
Crystallographic Fragment Screening of a Bifunctional Proline Catabolic Enzyme Reveals New Inhibitor Templates for Proline Dehydrogenase and L-Glutamate-γ-semialdehyde Dehydrogenase


Abstract

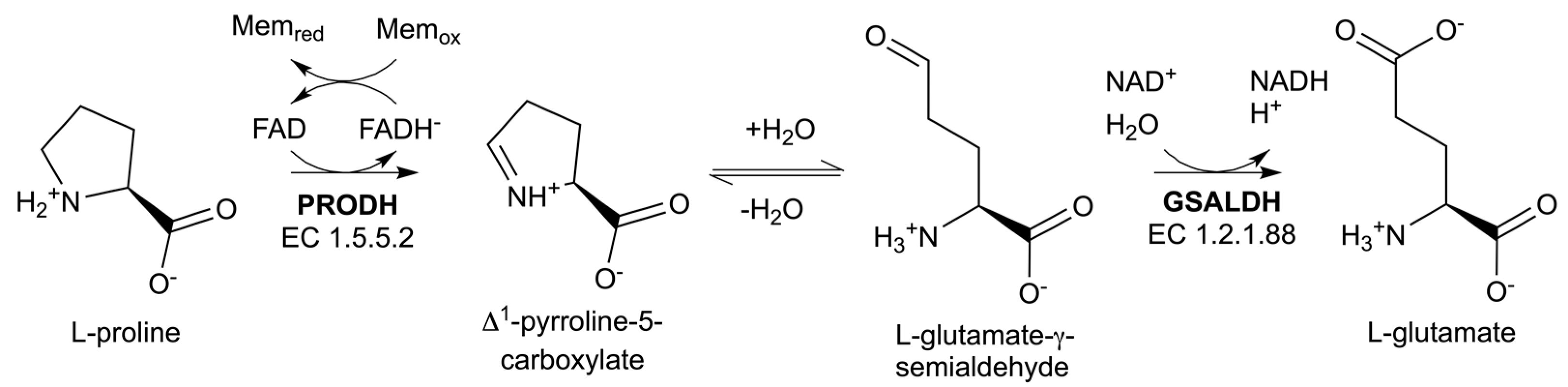
1. Introduction
2. Results
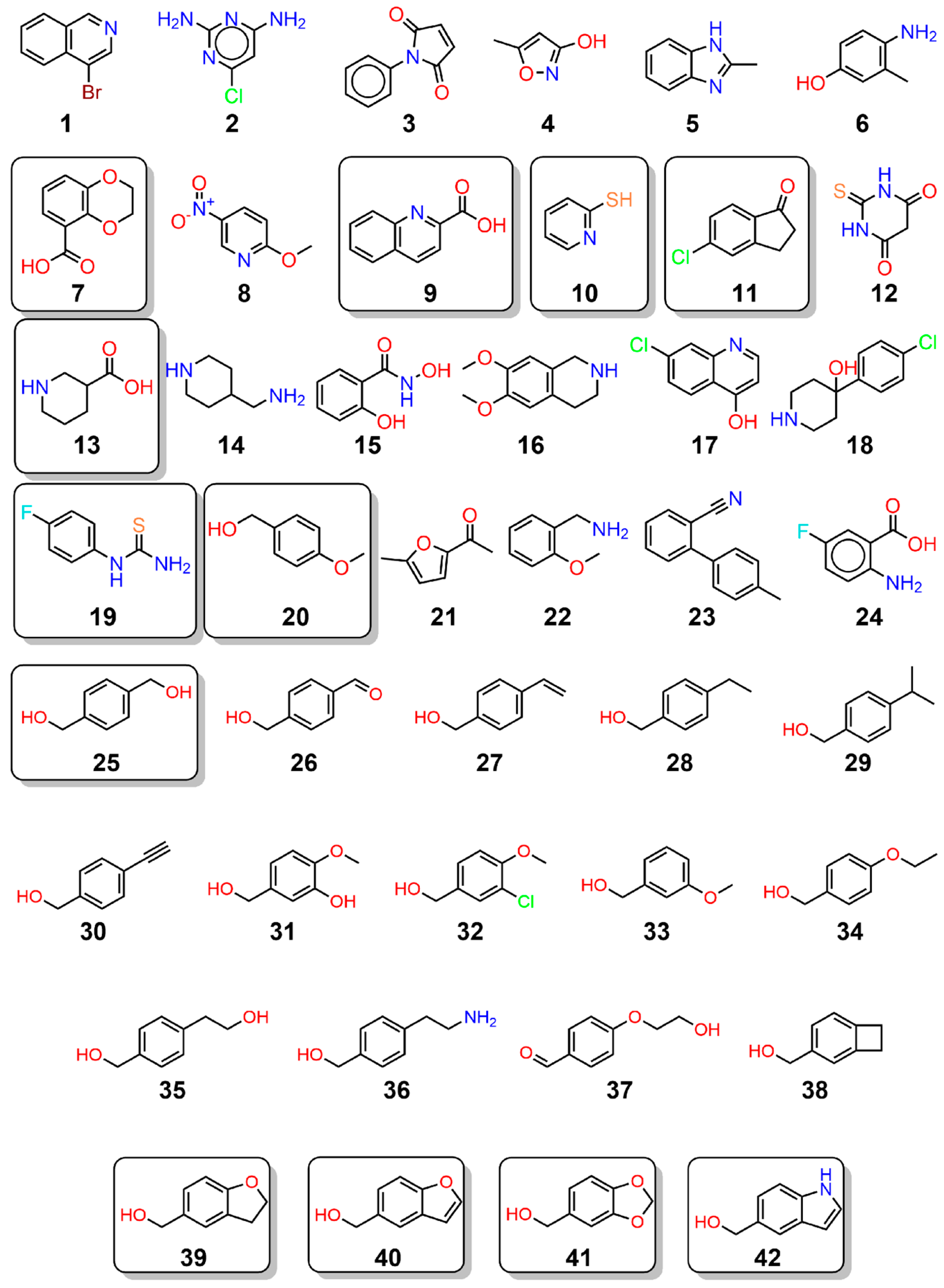
2.1. Crystallographic Fragment Screening and Validation
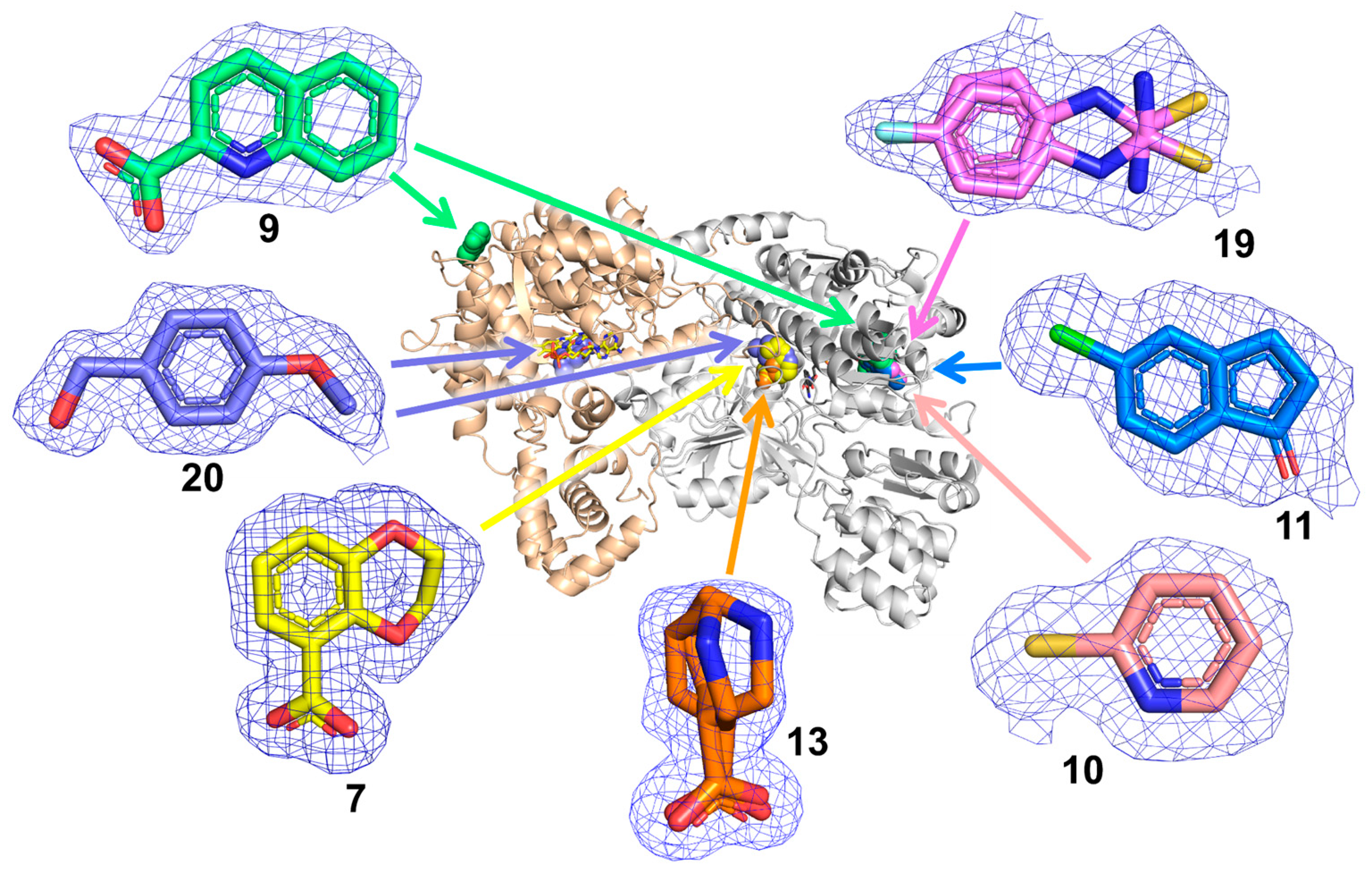
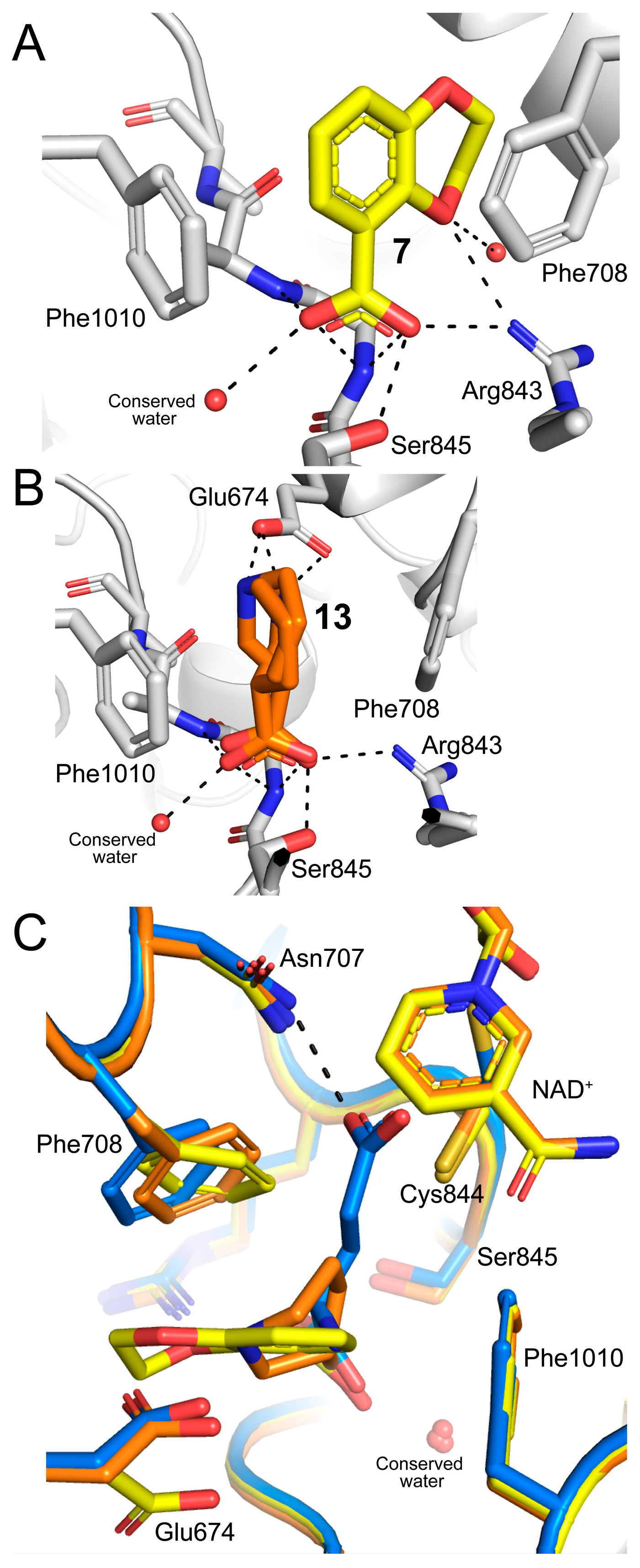
2.2. Poses and Interactions of Fragments 7, 13, and 20 Bound to the GSAL-Binding Site
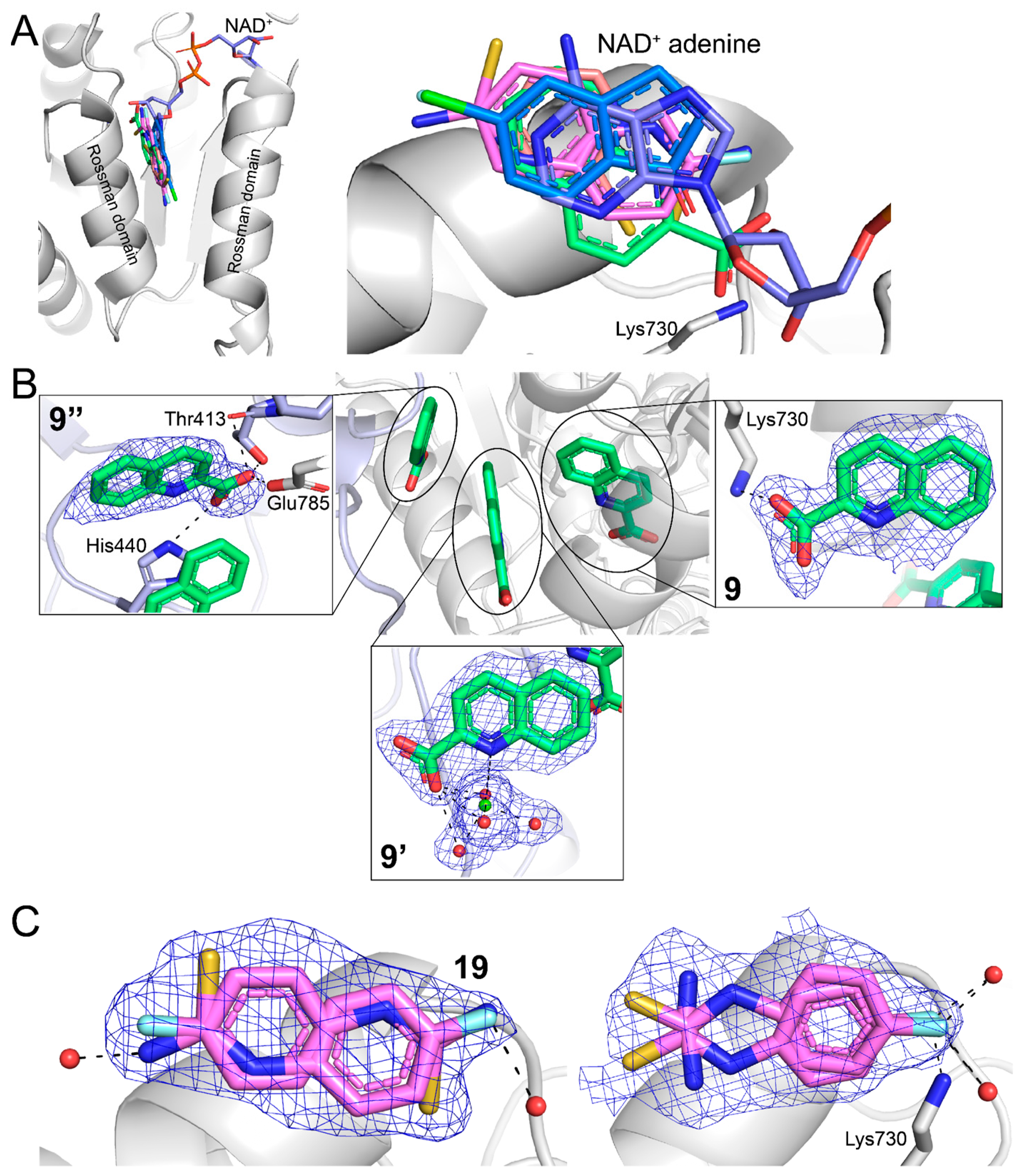
2.3. Poses and Interactions of Fragments 9, 10, 11, and 19 Bound to the GSALDH NAD+-Binding Site
2.4. Poses and Interactions of Fragment 9 Bound to a Surface Site
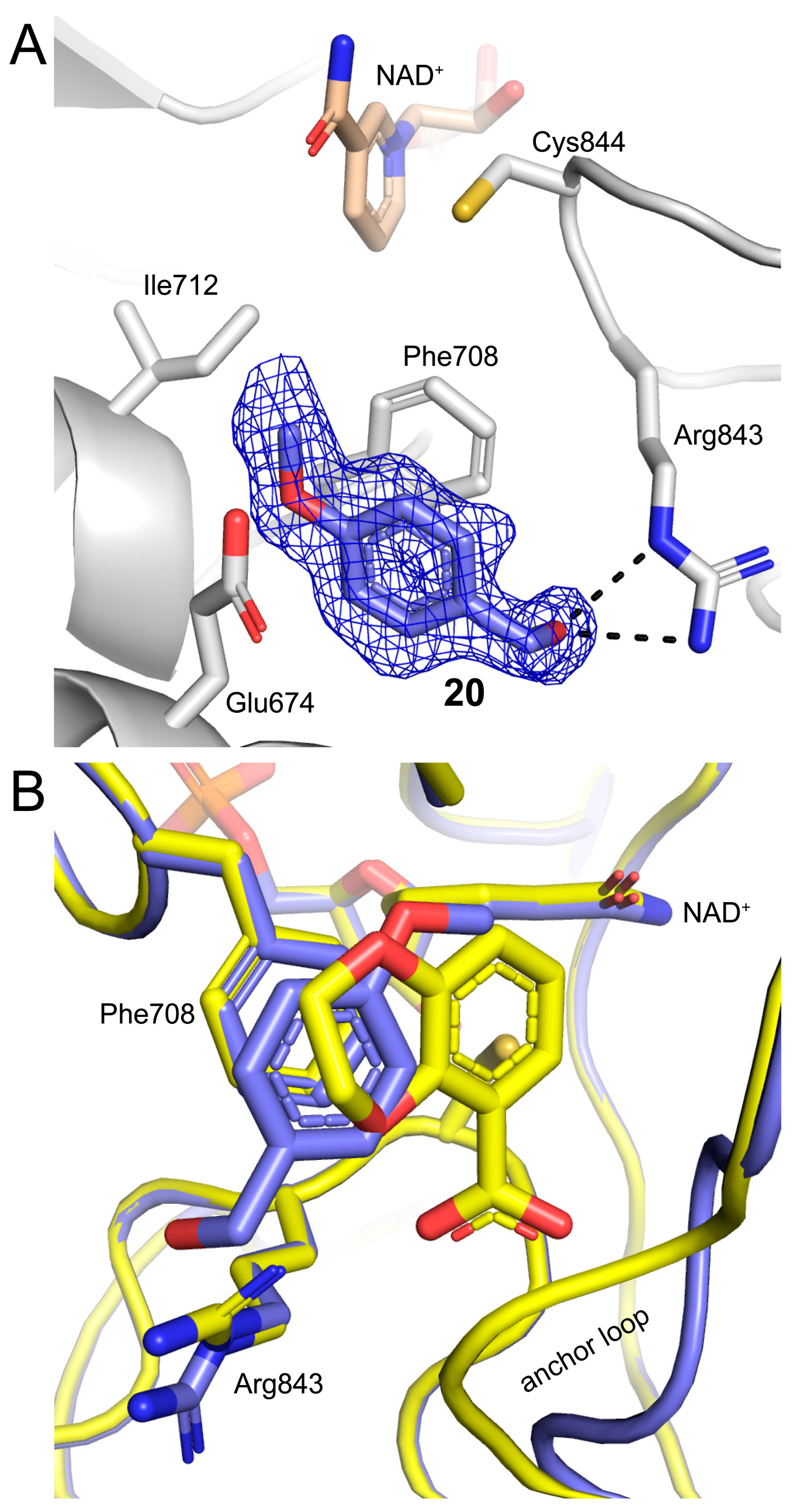
2.5. Poses and Interactions of Fragment 20 Bound to the PRODH Active Site
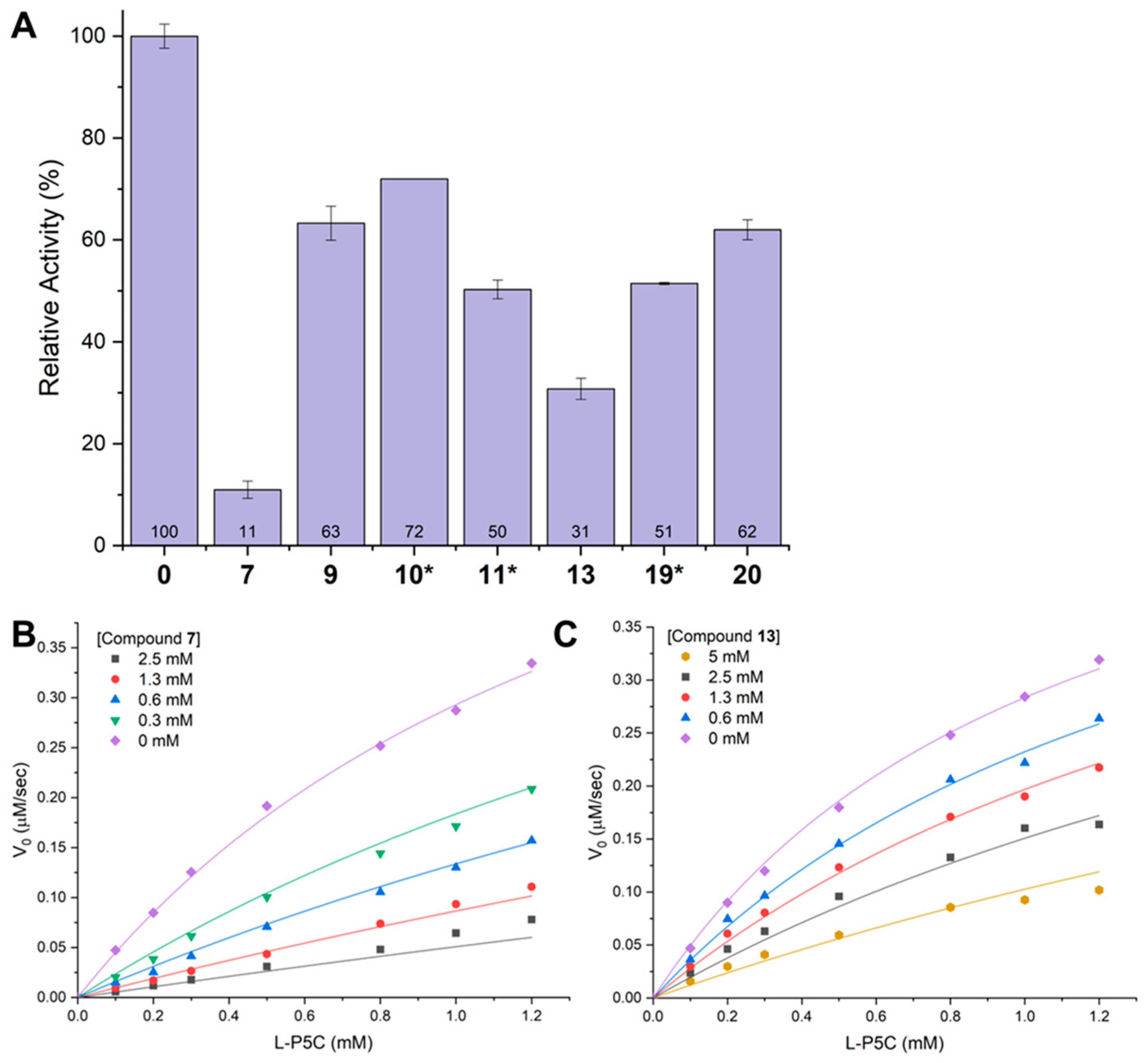
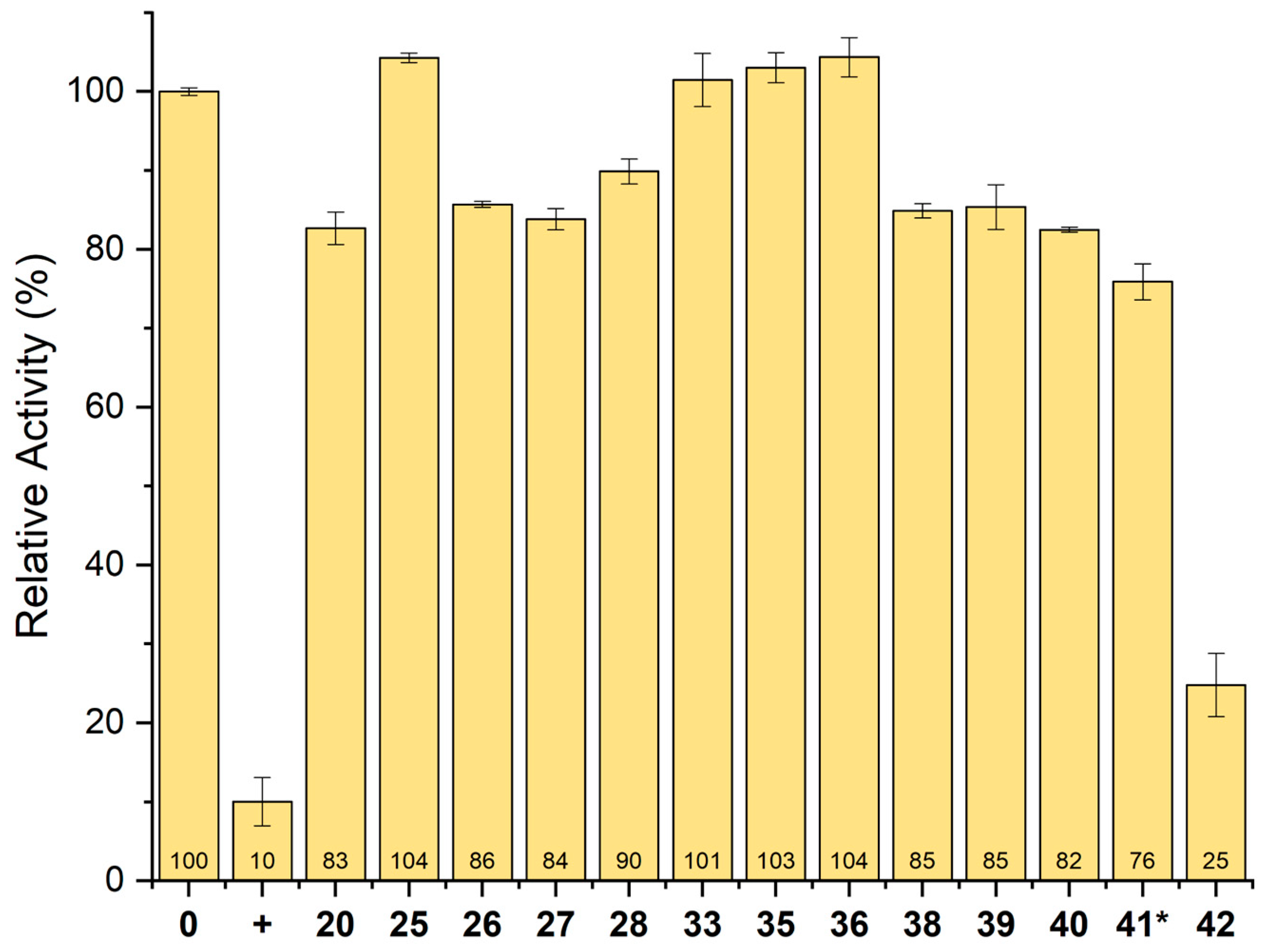
2.6. Inhibition of GSALDH Activity by Hits from Crystallographic Screening
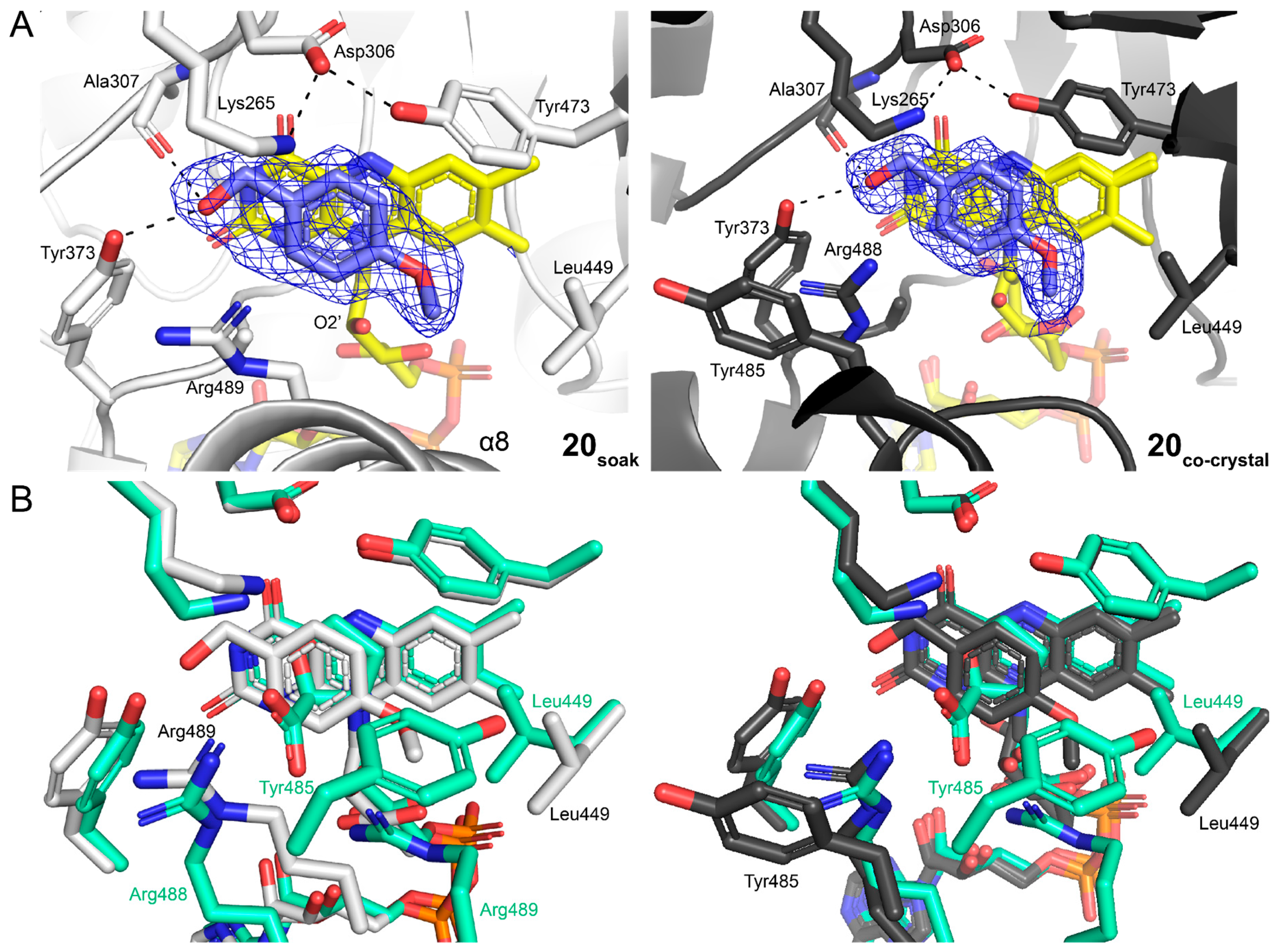
2.7. Crystal Structures of SmPutA Complexed with Analogs of 20
2.8. Inhibition of PRODH Activity by 20 and Its Analogs
3. Discussion
4. Materials and Methods
4.1. Protein Production
4.2. Crystallization, Crystal Soaking, and Co-Crystallization
4.3. X-Ray Crystal Structure Determination
