

Synthesis of 1,2,4-Oxadiazin-5(6H)-One Derivatives and Their Biological Investigation as Monoamine Oxidase Inhibitors

 , , , , ,
, , , , ,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
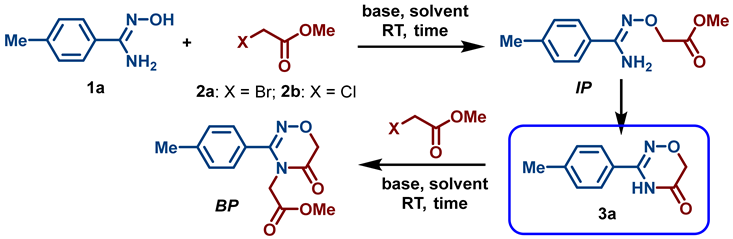
2.1. Chemistry
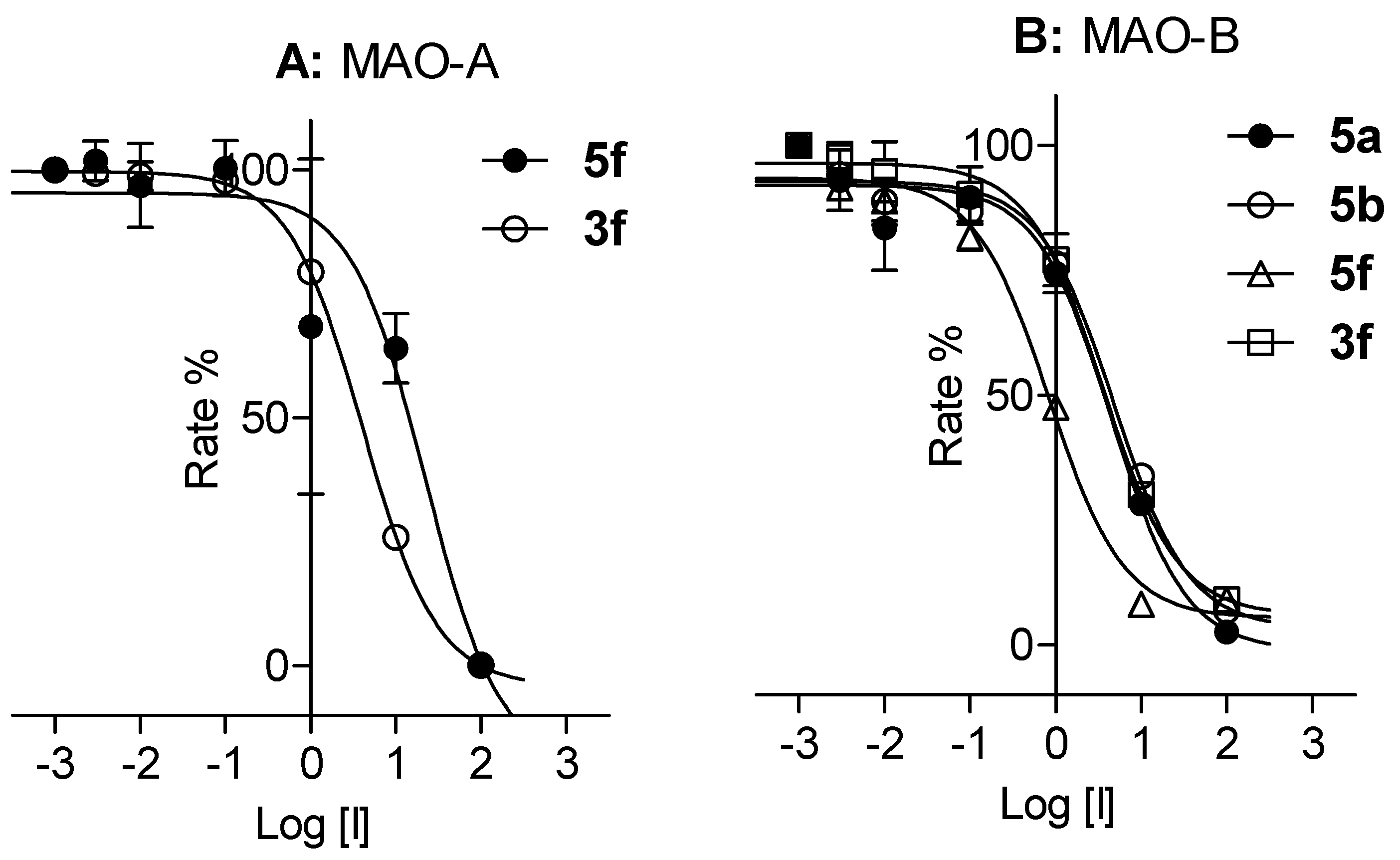
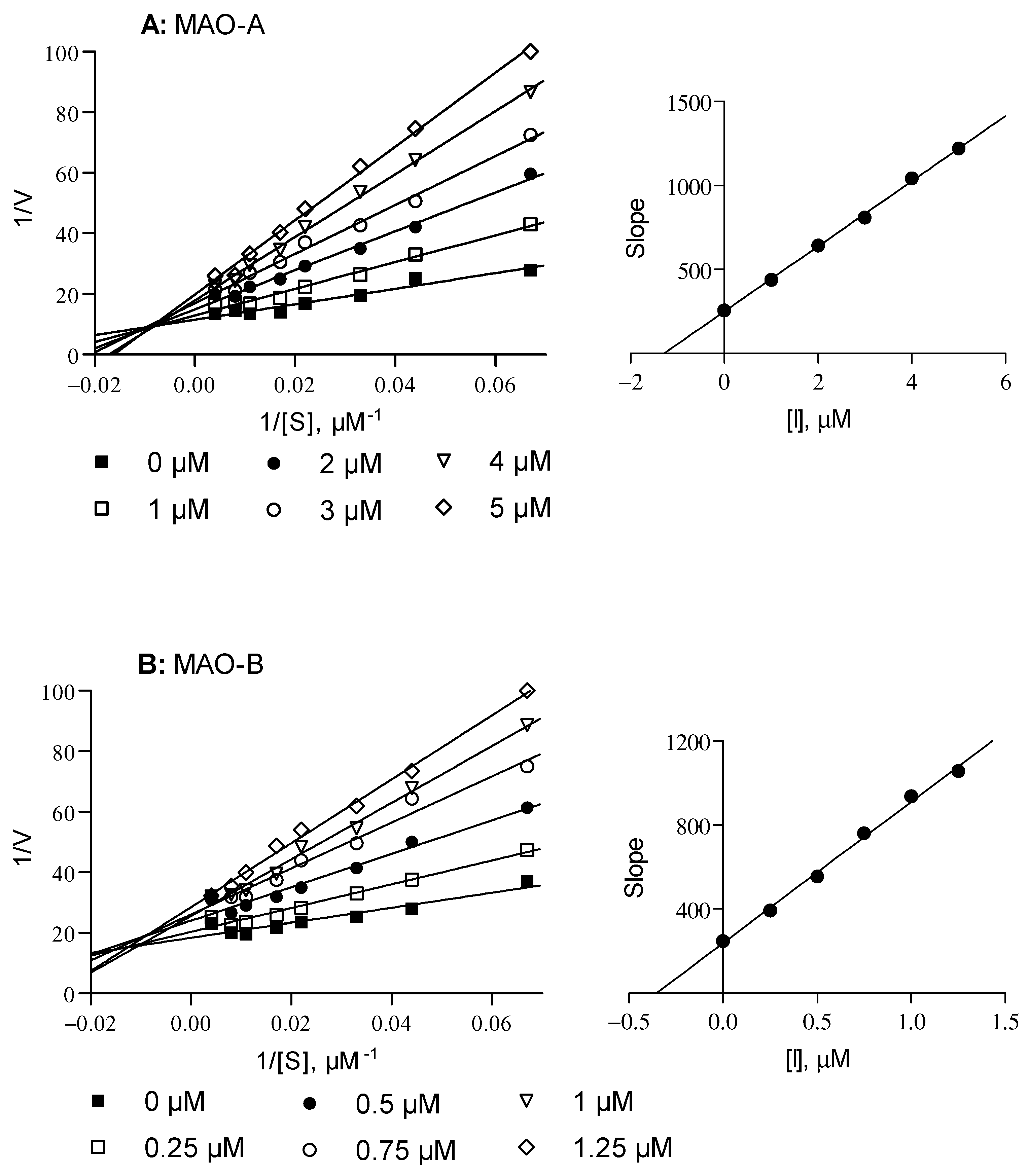
2.2. Biological Studies
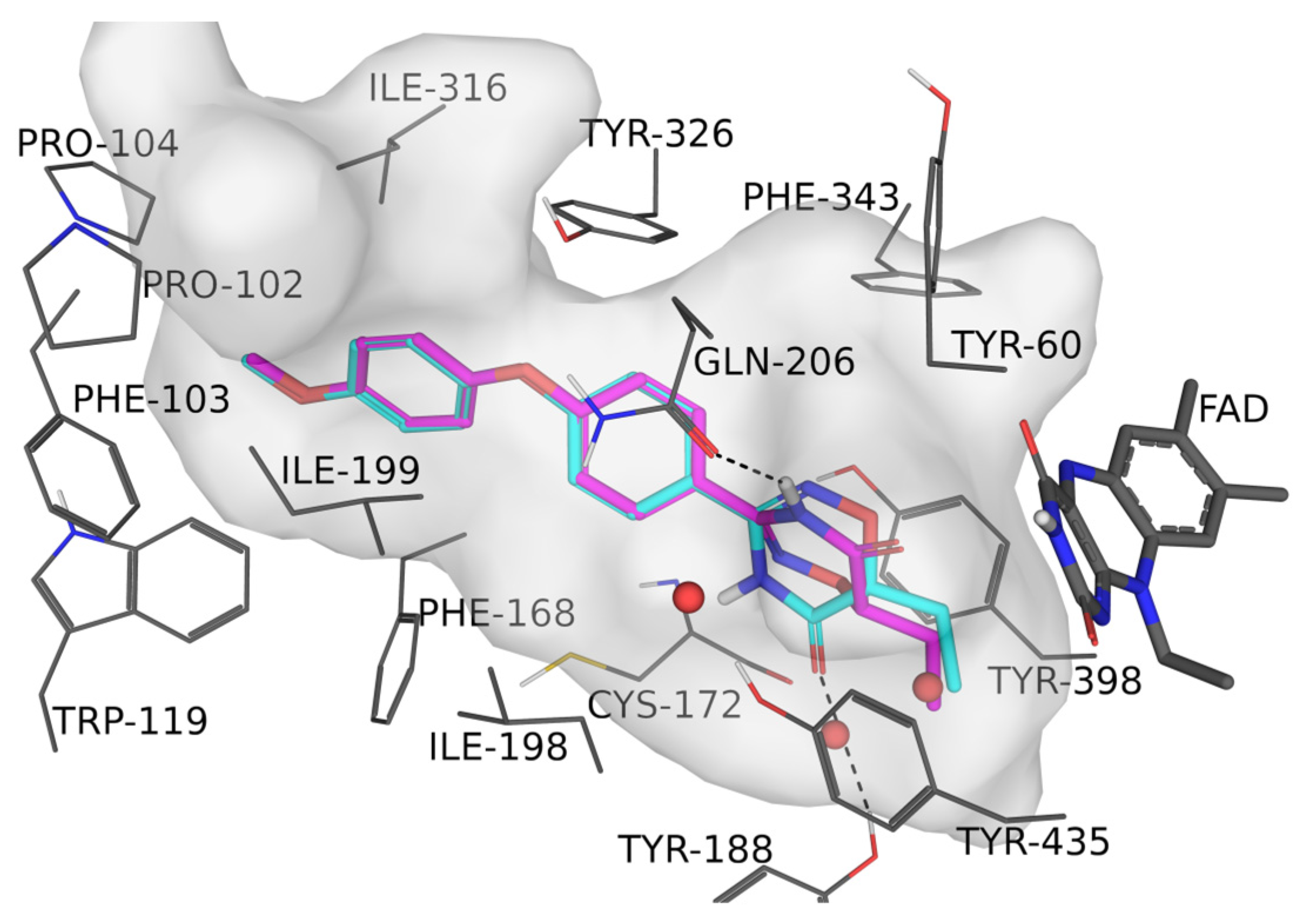
2.3. Molecular Docking
3. Experimental Section
3.1. Material and Methods
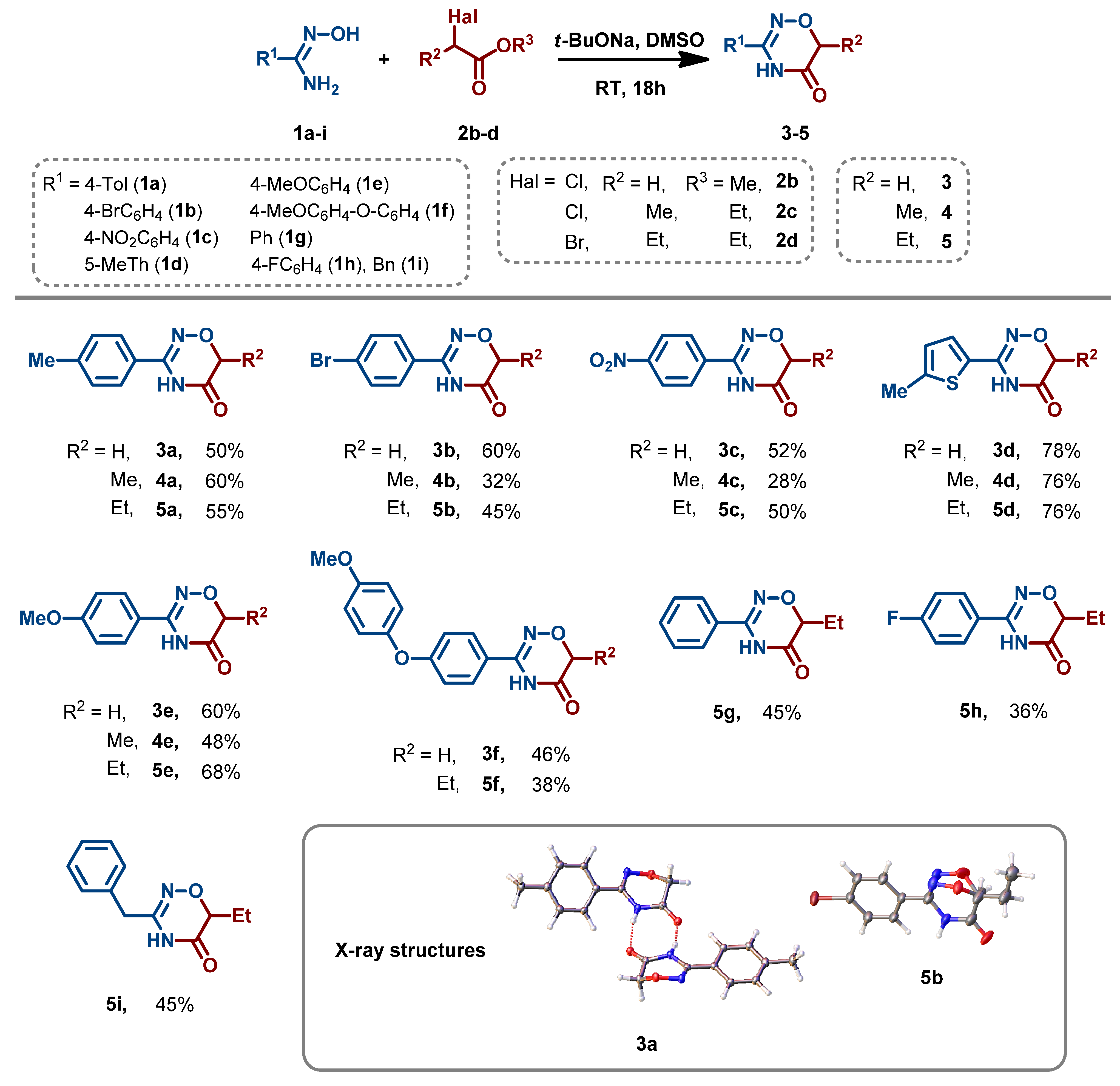
3.2. Synthesis and Characterization of 1,2,4-Oxadiazin-5(6H)-Ones
- General procedure for the synthesis of 1,2,4-oxadiazin-5(6H)-ones (3–5). To a solution of amidoxime 1 (2 mmol) in DMSO (3 mL), t-BuONa (384 mg, 4 mmol) was rapidly added at room temperature. The reaction mixture was stirred at room temperature for 10 min, and ester 2 (2.4 mmol) was added. The reaction mixture was stirred at room temperature for another 18 h and was then diluted with HCl (10% solution in water) (30 mL). The resulting precipitate was collected by filtration, washed with cold water (5 mL), and air-dried at 50 °C.
