
Stability of Selected Phenolic Acids Under Simulated and Real Extraction Conditions from Plants



Abstract
1. Introduction
2. Results and Discussion
2.1. Studies on the Stability of 5-CQA and 1,3-diCQA Acids in Simulated MASE Extraction Conditions—Identification of Derivatives
- 1-O-caffeoylquinic acid (1-CQA, tR = 8.5 min, peak no. 3);
- 3-O-caffeoylquinic acid (3-CQA, 9.9 min, peak no. 4);
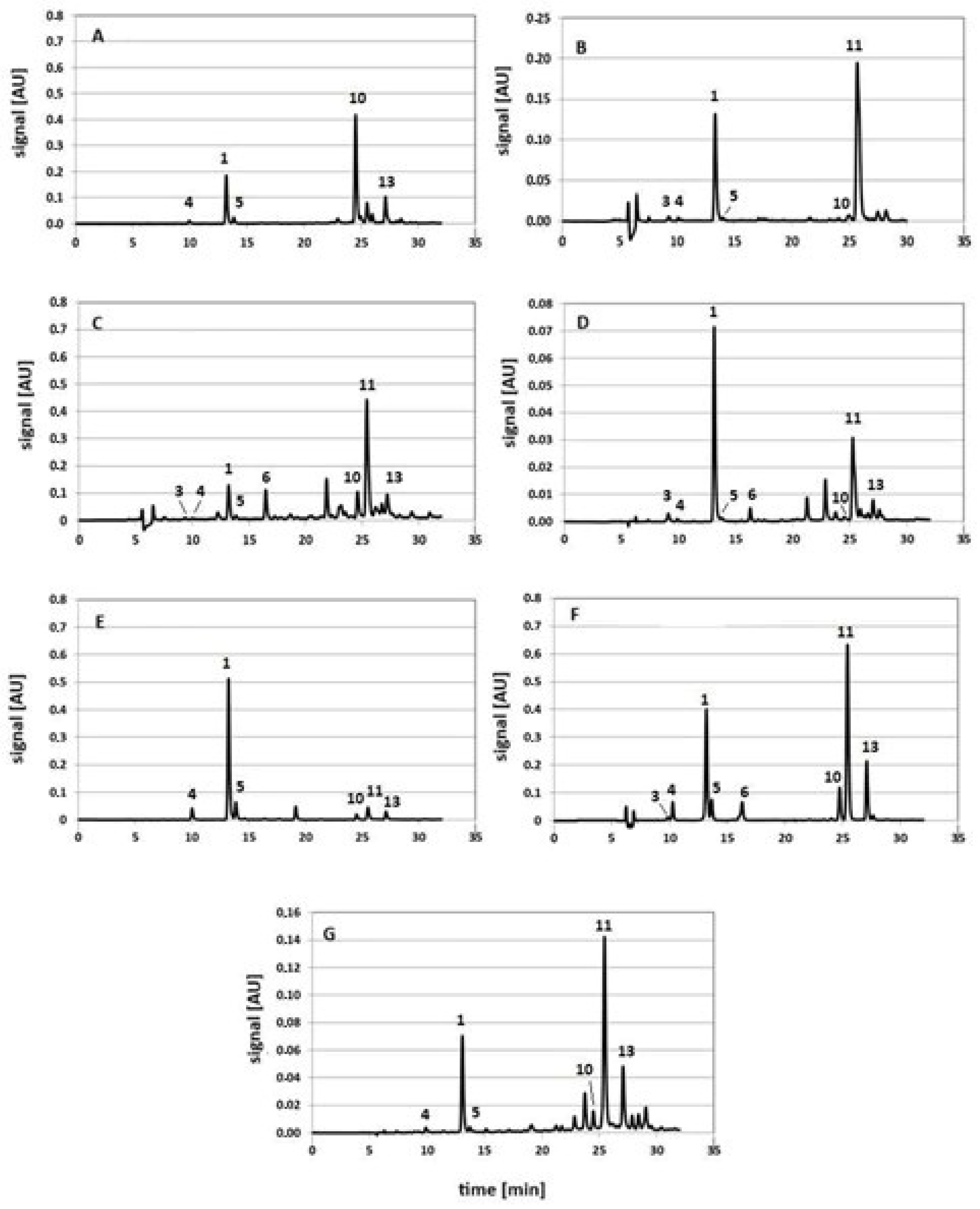
- 5-CQA (peak no. 1, see Figure 2D);
- 4-O-caffeoylquinic acid (4-CQA, 13.7 min, peak no. 5);
- cis isomer of 5-O-caffeoylquinic acid (cis-5-CQA, 16.3 min, peak no. 6);
- caffeic acid (CA, 17.3 min, peak no. 7);
- caffeic acid methyl ester (CME, 17.6 min, peak no. 8);
- 3,4-O-dicaffeoylquinic acid (3,4-diCQA, 23.7 min, peak no. 9);
- 1,5-O-dicaffeoylquinic acid (1,5-diCQA, 24.7 min, peak no. 10);
- 3,5-O-dicaffeoylquinic acid (3,5-diCQA, 25.3 min, peak no. 11);
- cis isomer of 1,5-O-dicaffeoylquinic acid (cis-1,5-diCQA, 25.6 min, peak no. 12); and
- 4,5-O-dicaffeoylquinic acid (4,5-diCQA, 26.9 min, peak no. 13).