4.4. Enzyme Inhibition Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tanner, J.J. Structural Biology of Proline Catabolic Enzymes. Antioxid. Redox Signal. 2019, 30, 650–673. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W. Proline metabolism and cancer. Front. Biosci. 2012, 17, 1835–1845. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C.; Christian, K.J. The proline regulatory axis and cancer. Front. Oncol. 2012, 2, 60. [Google Scholar] [CrossRef]
- Burke, L.; Guterman, I.; Palacios Gallego, R.; Britton, R.G.; Burschowsky, D.; Tufarelli, C.; Rufini, A. The Janus-like role of proline metabolism in cancer. Cell Death Discov. 2020, 6, 104. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, G.; Chen, Y.; Xu, W.; Liu, Y.; Ji, G.; Xu, H. Can proline dehydrogenase-a key enzyme involved in proline metabolism-be a novel target for cancer therapy? Front. Oncol. 2023, 13, 1254439. [Google Scholar] [CrossRef]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grunewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef]
- Scott, G.K.; Mahoney, S.; Scott, M.; Loureiro, A.; Lopez-Ramirez, A.; Tanner, J.J.; Ellerby, L.M.; Benz, C.C. N-Propargylglycine: A unique suicide inhibitor of proline dehydrogenase with anticancer activity and brain-enhancing mitohormesis properties. Amino Acids 2021, 53, 1927–1939. [Google Scholar] [CrossRef]
- Scott, G.K.; Yau, C.; Becker, B.C.; Khateeb, S.; Mahoney, S.; Jensen, M.B.; Hann, B.; Cowen, B.J.; Pegan, S.D.; Benz, C.C. Targeting Mitochondrial Proline Dehydrogenase with a Suicide Inhibitor to Exploit Synthetic Lethal Interactions with p53 Upregulation and Glutaminase Inhibition. Mol. Cancer Ther. 2019, 18, 1374–1385. [Google Scholar] [CrossRef]