- 3-(4-Methylphenyl)-4H-1,2,4-oxadiazin-5(6H)-one (3a). White powder; 50% yield; mp 140–141 °C. 1H NMR (400 MHz, CDCl3) δ 9.05 (s, 1H), 7.66 (d, J = 8.2 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 4.47 (s, 2H), 2.43 (s, 3H).13C NMR (101 MHz, CDCl3) δ 166.6, 151.0, 142.3, 129.8, 126.0, 125.7, 66.9, 21.5. HRMS (ESI), m/z: [M–H]− calcd for C10H9N2O2− 189.0669; found 189.0667.
- 3-(4-Bromophenyl)-4H-1,2,4-oxadiazin-5(6H)-one (3b). Pale beige powder; 59% yield; mp 180–181 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H), 7.70 (s, 4H), 4.38 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.4, 151.7, 132.4, 129.4, 129.0, 125.4, 67.2. HRMS (ESI), m/z: [M–H]− calcd for C9H6BrN2O2− 252.9618; found 252.9620.
- 3-(4-Nitrophenyl)-4H-1,2,4-oxadiazin-5(6H)-one (3c). Pale yellow powder; 52% yield; mp 248–250 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.36–8.30 (m, 2H), 8.06–8.00 (m, 2H), 4.44 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.0, 150.9, 149.4, 135.5, 128.7, 124.2, 67.0. HRMS (ESI), m/z: [M–H]− calcd for C9H6N3O4− 220.0364; found 220.0364.