- in the spectrum of cis-isomers, the absorption maximum is observed at shorter wavelengths of electromagnetic radiation compared to the spectrum of trans derivatives [24];
- the signal of cis-isomers in the chromatogram increases after the extension of the exposure of the 5-CQA and 1,5-diCQA standard solutions to UV–VIS [25].
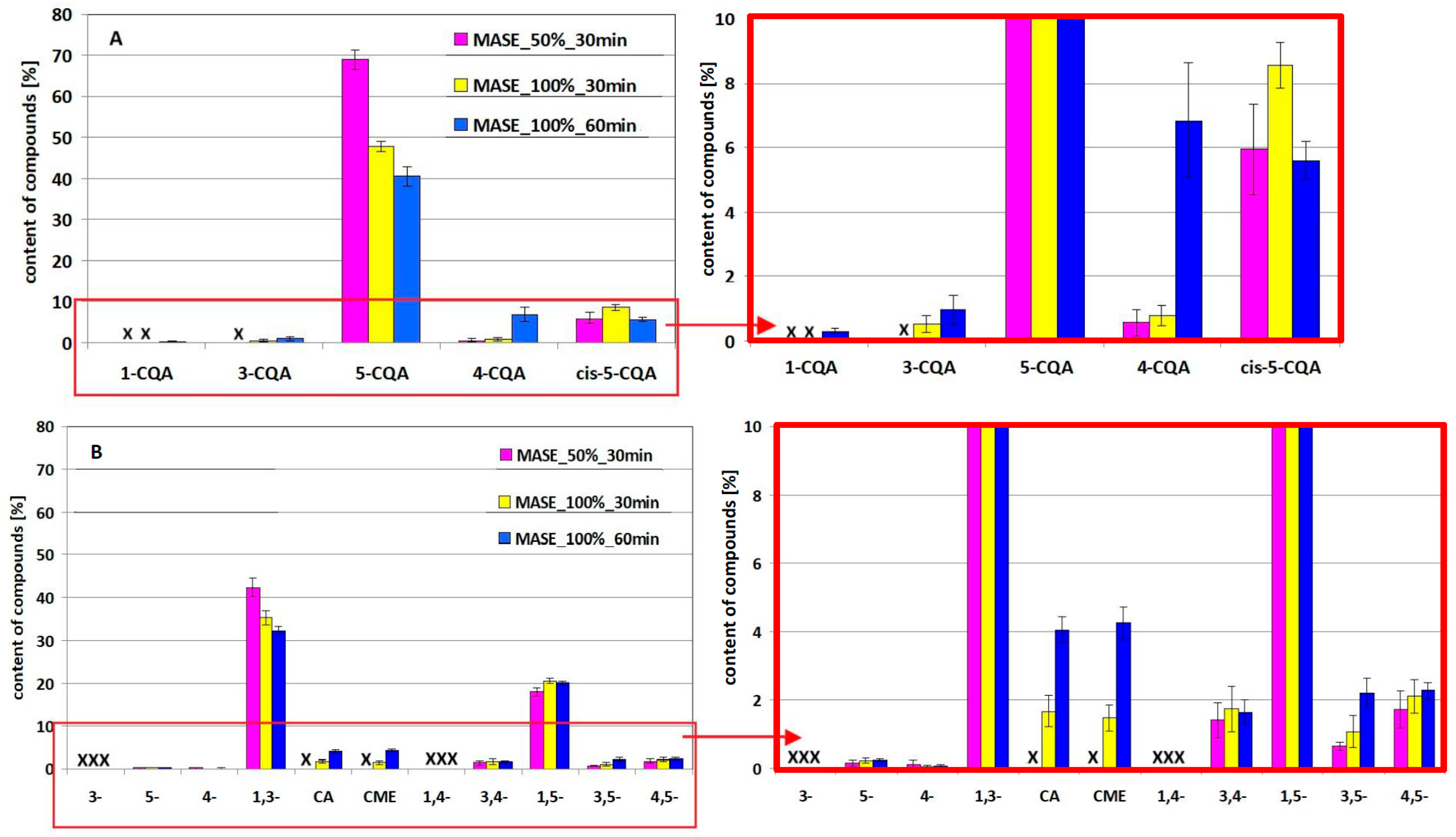
2.2. Assessment of the Amount of 5-CQA and 1,3-diCQA and Their Derivatives Under Simulated MASE Conditions
- 5-CQA is more stable than 1,3-diCQA under the same process conditions;
- changing the MASE conditions differentiates the loss of both compounds;
- increasing the power of the microwave generator, with the same exposure time of the standards, causes their greater transformation/degradation;
- extending the exposure time from 30 to 60 min at a given generator power (in this case 100% power) also causes greater losses.
