Wan, C.-W.; Ho, K.-Y.; Zhou, Z.-Y. Strongly luminescent metal-organic compounds: Spectroscopic properties andcrystal structure of substituted 1,8-naphthyridine and its zinc (II) complex. New. J. Chem. 2001, 25, 63–65. [Google Scholar] [CrossRef]
- Hikishima, S.; Minakawa, N.; Kuramoto, K.; Fujisawa, Y.; Ogawa, M.; Matsuda, A. Synthesis of 1,8-naphthyridine C-nucleosides and their base-pairing properties in oligodeoxynucleotides: Thermally stable naphthyridine:imidazopyridopyrimidine base-pairing motifs. Angew. Chem. Int. Ed. 2005, 44, 596–598. [Google Scholar] [CrossRef]
- Oliveras, J.M.; Puig de la Bellacasa, R.; Estrada-Tejedor, R.; Teixidó, J.; Borrell, J.I. 1,6-Naphthyridin-2(1H)-ones: Synthesis and Biomedical Applications. Pharmaceuticals 2021, 14, 1029. [Google Scholar] [CrossRef] [PubMed]
- Eldrup, A.B.; Christensen, C.; Haaima, G.; Nielsen, P.E. Substituted 1,8-Naphthyridin-2(1H)-ones are Superior to thymine in the recognition of adenine in duplex as well as triplex structures. J. Am. Chem. Soc. 2002, 124, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Füchtbauer, A.; Wranne, M.; Giraud, T.; Floyd, T.; Dumat, B.; Andersen, N.; El-Sagheer, A.; Brown, T.; Gradén, H.; et al. Synthesis, oligonucleotide incorporation and fluorescence properties in DNA of a bicyclic thymine analogue. Sci. Rep. 2018, 8, 13970. [Google Scholar] [CrossRef] [PubMed]
- Samaan, G.N.; Wyllie, M.K.; Cizmic, J.M.; Needham, L.M.; Nobis, D.; Ngo, K.; Andersen, S.; Magennis, S.W.; Lee, S.F.; Purse, B.W. Single-molecule fluorescence detection of a tricyclic nucleoside analogue. Chem. Sci. 2020, 12, 2623–2628. [Google Scholar] [CrossRef] [PubMed]
- Beghennou, A.; Gontard, G.; Dossmann, H.; Passador, K.; Thorimbert, S.; Corcé, V.; Botuha, C. 1,6-Naphthyridin-7(6H)-ones: Synthesis and optical properties. Org. Biomol. Chem. 2023, 21, 2976–2982. [Google Scholar] [CrossRef] [PubMed]
- Fantoni, N.Z.; El-Sagheer, A.H.; Brown, T.A. Hitchhiker’s guide to click-chemistry with nucleic acids. Chem. Rev. 2021, 121, 7122–7154. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Bose, P.; Jaiswal, M.K.; Rajkhowa, S.; Singh, A.S.; Hotha, S.; Mishra, N.; Tiwari, V.K. Cu(I)-Catalyzed Click Chemistry in Glycoscience and Their Diverse Applications. Chem. Rev. 2021, 121, 7638–7956. [Google Scholar] [CrossRef]
- Amblard, F.; Sari, O.; Boucle, S.; Khalil, A.; Schinazi, R.F. Modifications of Nucleosides, Nucleotides, and Nucleic Acids using Huisgen’s [3+2] Azide–Alkyne Cycloaddition: Opening Pandora’s Box. In Click Reactions in Organic Synthesis; Chandrasekaran, S., Ed.; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Ermolat’ev, D.; Mehta, V.; Van der Eycken, E. Synthesis of Furo[2,3-b]pyrazine Nucleoside Analogues with 1,2,3-Triazole Linkage. QSAR Comb. Sci. 2007, 26, 1266–1273. [Google Scholar] [CrossRef]
- Passays, J.; Rubay, C.; Marcélis, L.; Elias, B. Synthesis and Photophysical Properties of Triazolyl IrIII Nucleosides. Eur. J. Inorg. Chem. 2017, 3, 623–629. [Google Scholar] [CrossRef]
- Pandey, N.; Dwivedi, P.; Jyoti, S.M.; Kumar, D.; Tiwari, V.K.; Mishra, B.B. Click Chemistry Inspired Synthesis of Hydroxyanthracene Triazolyl Glycoconjugates. ACS Omega 2022, 7, 37112–37121. [Google Scholar] [CrossRef]
- Singh, S.K.; Kumar, S.; Yadav, M.S.; Tiwari, V.K. Pyridyl Glycosyl Triazole/CuI-Mediated Domino/Tandem Synthesis of Quinazolinones. J. Org. Chem. 2022, 87, 15389–15402. [Google Scholar] [CrossRef] [PubMed]