- 3-(5-Methylthiophen-2-yl)-4H-1,2,4-oxadiazin-5(6H)-one (3d). Pale beige powder; 77% yield; mp 216–218 °C. 1H NMR (400 MHz, CDCl3) δ 9.30 (s, 1H), 7.35 (d, J = 3.7 Hz, 1H), 6.80 (d, J = 3.5 Hz, 1H), 4.47 (s, 2H), 2.54 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.4, 148.4, 144.6, 129.7, 128.9, 126.8, 67.5, 15.7. HRMS (ESI), m/z: [M–H]− calcd for C8H8N2O2S− 195.0234; found 195.0235.
- 3-(4-Methoxyphenyl)-4H-1,2,4-oxadiazin-5(6H)-one (3e). White powder; 59% yield; mp 188–190 °C. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 7.68 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.9 Hz, 2H), 4.47 (s, 2H), 3.89 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.5, 162.0, 151.8, 128.7, 121.6, 114.5, 67.0, 55.8. HRMS (ESI), m/z: [M–H]− calcd for C10H9N2O3− 205.0619; found 205.0622.
- 3-(4-(4-Methoxyphenoxy)phenyl)-4H-1,2,4-oxadiazin-5(6H)-one (3f). White powder; 36% yield; mp 127–129 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 7.74 (d, J = 8.8 Hz, 2H), 7.10–6.94 (m, 6H), 4.35 (s, 2H), 3.78 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.4, 161.1, 156.7, 151.7, 148.8, 129.1, 123.4, 121.9, 117.1, 115.7, 67.0, 55.9. HRMS (ESI), m/z: [M–H]− calcd for C16H13N2O4− 297.0881; found 297.0880.