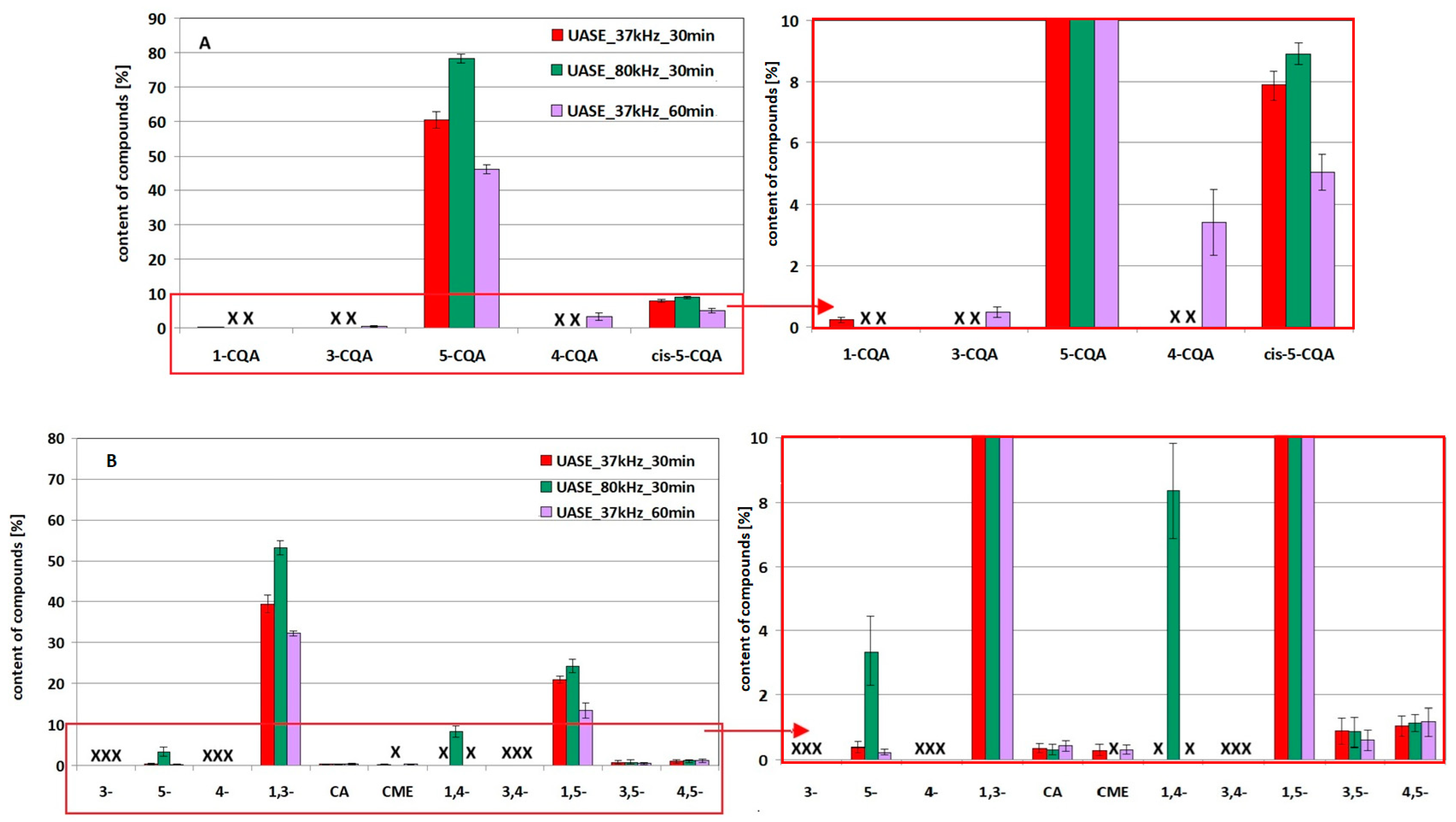
2.3. Stability/Quantity Studies of 5-CQA and 1,3-diCQA and Their Derivatives Under Simulated UASE Extraction Conditions
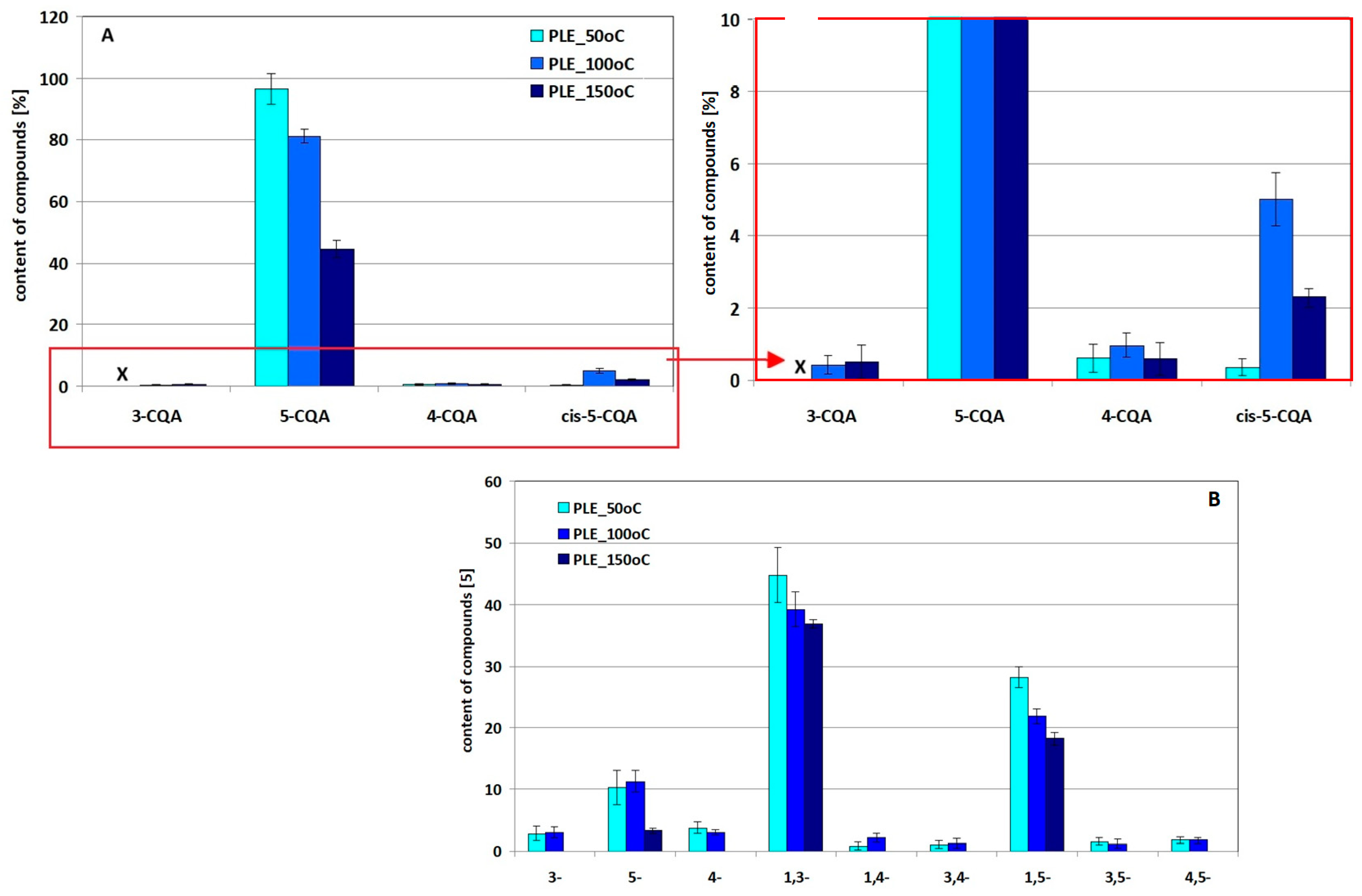
2.4. Stability/Quantity Studies of 5-CQA and 1,3-diCQA and Their Derivatives Under Simulated PLE Extraction Conditions
- there is an increase in the 5-CQA derivative signal (compare peak height 1 in Figures S2D and S3A);
- there are decreases in the 5-CQA, 1,3-diCQA and 1,5-diCQA signals with increasing extraction time;
- there are increases in the 3,4-diCQA and 4,5-diCQA signals with increased extraction time; and
- there appear 3-CQA and 4-CQA derivatives.
2.5. Studies on the Stability/Quantity of 5-CQA and 1,3-diCQA and Their Derivatives Under Simulated SSDM Extraction Conditions—Comparison of the Destructive Nature of Assisted Extraction Techniques
2.6. Studies on the Stability/Quantity of Chlorogenic Acids and Their Derivatives in Real Extraction Conditions from Diverse Plant Matrices—Comparison of Acid Content in Extracts Obtained by MASE, UASE, PLE and SSDM Techniques
- Compounds identified in model systems as degradation and/or transformation products of 5-CQA and 1,3-diCQA occur in real systems. Their number and content, however, varies and depends on the type of plant and its matrix, the extraction technique and the process conditions applied. An exception is CME identified in chamomile flower extracts under MASE and UASE conditions (see Table S16). Considering that this compound was not detected in the extract obtained using the SSDM technique, it should be considered an artifact.