- Štimac, A.; Jože, K. An improved preparation of 2,3,5-tri-O-acyl-β-d-ribofuranosyl azides by the Lewis acid-catalysed reaction of β-D-ribofuranosyl acetates and trimethylsilyl azide: An example of concomitant formation of the α anomer by trimethylsilyl triflate catalysis. Carbohydr. Res. 1992, 232, 359–365. [Google Scholar] [CrossRef]
- Luche, J.-L.; Gemal, A.L. Efficient synthesis of acetals catalyzed by rare earth chlorides. J. Chem. Soc. Chem. Commun. 1978, 22, 976–977. [Google Scholar] [CrossRef]
- Brunel, D.; Dumur, F. Recent advances in organic dyes and fluorophores comprising a 1,2,3-triazole moiety. New J. Chem. 2020, 44, 3546–3561. [Google Scholar] [CrossRef]
- Liese, D.; Haberhauer, G. Rotations in Excited ICT States—Fluorescence and its Microenvironmental Sensitivity. Isr. J. Chem. 2018, 58, 813–826. [Google Scholar] [CrossRef]
- Nisic, F.; Speciale, G.; Bernardi, A. Stereoselective synthesis of α- and β-glycofuranosyl amides by traceless ligation of glyco. furanosyl azides. Chem. Eur. J. 2012, 18, 6895–6906. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UV–Vis [b] | Fluorescence [c] | |||||
|---|---|---|---|---|---|---|
| Compd | Solvent [a] | λabs (ε) | λem | Stokes [d] Shifts in cm−1 | ΦF [e] | |
| TzNat A | CHCl3 CH2Cl2 THF acetone CH3CN DMF DMSO EtOH H2O PBS | 358 (8.8), 479 (5.8) 356 (10.3), 476 (4.7) 369 (6.4), 479 (0.6) 363 (5.3), 474 (0.5) 360 (5.1), 465 (1.9) 369 (5.2), 480 (1.1) 374 (3.6), 483 (1.1) 374 (3.8), 451(3.8) 427 (4.9) 427 (6.6) | 471 474 461 460 458 413, 478, 560 421, 475, 560 477 563 558 | 6701 6992 5809 5943 6179 6179 5685 5773 5657 5498 | <0.01 <0.01 0.32 0.20 0.11 0.01 <0.01 <0.01 <0.01 <0.01 | |
| TzNat B | CHCl3 CH2Cl2 THF acetone CH3CN DMF DMSO EtOH H2O PBS | 317 (17.2), 461 (6.7) 325 (16.3), 498 (4.9) 378 (7.6), 490 (0.4) 369 (8.1), 488 (1.6) 369 (7.6), 468 (3.1) 379 (9.5), 486 (2.2) 380 (7.1), 490 (1.9) 386 (3.2), 457 (4.0) 433 (8.1) 431 (7.3) | 492 499 489 492 494 499 502 -- -- -- | 11,220 10,729 6500 6775 6867 6345 6395 -- -- -- | <0.01 <0.01 0.34 0.22 0.18 0.14 0.12 -- -- -- | |
| TzNat C | CHCl3 CH2Cl2 THF acetone CH3CN DMF DMSO EtOH H2O PBS | 356 (12.5), 461 (6.4) 356 (9.8), 454 (6.1) 369 (8.6), 479 (1.1) 363 (8.4), 470 (1.7) 363 (5.4), 470 (2.1) 372 (9.2), 470 (1.7) 371 (9.2), 472 (2.7) 378 (4.6), 452 (5.5) 427 (7.5) 429 (7.1) | 470 467 460 459 458 476, 556 477, 558 479 561 561 | 6813 6676 5361 5761 5714 5873 5989 5578 5593 5484 | <0.01 <0.01 0.31 0.25 0.11 <0.01 <0.01 <0.01 <0.01 <0.01 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beghennou, A.; Rondot, O.; Corcé, V.; Botuha, C. 1H-1,2,3-triazolyl-1,6-naphthyridin-7(6H)-ones as Potential Fluorescent Nucleoside Analogues: Synthesis and Optical Properties. Molecules 2024, 29, 687. https://doi.org/10.3390/molecules29030687
Beghennou A, Rondot O, Corcé V, Botuha C. 1H-1,2,3-triazolyl-1,6-naphthyridin-7(6H)-ones as Potential Fluorescent Nucleoside Analogues: Synthesis and Optical Properties. Molecules. 2024; 29(3):687. https://doi.org/10.3390/molecules29030687
Chicago/Turabian StyleBeghennou, Anissa, Océane Rondot, Vincent Corcé, and Candice Botuha. 2024. "1H-1,2,3-triazolyl-1,6-naphthyridin-7(6H)-ones as Potential Fluorescent Nucleoside Analogues: Synthesis and Optical Properties" Molecules 29, no. 3: 687. https://doi.org/10.3390/molecules29030687
APA StyleBeghennou, A., Rondot, O., Corcé, V., & Botuha, C. (2024). 1H-1,2,3-triazolyl-1,6-naphthyridin-7(6H)-ones as Potential Fluorescent Nucleoside Analogues: Synthesis and Optical Properties. Molecules, 29(3), 687. https://doi.org/10.3390/molecules29030687