- 6-Methyl-3-(4-methylphenyl)-4H-1,2,4-oxadiazin-5(6H)-one (4a). White powder; 58% yield; mp 123–125 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 7.64 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 7.8 Hz, 2H), 4.32 (q, J = 6.6 Hz, 1H), 2.35 (s, 3H), 1.36 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 152.0, 141.5, 129.6, 127.0, 126.7, 72.1, 21.4, 13.6. HRMS (ESI), m/z: [M–H]− calcd for C11H11N2O2− 203.0826; found 203.0826.
- 3-(4-Bromophenyl)-6-methyl-4H-1,2,4-oxadiazin-5(6H)-one (4b). White powder; 31% yield; mp 192–194 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.37 (s, 1H), 7.70 (s, 4H), 4.35 (q, J = 6.7 Hz, 1H), 1.37 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.4, 151.6, 132.4, 129.4, 129.0, 125.4, 72.4, 13.8. HRMS (ESI), m/z: [M–H]− calcd for C10H8BrN2O2− 266.9775; found 266.9779.
- 6-Methyl-3-(4-nitrophenyl)-4H-1,2,4-oxadiazin-5(6H)-one (4c). White powder; 31% yield; mp 247–249 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 8.32 (d, J = 8.5 Hz, 2H), 8.02 (d, J = 8.5 Hz, 2H), 4.41 (q, J = 6.7 Hz, 1H), 1.39 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 151.1, 149.6, 135.7, 128.8, 124.4, 72.5, 13.8. HRMS (ESI), m/z: [M–H]− calcd for C11H11N2O3− 219.0775; found 219.0775.
- 6-Methyl-3-(5-methylthiophen-2-yl)-4H-1,2,4-oxadiazin-5(6H)-one (4d). Pale beige powder; 76% yield; mp 167–169 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H), 7.56 (d, J = 3.7 Hz, 1H), 6.85 (d, J = 2.4 Hz, 1H), 4.34 (q, J = 6.7 Hz, 1H), 2.45 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.3, 148.4, 144.5, 129.6, 129.0, 126.7, 72.7, 15.7, 13.8. HRMS (ESI), m/z: [M–H]− calcd for C9H9N2O2S− 209.0390; found 209.0391.
- 3-(4-Methoxyphenyl)-6-methyl-4H-1,2,4-oxadiazin-5(6H)-one (4e). White powder; 47% yield; mp 134–136 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.22 (s, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 4.31–4.29 (m, 1H), 3.80 (s, 3H), 1.36 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.7, 162.2, 152.0, 128.9, 121.9, 114.7, 72.3, 56.1, 13.8. HRMS (ESI), m/z: [M–H]− calcd for C11H11N2O3− 219.0775; found 219.0775.
- 6-Ethyl-3-(4-methylphenyl)-4H-1,2,4-oxadiazin-5(6H)-one (5a). White powder; 55% yield; mp 112–114 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 7.63 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H), 4.17 (dd, J = 7.7, 4.6 Hz, 1H), 2,35 (s, 3H), 1.91–1.82 (m, 1H), 1.76–1.65 (m, 1H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.1, 152.0, 141.7, 129.9, 127.2, 126.9, 76.7, 21.6, 21.5, 10.1. HRMS (ESI), m/z: [M–H]− calcd for C12H13N2O2− 217.0983; found 217.0983.
- 3-(4-Bromophenyl)-6-ethyl-4H-1,2,4-oxadiazin-5(6H)-one (5b). White powder; 45% yield; mp 176–178 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.37 (s, 1H), 7.69 (s, 4H), 4.20 (dd, J = 7.7, 4.7 Hz, 1H), 1.93–1.83 (m, 1H), 1.77–1.66 (m, 1H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.6, 151.2, 132.1, 129.1, 128.8, 125.1, 76.6, 21.2, 9.9. HRMS (ESI), m/z: [M–H]− calcd for C11H10BrN2O2− 280.9931; found 280.9931.
- 6-Ethyl-3-(4-nitrophenyl)-4H-1,2,4-oxadiazin-5(6H)-one (5c). White powder; 49% yield; mp 177–179 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 8.32 (d, J = 8.6 Hz, 2H), 8.02 (d, J = 8.6 Hz, 2H), 4.27 (dd, J = 7.7, 4.7 Hz, 1H), 1.96–1.85 (m, 1H), 1.80–1.69 (m, 1H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.4, 150.6, 149.3, 135.5, 128.6, 124.2, 76.7, 21.3, 9.8. HRMS (ESI), m/z: [M–H]− calcd for C11H10N3O4− 248.0677; found 248.0678.
- 6-Ethyl-3-(5-methylthiophen-2-yl)-4H-1,2,4-oxadiazin-5(6H)-one (5d). Pale beige powder; 61% yield; mp 151–153 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.40 (s, 1H), 7.55 (d, J = 3.7 Hz, 1H), 6.85 (d, J = 3.7 Hz, 1H), 4.19 (dd, J = 7.8, 4.6 Hz, 1H), 2.45 (s, 3H), 1.90–1.81 (m, 1H), 1.73–1.65 (m, 1H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.8, 148.2, 144.5, 129.6, 129.0, 126.7, 77.2, 21.5, 15.7, 10.0. HRMS (ESI), m/z: [M–H]− calcd for C10H11N2O2S− 223.0547; found 223.0554.
- 6-Ethyl-3-(4-methoxyphenyl)-4H-1,2,4-oxadiazin-5(6H)-one (5e). Pale beige powder; 67% yield; mp 139–141 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.21 (s, 1H), 7.69 (d, J = 8.5 Hz, 2H), 7.02 (d, J = 8.5 Hz, 2H), 4.15 (dd, J = 7.8, 4.6 Hz, 1H), 3,8 (s, 3H), 1.92–1.82 (m, 1H), 1.75–1.64 (m, 1H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.2, 162.1, 151.8, 128.9, 121.8, 114.7, 76.7, 56.1, 21.5, 10.1. HRMS (ESI), m/z: [M–H]− calcd for C12H13N2O3− 233.0932; found 233.0930.
- 6-Ethyl-3-(4-(4-methoxyphenoxy)phenyl)-4H-1,2,4-oxadiazin-5(6H)-one (5f). Pale beige powder; 38% yield; mp 117–119 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.26 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.09–6.92 (m, 6H), 4.16 (dd, J = 7.8, 4.6 Hz, 1H), 3.76 (s, 3H), 1.90–1.83 (m, 1H), 1.75–1.66 (m, 1H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 161.2, 156.9, 151.6, 149.0, 129.2, 123.7, 122.1, 117.4, 116.0, 76.8, 56.1, 21.5, 10.1. HRMS (ESI), m/z: [M–H]− calcd for C18H17N2O4− 325.1194; found 325.1219.
