
Quantum Chemical Model Calculations of Adhesion and Dissociation between Epoxy Resin and Si-Containing Molecules


Abstract

1. Introduction
2. Results
3. Calculation Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bello, S.A.; Agunsoye, J.O.; Hassan SE, A.; Kana, M.Z. Epoxy resin based composites, mechanical and tribological properties: A review. Tribol. Ind. 2015, 37, 500. [Google Scholar]
- Ramon, E.; Sguazzo, C.; Moreira, P.M.G.P. A review of recent research on bio-based epoxy systems for engineering applications and potentialities in the aviation sector. Aerospace 2018, 5, 110. [Google Scholar] [CrossRef]
- Alam, M.I.; Maraz, K.M.; Khan, R.A. A review on the application of high-performance fiber-reinforced polymer composite materials. GSC Adv. Res. Rev. 2022, 10, 20–36. [Google Scholar] [CrossRef]
- Xiang, Q.; Xiao, F. Applications of epoxy materials in pavement engineering. Constr. Build. Mater. 2020, 235, 117529. [Google Scholar] [CrossRef]
- Saba, N.; Jawaid, M.; Alothman, O.Y.; Paridah, M.; Hassan, A. Recent advances in epoxy resin, natural fiber-reinforced epoxy composites and their applications. J. Reinf. Plast. Compos. 2016, 35, 447–470. [Google Scholar] [CrossRef]
- Unnikrishnan, K.; Thachil, E.T. Toughening of epoxy resins. Des. Monomers Polym. 2006, 9, 129–152. [Google Scholar] [CrossRef]
- Xi, Y.; Fukuzawa, H.; Fukunaga, S.; Kikugawa, G.; Zhao, Y.; Kawagoe, Y.; Okabe, T.; Kishimoto, N. Development of cat-GRRM/MC/MD Method for the Simulation of Cross-linked Network Structure Formation with Molecular Autocatalysis. Mol. Catal. 2024, 552, 113680. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Wang, G.-T.; Mai, Y.-W.; Zeng, Y. On fracture toughness of nano-particle modified epoxy. Compos. Part B Eng. 2011, 42, 2170–2175. [Google Scholar] [CrossRef]