- Muzio, G.; Maggiora, M.; Paiuzzi, E.; Oraldi, M.; Canuto, R.A. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 2012, 52, 735–746. [Google Scholar] [CrossRef]
- Muralikrishnan, V.; Hurley, T.D.; Nephew, K.P. Targeting Aldehyde Dehydrogenases to Eliminate Cancer Stem Cells in Gynecologic Malignancies. Cancers 2020, 12, 961. [Google Scholar] [CrossRef] [PubMed]
- Bu, J.; Zhang, Y.; Wu, S.; Li, H.; Sun, L.; Liu, Y.; Zhu, X.; Qiao, X.; Ma, Q.; Liu, C.; et al. KK-LC-1 as a therapeutic target to eliminate ALDH(+) stem cells in triple negative breast cancer. Nat. Commun. 2023, 14, 2602. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.T.; Lippitt, J.M.; Guzman-Ramirez, N.; Hamdy, F.C.; Eaton, C.L.; Thalmann, G.N.; Cecchini, M.G.; et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010, 70, 5163–5173. [Google Scholar] [CrossRef] [PubMed]
- Convertino, M.; Das, J.; Dokholyan, N.V. Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. ACS Chem. Biol. 2016, 11, 1471–1489. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Yue, W.W. Chapter 13—Structure-guided discovery of pharmacological chaperones targeting protein conformational and misfolding diseases. In Protein Homeostasis Diseases; Academic Press: Cambridge, MA, USA, 2020; pp. 281–308. [Google Scholar]