- 6-Ethyl-3-phenyl-4H-1,2,4-oxadiazin-5(6H)-one (5g). White powder; 45% yield; mp 133–135 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 7.74 (d, J = 7.5 Hz, 2H), 7.56–7.46 (m, 3H), 4.20 (dd, J = 7.8, 4.6 Hz, 1H), 1.92–1.83 (m, 1H), 1.77–1.66 (m, 1H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 152.1, 131.8, 129.8, 129.3, 127.3, 76.8, 21.5, 10.1. HRMS (ESI), m/z: [M–H]− calcd for C11H11N2O2− 203.0826; found 203.0826.
- 6-Ethyl-3-(4-fluorophenyl)-4H-1,2,4-oxadiazin-5(6H)-one (5h). White powder; 36% yield; mp 153–155 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 7.89–7.73 (m, 2H), 7.35–7.31 (m, 2H), 4.19 (dd, J = 7.7, 4.6 Hz, 1H), 1.92–1.88 (m, 1H), 1.77–1.66 (m, 1H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.9, 165.6, 163.2, 151.4, 129.9 (d, J = 8.8 Hz, 1C), 126.3, 116.4 (d, J = 22.0 Hz, 1C), 76.8, 21.4, 10.1. 19F NMR (376 MHz, CDCl3) δ –107.49. HRMS (ESI), m/z: [M–H]− calcd for C11H10FN2O2− 221.0732; found 221.0732.
- 3-Benzyl-6-ethyl-4H-1,2,4-oxadiazin-5(6H)-one (5i). White powder; 33% yield; mp 97–99 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 7.35–7. 26 (m, 5H), 4.02 (dd, J = 7.8, 4.7 Hz, 1H), 3.54 (s, 2H), 1.79–1.69 (m, 1H), 1.63–1.52 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.0, 152.9, 136.1, 129.4, 129.2, 127.7, 76.4, 36.5, 21.5, 10.1. HRMS (ESI), m/z: [M–H]− calcd for C12H13N2O2− 217.0983; found 217.0983.
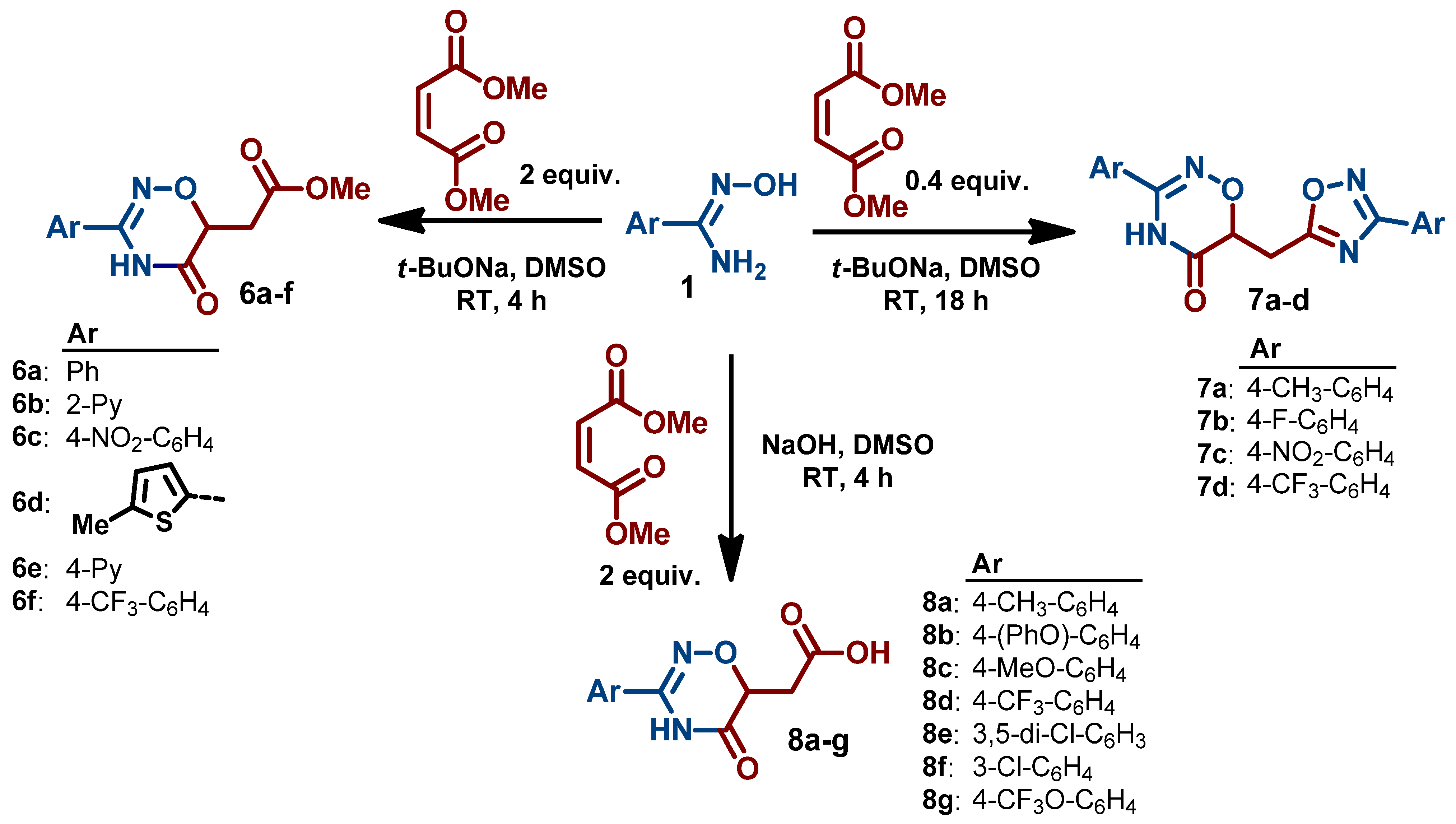
- 2-(3-(3-Chlorophenyl)-5-oxo-5,6-dihydro-4H-1,2,4-oxadiazin-6-yl)acetic acid (8f). Beige powder; 71% yield (380 mg); mp 211–212 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H), 11.45 (s, 1H), 7.80 (t, J = 1.9 Hz, 1H), 7.72 (dt, J = 7.9, 1.3 Hz, 1H), 7.62 (ddd, J = 8.1, 2.2, 1.1 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 4.59 (dd, J = 7.7, 4.5 Hz, 1H), 2.93 (dd, J = 16.8, 4.5 Hz, 1H), 2.68 (dd, J = 16.8, 7.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 166.4, 150.8, 133.4, 131.1, 131.0, 130.6, 126.7, 125.5, 72.5, 33.0. HRMS (ESI), m/z: [M+H]+ calcd. for C11H10ClN2O4+ 269.0324; found 269.0321.