- Haeri, S.; Ramezanzadeh, B.; Asghari, M. A novel fabrication of a high performance SiO2-graphene oxide (GO) nanohybrids: Characterization of thermal properties of epoxy nanocomposites filled with SiO2-GO nanohybrids. J. Colloid Interface Sci. 2017, 493, 111–122. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, M.; Pei, X.; Liu, S.; Luo, S.; Yan, M.; Shao, R.; Sun, Y.; Xu, W.; Xu, Z. Improving corrosion resistance of epoxy coating by optimizing the stress distribution and dispersion of SiO2 filler. Prog. Org. Coat. 2023, 179, 107522. [Google Scholar] [CrossRef]
- Anjum, N.; Prasad, S.L.A.; Suresha, B. Role of Silicon Dioxide Filler on Mechanical and Dry Sliding Wear Behaviour of Glass-Epoxy Composites. Adv. Tribol. 2013, 2013, 324952. [Google Scholar] [CrossRef]
- Wang, Z.; Cheng, Y.; Yang, M.; Huang, J.; Cao, D.; Chen, S.; Xie, Q.; Lou, W.; Wu, H. Dielectric properties and thermal conductivity of epoxy composites using core/shell structured Si/SiO2/Polydopamine. Compos. Part B Eng. 2018, 140, 83–90. [Google Scholar] [CrossRef]
- Li, S.; Lin, Z.; Zhou, Z.; Zhao, Y.; Ling, Z.; Zhang, Z.; Fang, X. Incorporating paraffin@SiO2 nanocapsules with abundant surface hydroxyl groups into polydimethylsiloxane to develop composites with enhanced interfacial heat conductance for chip heat dissipation. Nanoscale 2023, 15, 3419–3429. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cormack, A.N. Molecular Dynamics Simulation of the Structure and Hydroxylation of Silica Glass Surfaces. J. Am. Ceram. Soc. 2005, 88, 2532–2539. [Google Scholar] [CrossRef]