- In general, the highest number of tested compounds was estimated for coffee (see Table S8), and the lowest for chamomile (Table S16). Artichoke (leaves) is characterized by the highest numbers of identified 5-CQA and 1,3-diCQA transformation/degradation products (Table S10). In the hearts of artichoke inflorescence buds, i.e., in the youngest elements of the plant, in contrast to artichoke leaves, the presence of CA and 1,4-diCQA was not confirmed (see Table S12). This fact can be related to the age of individual morphological elements of the plant (“aging” of the plant) and/or the storage time of the collected raw material.
- Among the mono-caffeoylquinic acids, the dominant compound in all plants is the 5-CQA isomer. The contents of 4-CQA and 3-CQA are lower, with the amount of 4-CQA exceeding the amount of 3-CQA. 1-CQA and cis-5-CQA are relatively rare compounds. Their presence was confirmed for artichoke, yarrow and tansy and coffee and artichoke, respectively.
- Among the di-caffeoylquinic acids, the most popular isomer is 1,5-diCQA and the rarest is 1,3-diCQA (this compound was identified only in artichoke extracts, it should be added that it is not a dominant compound although it is considered characteristic of this plant). Generally, 1,5-diCQA is the dominant representative of di-CQA acids. Only in the case of coltsfoot the dominant representative of this group of compounds is the 3,4-diCQA isomer.