- 2-(5-Oxo-3-(4-(trifluoromethoxy)phenyl)-5,6-dihydro-4H-1,2,4-oxadiazin-6-yl)acetic acid (8g). White powder; 77% yield (488 mg); mp 208–211 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 7.96–7.77 (m, 2H), 7.49 (d, J = 8.4 Hz, 2H), 4.59 (dd, J = 7.7, 4.5 Hz, 1H), 2.93 (dd, J = 16.9, 4.4 Hz, 1H), 2.68 (dd, J = 16.7, 7.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 186.2, 171.4, 167.11, 151.6, 150.9, 129.8, 128.9, 121.9, 120.6 120.6 (q, J = 257.1 Hz), 73.1, 33.6. HRMS (ESI), m/z: [M+H]+ calcd. for C12H10F3N2O5+ 319.0536; found 319.0539.
3.3. X-Ray Studies
3.4. MAO Inhibition Studies
3.5. Molecular Docking Studies Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edmondson, D.E.; Binda, C.; Wang, J.; Upadhyay, A.K.; Mattevi, A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 2009, 48, 4220–4230. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R. Monoamine oxidases: The biochemistry of the proteins as targets in medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2012, 12, 2189–2209. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubálek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2 Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S287–S296. [Google Scholar] [CrossRef]
- Fowler, J.S.; Volkow, N.D.; Wang, G.-J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol. Aging 1997, 18, 431–435. [Google Scholar] [CrossRef]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef]
- Maggiorani, D.; Manzella, N.; Edmondson, D.E.; Mattevi, A.; Parini, A.; Binda, C.; Mialet-Perez, J. Monoamine oxidases, oxidative stress, and altered mitochondrial dynamics in cardiac ageing. Oxid. Med. Cell. Longev. 2017, 2017, 3017947. [Google Scholar] [CrossRef]
- Wu, J.B.; Shao, C.; Li, X.; Li, Q.; Hu, P.; Shi, C.; Li, Y.; Chen, Y.-T.; Yin, F.; Liao, C.-P.; et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J. Clin. Investig. 2014, 124, 2891–2908. [Google Scholar] [CrossRef]
- Flamand, V.; Zhao, H.; Peehl, D.M. Targeting monoamine oxidase A in advanced prostate cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1761–1771. [Google Scholar] [CrossRef]
- Guglielmi, P.; Carradori, S.; D’Agostino, I.; Campestre, C.; Petzer, J.P. An updated patent review on monoamine oxidase (MAO) inhibitors. Expert Opin. Ther. Pat. 2022, 32, 849–883. [Google Scholar] [CrossRef]
- Robakis, D.; Fahn, S. Defining the role of the monoamine oxidase-B inhibitors for Parkinson’s disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef]
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase A levels in the brain. Arch. Gen. Psychiatry 2006, 63, 1209–1216. [Google Scholar] [CrossRef]
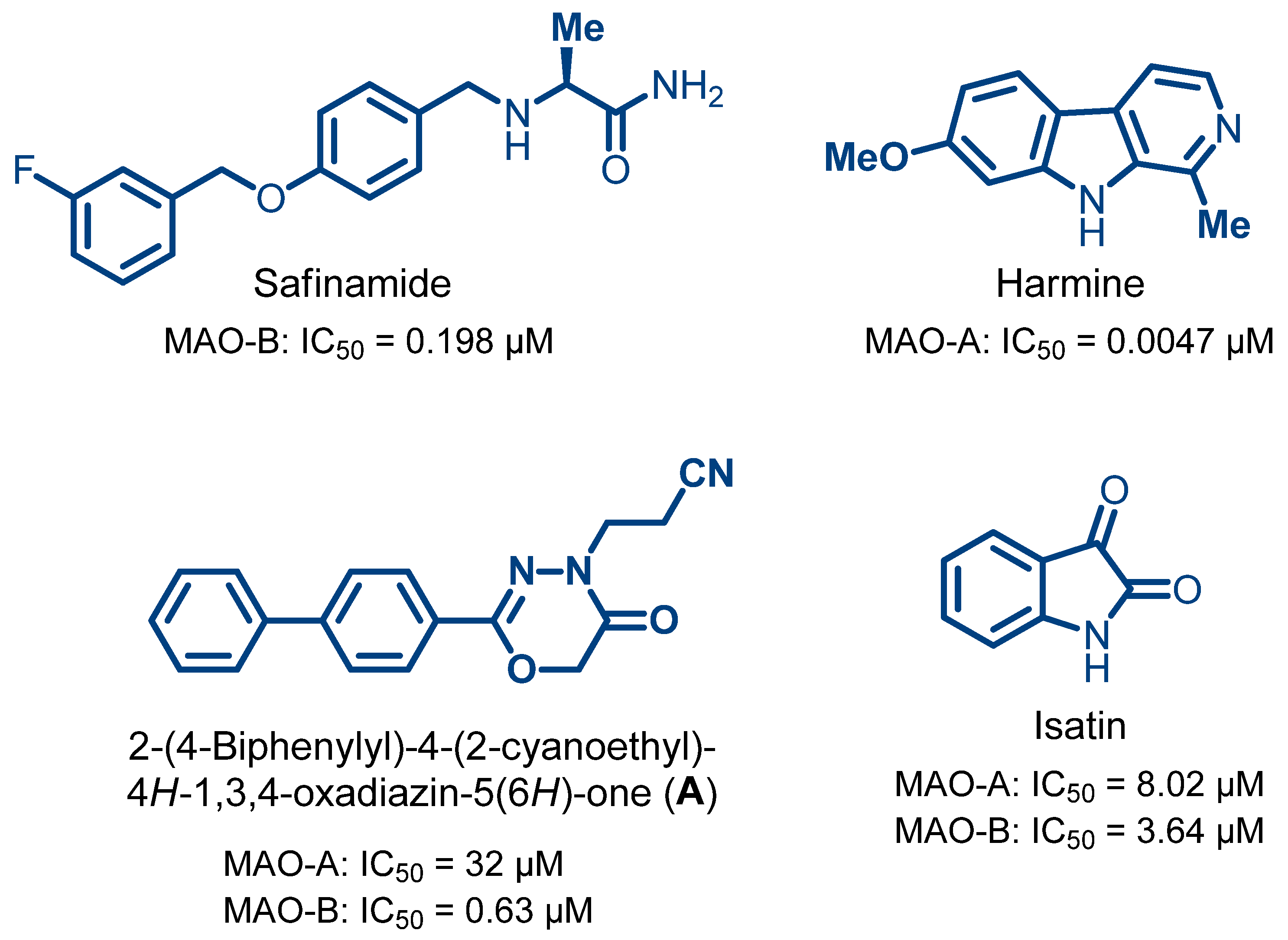
- Prinsloo, I.F.; Petzer, J.P.; Cloete, T.T.; Petzer, A. The evaluation of isatin analogues as inhibitors of monoamine oxidase. Chem. Biol. Drug Des. 2023, 102, 1067–1074. [Google Scholar] [CrossRef]
- Hubálek, F.; Binda, C.; Khalil, A.; Li, M.; Mattevi, A.; Castagnoli, N.; Edmondson, D.E. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J. Biol. Chem. 2005, 280, 15761–15766. [Google Scholar] [CrossRef]
- Mazouz, F.; Lebreton, L.; Milcent, R.; Burstein, C. Inhibition of monoamine oxidase types A and B by 2-aryl-4H-1,3,4-oxadiazin-5(6H)-one derivatives. Eur. J. Med. Chem. 1988, 23, 441–451. [Google Scholar] [CrossRef]
- Zainal Abidin, A.; Norrrahim, M.N.F.; Mohamed Shakrin, N.N.S.; Ibrahim, B.; Abdullah, N.; Abdul Rashid, J.I.; Mohd Kasim, N.A.; Ahmad Shah, N.A. Amidine containing compounds: Antimicrobial activity and its potential in combating antimicrobial resistance. Heliyon 2024, 10, e32010. [Google Scholar] [CrossRef]
- Krasavin, M. N-(Hetero)aryl-2-imidazolines: An emerging privileged motif for contemporary drug design. Chem. Heterocycl. Compd. 2017, 53, 240–255. [Google Scholar] [CrossRef]