- Geraghty, M.T.; Vaughn, D.; Nicholson, A.J.; Lin, W.W.; Jimenez-Sanchez, G.; Obie, C.; Flynn, M.P.; Valle, D.; Hu, C.A. Mutations in the Delta1-pyrroline 5-carboxylate dehydrogenase gene cause type II hyperprolinemia. Hum. Mol. Genet. 1998, 7, 1411–1415. [Google Scholar] [CrossRef]
- Phang, J.M.; Hu, C.A.; Valle, D. Disorders of proline and hydroxyproline metabolism. In Metabolic and Molecular Basis of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw Hill: New York, NY, USA, 2001; pp. 1821–1838. [Google Scholar]
- Riveros-Rosas, H.; Gonzalez-Segura, L.; Julian-Sanchez, A.; Diaz-Sanchez, A.G.; Munoz-Clares, R.A. Structural determinants of substrate specificity in aldehyde dehydrogenases. Chem. Biol. Interact. 2013, 202, 51–61. [Google Scholar] [CrossRef]
- Pemberton, T.A.; Tanner, J.J. Structural basis of substrate selectivity of Delta(1)-pyrroline-5-carboxylate dehydrogenase (ALDH4A1): Semialdehyde chain length. Arch. Biochem. Biophys. 2013, 538, 34–40. [Google Scholar] [CrossRef]
- Luo, M.; Gamage, T.T.; Arentson, B.W.; Schlasner, K.N.; Becker, D.F.; Tanner, J.J. Structures of Proline Utilization A (PutA) Reveal the Fold and Functions of the Aldehyde Dehydrogenase Superfamily Domain of Unknown Function. J. Biol. Chem. 2016, 291, 24065–24075. [Google Scholar] [CrossRef]
- Harding, M.M. The architecture of metal coordination groups in proteins. Acta Cryst. 2004, D60 Pt 5, 849–859. [Google Scholar] [CrossRef]