- By comparing the amounts of native plant components, i.e., compounds present in the SSDM extract with the amounts of these compounds estimated in extracts obtained using assisted extraction techniques, it can be concluded that their content is a consequence of the success of the isolation process (its efficiency) and the degree of degradation/transformation of the main components during assisted extraction techniques. The greatest differences in the total amount of analyzed compounds, estimated in relation to the amounts revealed for each plant in the SSDM process, are observed for the MASE technique, UASE at an ultrasound frequency of 37 kHz and PLE conducted at 150 °C.
- When analyzing the contents of model compounds (mainly 5-CQA due to their common occurrence) with the amounts of their identified derivatives in extracts of individual plants, it is difficult to find unambiguous correlations. This may indicate differences in their metabolic pathways leading to the synthesis of other acids/compounds.
3. Materials and Methods
3.1. Materials
3.2. Extraction Methods Used During the Studies
3.2.1. MASE
- microwave generator power: 50 and/or 100% of maximum power (800 W);
- extraction time: 30 and/or 60 min in a single extraction cycle regardless of the plant matrix and analyte.
3.2.2. UASE
- frequency: 37 kHz and/or 80 kHz;
- ultrasonic generator power: 50% and/or 100% of the maximum power (720 W);
- extraction time: 30 or 60 min;
- temperature: 25 °C.
3.2.3. PLE
- extraction time: 10 and/or 20 min;
- temperature: 50, 100 and 150 °C;
- pressure: 40 bar.
3.2.4. SSDM
3.3. HPLC
3.4. LC–MS
3.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reshi, Z.A.; Ahmad, W.; Lukatkin, A.S.; Javed, S.B. From Nature to Lab: A Review of Secondary Metabolite Biosynthetic Pathways, Environmental Influences, and In Vitro Approaches. Metabolites 2023, 13, 895. [Google Scholar] [CrossRef] [PubMed]
- Crozier, A.; Yokota, T.; Jaganath, I.B.; Marks, S.; Saltmarsh, M.; Clifford, M.N. Secondary Metabolites in Fruits, Vegetables, Beverages and Other Plant-based Dietary Components. In Plant Secondary Metabolites; Blackwell Publishing Ltd.: Oxford, UK, 2007; pp. 208–302. [Google Scholar] [CrossRef]
- Choi, Y.E.; Choi, S.I.; Han, X.; Men, X.; Jang, G.W.; Kwon, H.Y.; Kang, S.R.; Han, J.S.; Lee, O.A. Radical Scavenging-Linked Anti-Adipogenic Activity of Aster scaber Ethanolic Extract and Its Bioactive Compound. Antioxidants 2020, 9, 1290. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G. Protection against developing type 2 diabetes by coffee consumption: Assessment of the role of chlorogenic acid and metabolites on glycaemic responses. Food Funct. 2020, 11, 4826–4833. [Google Scholar] [CrossRef]
- Kusumah, J.; Gonzalez de Mejia, E. Coffee constituents with antiadipogenic and antidiabetic potentials: A narrative review. Food Chem. Toxicol. 2022, 161, 112821. [Google Scholar] [CrossRef]
- Ludwig, I.A.; Clifford, M.N.; Lean, M.E.J.; Ashihara, H.; Crozier, A. Coffee: Biochemistry and potential impact on health. Food Funct. 2014, 5, 1695–1717. [Google Scholar] [CrossRef]
- Wianowska, D.; Gil, M. Recent advances in extraction and analysis procedures of natural chlorogenic acids. Phytochem. Rev. 2019, 18, 173–302. [Google Scholar] [CrossRef]