- Krasavin, M. Biologically active compounds based on the privileged 2-imidazoline scaffold: The world beyond adrenergic/imidazoline receptor modulators. Eur. J. Med. Chem. 2015, 97, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Shetnev, A.; Baykov, S.; Kalinin, S.; Belova, A.; Sharoyko, V.; Rozhkov, A.; Zelenkov, L.; Tarasenko, M.; Sadykov, E.; Korsakov, M.; et al. 1,2,4-Oxadiazole/2-imidazoline hybrids: Multi-target-directed compounds for the treatment of infectious diseases and cancer. Int. J. Mol. Sci. 2019, 20, 1699. [Google Scholar] [CrossRef] [PubMed]
- Shetnev, A.; Osipyan, A.; Baykov, S.; Sapegin, A.; Chirkova, Z.; Korsakov, M.; Petzer, A.; Engelbrecht, I.; Petzer, J.P. Novel monoamine oxidase inhibitors based on the privileged 2-imidazoline molecular framework. Bioorg. Med. Chem. Lett. 2019, 29, 40–46. [Google Scholar] [CrossRef]
- Baykov, S.; Semenov, A.; Tarasenko, M.; Boyarskiy, V.P. Application of amidoximes for the heterocycles synthesis. Tetrahedron Lett. 2020, 61, 152403. [Google Scholar] [CrossRef]
- Bursavich, M.G.; Harrison, B.A.; Acharya, R.; Costa, D.E.; Freeman, E.A.; Hrdlicka, L.A.; Jin, H.; Kapadnis, S.; Moffit, J.S.; Murphy, D.; et al. Discovery of the Oxadiazine FRM-024: A potent CNS-penetrant gamma secretase modulator. J. Med. Chem. 2021, 64, 14426–14447. [Google Scholar] [CrossRef] [PubMed]
- Eşme, A.; Kara, Y.S. 4-(4-Methoxyphenyl)-6-methyl-3-phenyl-4H-1,2,4-oxadiazin-5(6H)-one: Synthesis, crystal structure, Hirshfeld surface analysis, noncovalent, ADMET studies and biological evaluation. J. Mol. Struct. 2023, 1282, 135197. [Google Scholar] [CrossRef]
- Kara, Y.S.; Yıldız, B. Synthesis and substituent effect study on 13C NMR chemical shifts of 4-(substitue-phenyl)-6-methyl-3-phenyl-4H-1,2,4-oxadiazin-5(6H)-one. J. Mol. Struct. 2022, 1250, 131787. [Google Scholar] [CrossRef]
- Baykov, S.V.; Shetnev, A.A.; Semenov, A.V.; Baykova, S.O.; Boyarskiy, V.P. Room temperature synthesis of bioactive 1,2,4-oxadiazoles. Int. J. Mol. Sci. 2023, 24, 5406. [Google Scholar] [CrossRef]
- Presnukhina, S.I.; Tarasenko, M.V.; Geyl, K.K.; Baykova, S.O.; Baykov, S.V.; Shetnev, A.A.; Boyarskiy, V.P. Unusual formation of 1,2,4-oxadiazine core in reaction of amidoximes with maleic or fumaric esters. Molecules 2022, 27, 7508. [Google Scholar] [CrossRef]
- Shetnev, A.A.; Vasilieva, E.A.; Proskurina, I.K.; Forostyanko, A.S.; Presnukhina, S.I.; Tarasenko, M.V.; Lebedev, A.S.; Ivanovskii, S.A.; Kotov, A.D. Synthesis and biological activity of 3-aryl-5-(aryloxymethyl)-1,2,4-oxadiazoles. Russ. J. Org. Chem. 2022, 58, 306–314. [Google Scholar] [CrossRef]
- Weissbach, H.; Smith, T.E.; Daly, J.W.; Witkop, B.; Udenfriend, S. A rapid spectrophotometric assay of mono-amine oxidase based on the rate of disappearance of kynuramine. J. Biol. Chem. 1960, 235, 1160–1163. [Google Scholar] [CrossRef]
- Mostert, S.; Petzer, A.; Petzer, J.P. Indanones as high-potency reversible inhibitors of monoamine oxidase. ChemMedChem 2015, 10, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Sharonova, T.; Pankrat’eva, V.; Savko, P.; Baykov, S.; Shetnev, A. Facile room-temperature assembly of the 1,2,4-oxadiazole core from readily available amidoximes and carboxylic acids. Tetrahedron Lett. 2018, 59, 2824–2827. [Google Scholar] [CrossRef]
- Tarasenko, M.; Duderin, N.; Sharonova, T.; Baykov, S.; Shetnev, A.; Smirnov, A.V. Room-temperature synthesis of pharmaceutically important carboxylic acids bearing the 1,2,4-oxadiazole moiety. Tetrahedron Lett. 2017, 58, 3672–3677. [Google Scholar] [CrossRef]
- Srivastava, R.M.; Pereira, M.C.; Faustino, W.W.M.; Coutinho, K.; dos Anjos, J.V.; de Melo, S.J. Synthesis, mechanism of formation, and molecular orbital calculations of arylamidoximes. Monatshefte Für Chem. Chem. Mon. 2009, 140, 1319–1324. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2 : A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Crystallogr. Sect. B Struct. Sci. 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Compound 2 (Equiv.) | X | Base (Equiv.) | Solvent | Time, h | Isolated Yield, % |