- Lin, G.Y.; Su, Y.C.; Huang, Y.L.; Hsin, K.Y. MESPEUS: A database of metal coordination groups in proteins. Nucleic Acids Res 2024, 52, D483–D493. [Google Scholar] [CrossRef]
- Campbell, A.C.; Becker, D.F.; Gates, K.S.; Tanner, J.J. Covalent Modification of the Flavin in Proline Dehydrogenase by Thiazolidine-2-Carboxylate. ACS Chem. Biol. 2020, 15, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 2017, 73 Pt 2, 148–157. [Google Scholar] [CrossRef]
- Schiebel, J.; Radeva, N.; Krimmer, S.G.; Wang, X.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Metz, A.; Huschmann, F.U.; Weiss, M.S.; et al. Six Biophysical Screening Methods Miss a Large Proportion of Crystallographically Discovered Fragment Hits: A Case Study. ACS Chem. Biol. 2016, 11, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Meeks, K.R.; Ji, J.; Protopopov, M.V.; Tarkhanova, O.O.; Moroz, Y.S.; Tanner, J.J. Novel Fragment Inhibitors of PYCR1 from Docking-Guided X-ray Crystallography. J. Chem. Inf. Model 2024, 64, 1704–1718. [Google Scholar] [CrossRef]
- Campbell, A.C.; Bogner, A.N.; Mao, Y.; Becker, D.F.; Tanner, J.J. Structural analysis of prolines and hydroxyprolines binding to the l-glutamate-gamma-semialdehyde dehydrogenase active site of bifunctional proline utilization A. Arch. Biochem. Biophys. 2021, 698, 108727. [Google Scholar] [CrossRef]