- Jaiswal, R.; Kiprotich, J.; Kuhnert, N. Determination of the hydroxycinnamate profile of 12 members of the Asteraceae family. Phytochemistry 2011, 72, 781–790. [Google Scholar] [CrossRef]
- Wu, J.; Qian, Y.; Mao, P.; Chen, L.; Lu, Y.; Wang, H. Separation and identification of phenolic compounds in canned artichoke by LC/DAD/ESI-MS using core-shell C18 column: A comparative study. J. Chromatogr. B Biomed. Appl. 2013, 927, 173–180. [Google Scholar] [CrossRef]
- Yang, M.; Ma, Y.; Wang, Z.; Khan, A.; Zhou, W.; Zhao, T.; Cao, J.; Cheng, G.; Cai, S. Phenolic constituents, antioxidant and cytoprotective activities of crude extract and fractions from cultivated artichoke inflorescence. Ind. Crops Prod. 2020, 143, 111433. [Google Scholar] [CrossRef]
- Tena, M.T.; Martínez-Moral, M.P.; Cardozo, P.W. Determination of caffeoylquinic acids in feed and related products by focused ultrasound solid-liquid extraction and ultra-high performance liquid chromatography-mass spectrometry. J. Chromatogr. A 2015, 1400, 1–9. [Google Scholar] [CrossRef]
- Haddouchi, F.; Chaouche, T.M.; Ksouri, R.; Larbat, R. Leafy Stems of Phagnalon saxatile subsp. Saxatile from Algeriaas a Source of Chlorogenic Acids and Flavonoids with Antioxidant Activity: Characterization and Quantification Using UPLC-DAD-ESI-MSn. Metabolites 2021, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Müller, H.; Müller, A.; Karar, M.G.E.; Kuhnert, N. Identification and characterization of chlorogenic acids, chlorogenic acid glycosides and flavonoids from Lonicera henryi L. (Caprifoliaceae) leaves by LC-MSn. Phytochemistry 2014, 108, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, S.C.; Castilho, P.C. Validation of a HPLC-DAD-ESI/MS n method for caffeoylquinic acids separation, quantification and identification in medicinal Helichrysum species from Macaronesia. Food Res. Int. 2012, 45, 362–368. [Google Scholar] [CrossRef]
- Dias, M.I.; Barros, L.; Dueñas, M.; Pereira, E.; Carvalho, A.M.; Alves, R.C.; Oliveira, M.B.P.P.; Santos-Buelga, C.; Ferreira, I.C.F.R. Chemical composition of wild and commercial Achillea millefolium L. and bioactivity of the methanolic extract, infusion and decoction. Food Chem. 2013, 141, 4152–4160. [Google Scholar] [CrossRef]
- Wianowska, D.; Typek, R.; Dawidowicz, A.L. How to eliminate the formation of chlorogenic acids artefacts during plants analysis? Sea sand disruption method (SSDM) in the HPLC analysis of chlorogenic acids and their native derivatives in plants. Phytochemistry 2015, 117, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Wianowska, D.; Typek, R.; Dawidowicz, A.L. Chlorogenic acid stability in pressurized liquid extraction conditions. J. AOAC Int. 2015, 98, 415–421. [Google Scholar] [CrossRef]
- Dawidowicz, A.L.; Typek, R. Transformation of chlorogenic acids during the coffee beans roasting process. Eur. Food Res. Technol. 2017, 243, 379–390. [Google Scholar] [CrossRef]
- Wianowska, D.; Dawidowicz, A.L.; Bernacik, K.; Typek, R. Determining the true content of quercetin and its derivatives in plants employing SSDM and LC–MS analysis. Eur. Food Res. Technol. 2017, 243, 27–40. [Google Scholar] [CrossRef]