| 1 | 2a (1.2) | Br | NaOH (1.2) | DMSO | 4 | – b |
| 2 | 2a (1.2) | Br | NaOH (1.5) | DMSO | 4 | – b |
| 3 | 2a (1.2) | Br | NaOH (2.0) | DMSO | 18 | 29 |
| 4 | 2a (1.2) | Br | t-BuONa (1.2) | DMSO | 4 | – b |
| 5 | 2a (1.2) | Br | t-BuONa (1.5) | DMSO | 4 | – c |
| 6 | 2a (1.2) | Br | t-BuONa (2.0) | DMSO | 18 | 42 |
| 7 | 2b (1.2) | Cl | NaOH (2.0) | DMSO | 18 | 35 |
| 8 | 2b (1.2) | Cl | KOH (2.0) | DMSO | 18 | 38 |
| 9 | 2b (1.2) | Cl | t-BuONa (2.0) | DMSO | 18 | 50 |
| 10 | 2b (1.2) | Cl | t-BuOK (2.0) | DMSO | 18 | 32 |
| 11 | 2b (1.2) | Cl | t-BuONa (2.0) | DMAA | 18 | 37 |
| 12 | 2b (1.2) | Cl | t-BuONa (2.0) | DMF | 18 | 43 |
| 13 | 2b (1.2) | Cl | t-BuONa (2.0) | DMSO | 48 | 45 |
| 14 | 2b (1.5) | Cl | t-BuONa (2.0) | DMSO | 18 | 13 |
| IC50 (µM ± SD) | IC50 (µM ± SD) | ||||
|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | ||
| 3a | 36.8 ± 1.83 | 37.2 ± 0.071 | 6a | NI c | NI c |
| 3b | 14.2 ± 0.940 | 13.6 ± 6.63 | 6b | 34.2 ± 6.15 | 23.6 ± 1.11 |
| 3c | 17.3 ± 0.594 | 24.4 ± 2.48 | 6c | NI c | 52.3 ± 12.7 |
| 3d | 34.6 ± 5.47 | 16.6 ± 1.67 | 6d | 40.7 ± 8.83 | 33.9 ± 1.44 |
| 3e | 27.1 ± 3.98 | 37.6 ± 9.47 | 6e | NI c | NI c |
| 3f | 3.75 ± 0.212 | 3.98 ± 0.518 | 6f | NI c | 22.9 ± 5.27 |
| 4a | 67.0 ± 0.007 | 24.5 ± 0.198 | 7a | 54.3 ± 14.3 | 166 ± 49.4 |
| 4b | 37.4 ± 4.18 | 27.1 ± 3.37 | 7b | NI c | 14.3 ± 3.27 |
| 4c | 58.5 ± 4.17 | 14.6 ± 0.092 | 7c | 12.9 ± 0.014 | 0.371 ± 0.037 |
| 4d | 46.2 ± 9.22 | 22.3 ± 0.940 | 7d | NI c | 46.3 ± 17.8 |
| 4e | 51.2 ± 15.07 | 20.0 ± 0.643 | 8a | NI c | 109 ± 0.566 |
| 5a | NI c | 4.28 ± 0.318 | 8b | 12.7 ± 1.34 | NI c |
| 5b | 35.4 ± 1.54 | 5.11 ± 0.308 | 8c | NI c | NI c |
| 5c | 57.9 ± 9.84 | 21.0 ± 4.09 | 8d | NI c | NI c |
| 5d | 58.1 ± 5.28 | 6.14 ± 0.137 | 8e | NI c | 62.2 ± 15.7 |
| 5e | 44.1 ± 6.05 | 6.22 ± 0.596 | 8f | NI c | NI c |
| 5f | 15.4 ± 1.98 | 0.900 ± 0.050 | 8g | NI c | NI c |
| 5g | NI c | NI c | |||
| 5h | 93.0 ± 3.23 | 30.7 ± 2.02 | |||
| 5i | NI c | NI c | |||
| Isatin d | 8.02 ± 1.69 | 3.64 ± 0.476 | |||
| Harmine d | 0.0047 ± 0.0001 | NI c | |||
| Safinamide d | NI c | 0.198 ± 0.014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Presnukhina, S.I.; Kotlyarova, V.D.; Shetnev, A.A.; Baykov, S.V.; Turmanov, R.; Appazov, N.; Zhapparbergenov, R.; Zhussupova, L.; Togyzbayeva, N.; Cloete, S.J.; et al. Synthesis of 1,2,4-Oxadiazin-5(6H)-One Derivatives and Their Biological Investigation as Monoamine Oxidase Inhibitors. Molecules 2024, 29, 5550. https://doi.org/10.3390/molecules29235550
Presnukhina SI, Kotlyarova VD, Shetnev AA, Baykov SV, Turmanov R, Appazov N, Zhapparbergenov R, Zhussupova L, Togyzbayeva N, Cloete SJ, et al. Synthesis of 1,2,4-Oxadiazin-5(6H)-One Derivatives and Their Biological Investigation as Monoamine Oxidase Inhibitors. Molecules. 2024; 29(23):5550. https://doi.org/10.3390/molecules29235550
Chicago/Turabian StylePresnukhina, Sofia I., Valentina D. Kotlyarova, Anton A. Shetnev, Sergey V. Baykov, Rakhymzhan Turmanov, Nurbol Appazov, Rakhmetulla Zhapparbergenov, Leilya Zhussupova, Nurila Togyzbayeva, Stephanus J. Cloete, and et al. 2024. "Synthesis of 1,2,4-Oxadiazin-5(6H)-One Derivatives and Their Biological Investigation as Monoamine Oxidase Inhibitors" Molecules 29, no. 23: 5550. https://doi.org/10.3390/molecules29235550
APA StylePresnukhina, S. I., Kotlyarova, V. D., Shetnev, A. A., Baykov, S. V., Turmanov, R., Appazov, N., Zhapparbergenov, R., Zhussupova, L., Togyzbayeva, N., Cloete, S. J., Korsakov, M. K., Boyarskiy, V. P., Petzer, A., & Petzer, J. P. (2024). Synthesis of 1,2,4-Oxadiazin-5(6H)-One Derivatives and Their Biological Investigation as Monoamine Oxidase Inhibitors. Molecules, 29(23), 5550. https://doi.org/10.3390/molecules29235550