- Muller, J.; Klein, R.; Tarkhanova, O.; Gryniukova, A.; Borysko, P.; Merkl, S.; Ruf, M.; Neumann, A.; Gastreich, M.; Moroz, Y.S.; et al. Magnet for the Needle in Haystack: “Crystal Structure First” Fragment Hits Unlock Active Chemical Matter Using Targeted Exploration of Vast Chemical Spaces. J. Med. Chem. 2022, 65, 15663–15678. [Google Scholar] [CrossRef]
- Vasiliou, V.; Thompson, D.C.; Smith, C.; Fujita, M.; Chen, Y. Aldehyde dehydrogenases: From eye crystallins to metabolic disease and cancer stem cells. Chem. Biol. Interact. 2013, 202, 2–10. [Google Scholar] [CrossRef]
- Vasiliou, V.; Nebert, D.W. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum. Genomics 2005, 2, 138–143. [Google Scholar] [CrossRef]
- Sophos, N.A.; Vasiliou, V. Aldehyde dehydrogenase gene superfamily: The 2002 update. Chem. Biol.Interact. 2003, 143–144, 5–22. [Google Scholar] [CrossRef]
- Lavudi, K.; Nuguri, S.M.; Pandey, P.; Kokkanti, R.R.; Wang, Q.E. ALDH and cancer stem cells: Pathways, challenges, and future directions in targeted therapy. Life Sci. 2024, 356, 123033. [Google Scholar] [CrossRef]
- Bogner, A.N.; Tanner, J.J. Structure-affinity relationships of reversible proline analog inhibitors targeting proline dehydrogenase. Org. Biomol. Chem. 2022, 20, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Meeks, K.R.; Bogner, A.N.; Tanner, J.J. Screening a knowledge-based library of low molecular weight compounds against the proline biosynthetic enzyme 1-pyrroline-5-carboxylate 1 (PYCR1). Protein Sci. 2024, 33, e5072. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Coyne, A.G.; Hudson, S.A.; Abell, C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. [Google Scholar] [CrossRef]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672. [Google Scholar] [CrossRef]
- Campbell, A.C.; Prater, A.R.; Bogner, A.N.; Quinn, T.P.; Gates, K.S.; Becker, D.F.; Tanner, J.J. Photoinduced Covalent Irreversible Inactivation of Proline Dehydrogenase by S-Heterocycles. ACS Chem. Biol. 2021, 16, 2268–2279. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 125–132. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62 Pt 1, 72–82. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69 Pt 7, 1204–1214. [Google Scholar] [CrossRef]
- Evans, P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 282–292. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 Pt 4, 352–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment | Resolution (Å) | Binding Location | PDB ID |
|---|---|---|---|
| 7 | 1.55 | GSAL site | 9DL2 |
| 9 | 1.77 | NAD+ adenine site, surface | 9DL3 |
| 10 | 1.80 | NAD+ adenine site | 9DL4 |
| 11 | 1.77 | NAD+ adenine site | 9DL5 |
| 13 | 1.42 | GSAL site | 9DL6 |
| 19 | 1.72 | NAD+ adenine site | 9DL7 |
| 20 (replicate 1) | 1.64 | PRODH active site | 9DL8 |
| 20 (replicate 2) | 1.32 | PRODH active site, GSAL site | 9DL9 |
| 25 | 1.39 | PRODH active site | 9E0A |
| 39 | 1.47 | PRODH active site | 9E0B |