- Deshpande, S.; Jaiswal, R.; Matei, M.F.; Kuhnert, N. Investigation of acyl migration in mono- and dicaffeoylquinic acids under aqueous basic, aqueous acidic, and dry roasting conditions. J. Agric. Food Chem. 2014, 62, 9160–9170. [Google Scholar] [CrossRef]
- Yang, J.; Yao, L.; Gong, K.; Li, K.; Sun, L.; Cai, W. Identification and Quantification of Chlorogenic Acids from the Root Bark of Acanthopanax gracilistylus by UHPLC-Q-Exactive Orbitrap Mass Spectrometry. ACS Omega 2022, 7, 25675–25685. [Google Scholar] [CrossRef]
- Ramabulana, A.T.; Steenkamp, P.; Madala, N.; Duber, I.A. Profiling of Chlorogenic Acids from Bidens pilosa and Differentiation of Closely Related Positional Isomers with the Aid of UHPLC-QTOF-MS/MS-Based In-Source Collision-Induced Dissociation. Metabolites 2020, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.; Burgos, V.; Marín, V.; Camins, A.; Olloquequi, J.; González-Chavarría, I.; Ulrich, H.; Wyneken, U.; Luarte, A.; Ortiz, L.; et al. Caffeic Acid Phenethyl Ester (CAPE): Biosynthesis, Derivatives and Formulations with Neuroprotective Activities. Antioxidants 2023, 12, 1500. [Google Scholar] [CrossRef] [PubMed]
- Clifford, M.N.; Kirkpatrick, J.; Kuhnert, N.; Roozendaal, H.; Salgado, P.R. LC-MSn analysis of the cis isomers of chlorogenic acids. Food Chem. 2008, 106, 379–385. [Google Scholar] [CrossRef]
- Cangeloni, L.; Bonechi, C.; Leone, G.; Consumi, M.; Andreassi, M.; Magnani, A.; Rossi, C.; Tamasi, G. Characterization of Extracts of Coffee Leaves (Coffea arabica L.) by Spectroscopic and Chromatographic/Spectrometric Techniques. Foods 2022, 11, 2495. [Google Scholar] [CrossRef]
- Cirkva, V.; Relich, S. Microwave Photochemistry and Photocatalysis. Part 1: Principles and Overview. Curr. Org. Chem. 2010, 15, 248–264. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
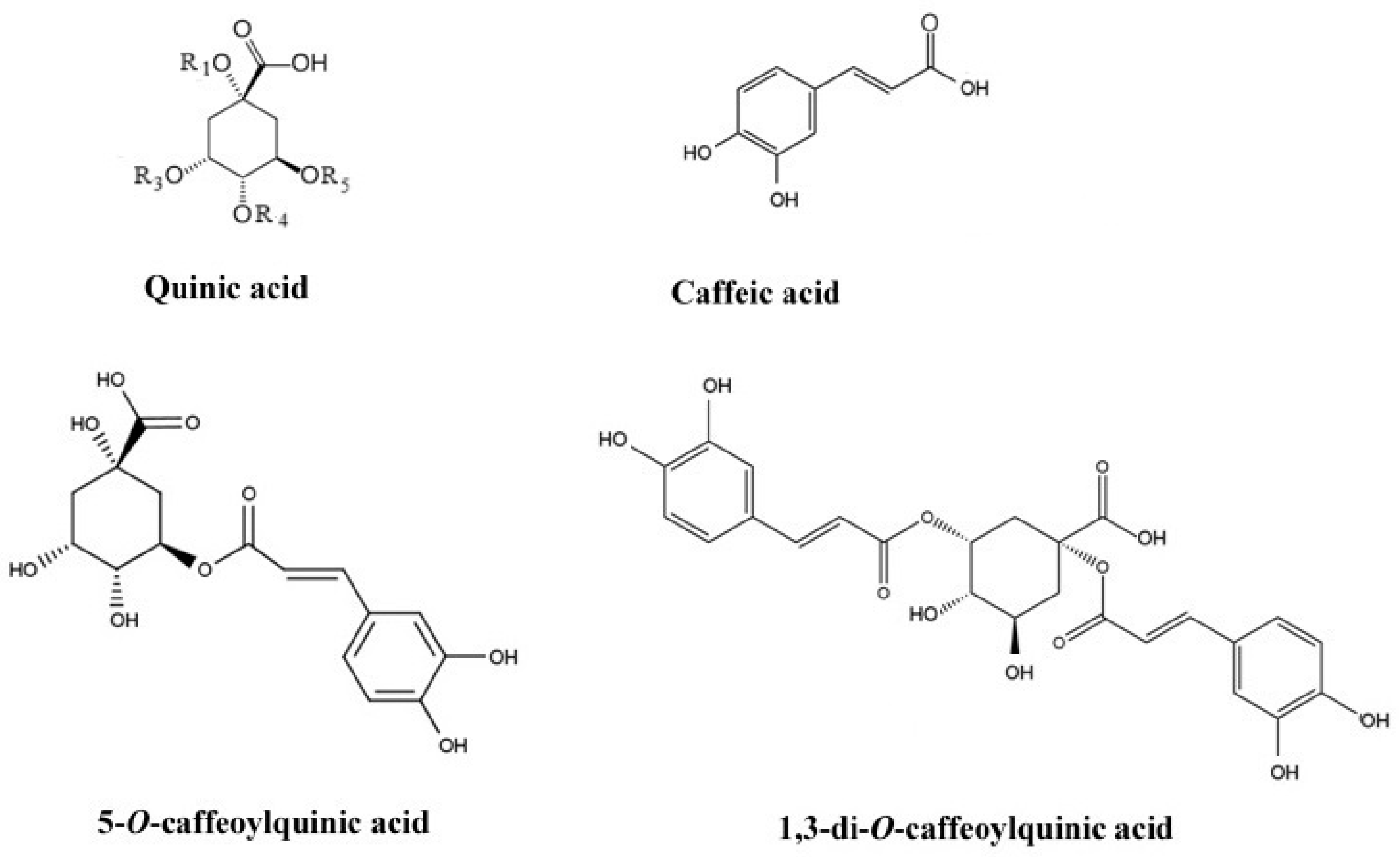
| No. | Compound Name | Abbreviation | R1 | R3 | R4 | R5 |
|---|---|---|---|---|---|---|
| 1 | 1-O-caffeoylquinic acid | 1-CQA | CA | H | H | H |
| 2 | 3-O-caffeoylquinic acid (neochlorogenic acid) | 3-CQA | H | CA | H | H |
| 3 | 5-O-caffeoylquinic acid (chlorogenic acid) | 5-CQA | H | H | H | CA |
| 4 | 4-O-caffeoylquinic acid (chlorogenic acid) | 4-CQA | H | H | CA | H |
| 5 | 1,3-di-O-caffeoylquinic acid | 1,3-di-CQA | CA | CA | H | H |
| 6 | 1,4-di-O-caffeoylquinic acid | 1,4-di-CQA | CA | H | CA | H |
| 7 | 1,5-di-O-caffeoylquinic acid | 1,5-di-CQA | CA | H | H | CA |
| 8 | 3,4-di-O-caffeoylquinic acid | 3,4-di-CQA | H | CA | CA | H |
| 9 | 3,5-di-O-caffeoylquinic acid | 3,5-di-CQA | H | CA | H | CA |
| 10 | 4,5-di-O-caffeoylquinic acid | 4,5-di-CQA | H | H | CA | CA |
| Compound Number | tR [min] | λmax [nm] | MS1 Parent Ion [M-H]− m/z | MS2 Characteristic Ions (Intensity, %) m/z (%) | Compound Shortcut |
|---|---|---|---|---|---|
| 1 | 13.1 | 300*, 326 | 353 | 191 (100), 179 (5) | trans-5-CQA (5-CQA) |
| 2 | 16.2 | 300*, 322 | 515 | 353 | 1,3-diCQA |
| 3 | 8.5 | 300*, 325 | 353 | 191 (100) | 1-CQA |
| 4 | 9.9 | 300*, 328 | 353 | 191 (100), 179 (91), 173 (1);135 (14) | 3-CQA |
| 5 | 13.7 | 300*, 328 | 353 | 179 (100), 191 (15), 173 (88), 135 (9) | 4-CQA |