| 40 | 1.32 | PRODH active site | 9E0C |
| 41 | 1.33 | PRODH active site | 9E0D |
| 42 | 1.37 | PRODH active site | 9E0E |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meeks, K.R.; Bogner, A.N.; Nix, J.C.; Tanner, J.J. Crystallographic Fragment Screening of a Bifunctional Proline Catabolic Enzyme Reveals New Inhibitor Templates for Proline Dehydrogenase and L-Glutamate-γ-semialdehyde Dehydrogenase. Molecules 2024, 29, 5408. https://doi.org/10.3390/molecules29225408
Meeks KR, Bogner AN, Nix JC, Tanner JJ. Crystallographic Fragment Screening of a Bifunctional Proline Catabolic Enzyme Reveals New Inhibitor Templates for Proline Dehydrogenase and L-Glutamate-γ-semialdehyde Dehydrogenase. Molecules. 2024; 29(22):5408. https://doi.org/10.3390/molecules29225408
Chicago/Turabian StyleMeeks, Kaylen R., Alexandra N. Bogner, Jay C. Nix, and John J. Tanner. 2024. "Crystallographic Fragment Screening of a Bifunctional Proline Catabolic Enzyme Reveals New Inhibitor Templates for Proline Dehydrogenase and L-Glutamate-γ-semialdehyde Dehydrogenase" Molecules 29, no. 22: 5408. https://doi.org/10.3390/molecules29225408
APA StyleMeeks, K. R., Bogner, A. N., Nix, J. C., & Tanner, J. J. (2024). Crystallographic Fragment Screening of a Bifunctional Proline Catabolic Enzyme Reveals New Inhibitor Templates for Proline Dehydrogenase and L-Glutamate-γ-semialdehyde Dehydrogenase. Molecules, 29(22), 5408. https://doi.org/10.3390/molecules29225408