| 6 | 16.3 | 300, 315 | 353 | 191 (100), 179 (2) | cis-5-CQA |
| 7 | 17.1 | 300*, 321 | 179 | 135 (100) | CA |
| 8 | 17.3 | 280 | 194 | 161 (100) | CME |
| 9 | 23.9 | 300*, 323 | 515 | 353 | 3,4-diCQA |
| 10 | 24.6 | 300*, 328 | 515 | 353 | 1,5-diCQA |
| 11 | 25.3 | 300, 323 | 515 | 353 | 3,5-diCQA |
| 12 | 25.7 | 300*, 320 | 515 | 353 | cis-1,5-diCQA |
| 13 | 27.0 | 300*, 328 | 515 | 353 | 4,5-diCQA |
| 14 | 21.2 | 300*, 323 | 515 | 353 | 1,4-diCQA |
| Compound Shortcut | MASE | UASE | PLE | SSDM | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 50% 30 min | 100% 30 min | 100% 60 min | 37 kHz 30 min | 80 kHz 30 min | 37 kHz 60 min | 50 °C | 100 °C | 150 °C | ||
| 5-CQA | 6.89 ± 0.28 | 4.75 ± 0.18 | 4.05 ± 0.24 | 6.03 ± 0.23 | 7.83 ± 0.12 | 4.60 ± 0.14 | 9.64 ± 0.49 | 8.12 ± 0.29 | 4.45 ± 0.23 | 9.79 ± 0.13 |
| 1,3-diCQA | 4.24 ± 0.21 | 3.53 ± 0.18 | 3.24 ± 0.07 | 3.95 ± 0.22 | 5.32 ± 0.18 | 3.22 ± 0.07 | 4.48 ± 0.45 | 3.92 ± 0.28 | 3.68 ± 0.07 | 9.98 ± 0.49 |
| Compound Shortcut | MASE/SSDM | UASE/SSDM | PLE/SSDM | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 50%_30 min | 100%_30 min | 100%_60 min | 37 kHz_30 min | 80 kHz_30 min | 37 kHz_60 min | 50 °C | 100 °C | 150 °C | |
| F-Value (p) | F-Value (p) | F-Value (p) | F-Value (p) | F-Value (p) | F-Value (p) | F-Value (p) | F-Value (p) | F-Value (p) | |
| 5-CQA | 270.75 (8.00 × 10−5) | 1587.16 (2.37 × 10−6) | 1345.38 (3.30 × 10−6) | 614.71 (1.57 × 10−2) | 386.98 (3.94 × 10−5) | 2259.23 (1.17 × 10−6) | 0.250 (0.644) | 125.06 (3.64 × 10−4) | 857.20 (8.08 × 10−6) |
| 1,3-diCQA | 347.23 (4.88 × 10−5) | 461.42 (2.78 × 10−5) | 561.08 (1.88 × 10−5) | 384.30 (4.00 × 10−5) | 241.12 (1.00 × 10−4) | 563.69 (1.87 × 10−6) | 206.00 (1.37 × 10−4) | 348.27 (4.85 × 10−5) | 489.81 (2.47 × 10−5) |
| Compound Shortcut | Slope (a) | Intercept (b) | R2 | Slope Estimation Error | Intercept Estimation Error | Calibration Curve Error |
|---|---|---|---|---|---|---|
| 5-CQA | 2464.72 | −0.3093 | 0.9999 | 15.4832 | 0.34853 | 0.09853 |
| 1,3-di-CQA | 3077.74 | 1.0663 | 0.9997 | 113.546 | 0.82738 | 0.67254 |
| 1,5-di-CQA | 2560.02 | 0.2908 | 0.9996 | 89.5472 | 0.89751 | 0.86738 |
| CA | 0.22433 | −0.0080 | 0.9958 | 0.01282 | 0.04966 | 0.01959 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olszowy-Tomczyk, M.; Paprotny, Ł.; Wianowska, D. Stability of Selected Phenolic Acids Under Simulated and Real Extraction Conditions from Plants. Molecules 2024, 29, 5861. https://doi.org/10.3390/molecules29245861
Olszowy-Tomczyk M, Paprotny Ł, Wianowska D. Stability of Selected Phenolic Acids Under Simulated and Real Extraction Conditions from Plants. Molecules. 2024; 29(24):5861. https://doi.org/10.3390/molecules29245861
Chicago/Turabian StyleOlszowy-Tomczyk, Małgorzata, Łukasz Paprotny, and Dorota Wianowska. 2024. "Stability of Selected Phenolic Acids Under Simulated and Real Extraction Conditions from Plants" Molecules 29, no. 24: 5861. https://doi.org/10.3390/molecules29245861
APA StyleOlszowy-Tomczyk, M., Paprotny, Ł., & Wianowska, D. (2024). Stability of Selected Phenolic Acids Under Simulated and Real Extraction Conditions from Plants. Molecules, 29(24), 5861. https://doi.org/10.3390/molecules29245861