- Gomes, O.P.; Rheinheimer, J.P.; Dias, L.F.; Batagin-Neto, A.; Lisboa-Filho, P.N. Revisiting the hydroxylation phenomenon of SiO2: A study through “hard-hard” and “soft–soft” interactions. J. Mol. Model. 2022, 28, 115. [Google Scholar] [CrossRef]
- Wang, Z.; Lv, Q.; Chen, S.; Li, C.; Sun, S.; Hu, S. Effect of interfacial bonding on interphase properties in SiO2/epoxy nanocomposite: A molecular dynamics simulation study. ACS Appl. Mater. Interfaces 2016, 8, 7499–7508. [Google Scholar] [CrossRef]
- Li, W.; Zhang, L.; Zhang, M.; Dou, W.; Zhang, X.; Chen, S. The effects of interfacial water and SiO2 surface wettability on the adhesion properties of SiO2 in epoxy nanocomposites. Appl. Surf. Sci 2020, 502, 144151. [Google Scholar] [CrossRef]
- Oya, Y.; Nakazawa, M.; Shirasu, K.; Hino, Y.; Inuyama, K.; Kikugawa, G.; Li, J.; Kuwahara, R.; Kishimoto, N.; Waizumi, H.; et al. Molecular dynamics simulation of cross-linking processes and material properties for epoxy resins using first-principle calculation combined with global reaction route mapping algorithms. Chem. Phys. Lett. 2021, 762, 138104. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K.; Morokuma, K. Systematic exploration of the mechanism of chemical reactions: The global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Taketsugu, T.; Morokuma, K. Artificial Force Induced Reaction (AFIR) Method for Exploring Quantum Chemical Potential Energy Surfaces. Chem. Rec. 2016, 16, 2232–2248. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Saita, K.; Suzuki, K.; Ichino, T.; Sumiya, Y.; Sugiyama, K.; Ono, Y. Implementation and Performance of the Artificial Force Induced Reaction Method in the GRRM17 Program. J. Comput. Chem. 2018, 39, 233–250. [Google Scholar] [CrossRef] [PubMed]
- AFIR Manual Website. Available online: https://afir.sci.hokudai.ac.jp/manual/grrm17/grrm17_50.html (accessed on 24 September 2024).


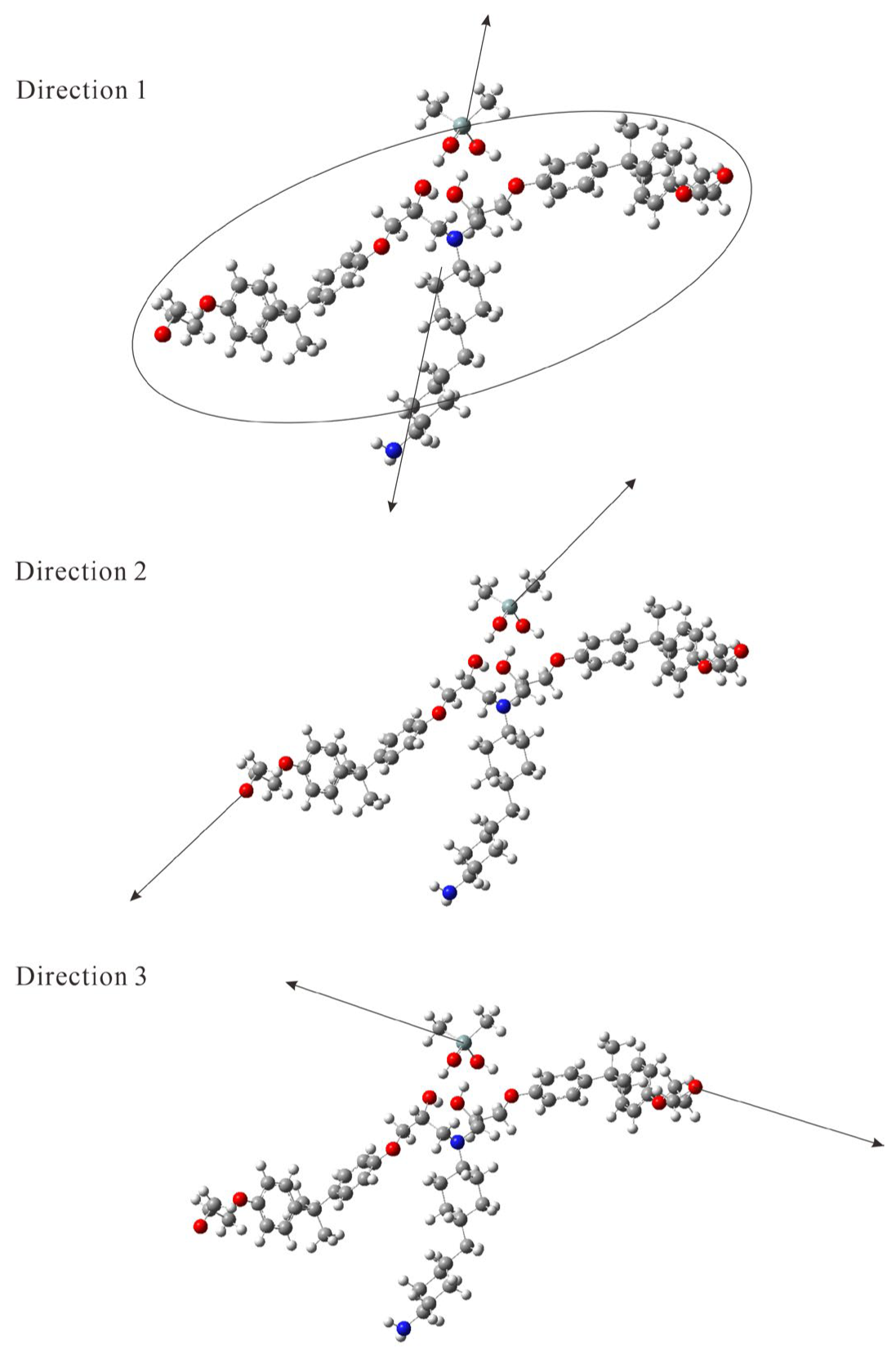
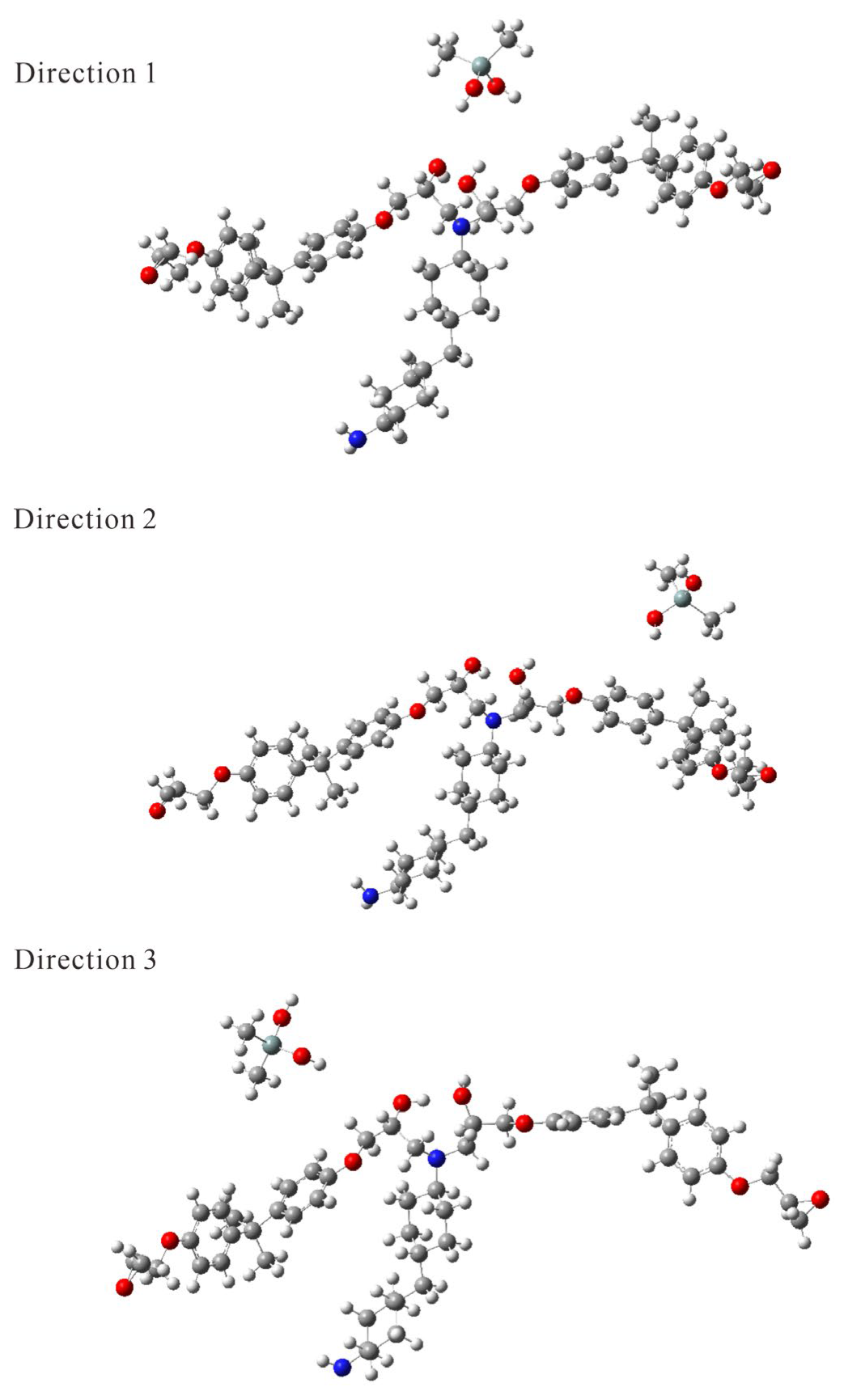

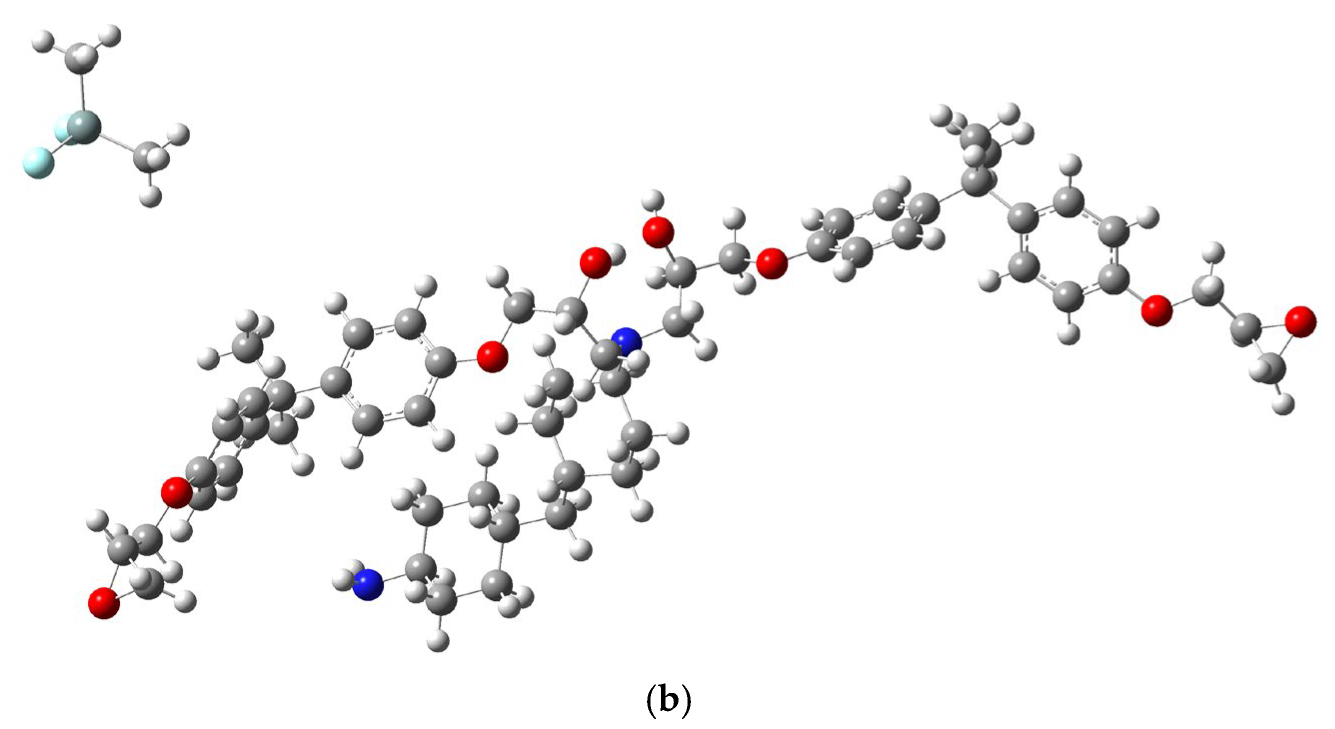

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Direction 1 | Direction 2 | Direction 3 | ||
|---|---|---|---|---|
| Si(CH3)2(OH)2 | Structure 1 | 35 * | 30 * | 25 * |
| Structure 2 | 25 * | 12.5 * | 17.5 * | |
| Si(CH3)4 | <2.5 ** | <2.5 ** | <2.5 ** | |
| (CH3)2SiF2 | 7.5 * | 5 * | 10 * | |
| Direction 1 | Direction 2 | Direction 3 | ||
|---|---|---|---|---|
| Si(CH3)2(OH)2 | Structure 1 | 35 * | 30 * | 25 * |
| Structure 2 | 25 * | 12.5 * | 20 * | |
| Si(CH3)4 | <2.5 ** | <2.5 ** | <2.5 ** | |
| (CH3)2SiF2 | 7.5 * | 5 * | 15 * | |
| Direction 1 | Direction 2 | Direction 3 | ||
|---|---|---|---|---|
| Si(CH3)2(OH)2 | Structure 1 | 22.5 * | 20 * | 7.5 * |
| Structure 2 | 12.5 * | 5 * | 7.5 * | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, H.; Xi, Y.; Kishimoto, N. Quantum Chemical Model Calculations of Adhesion and Dissociation between Epoxy Resin and Si-Containing Molecules. Molecules 2024, 29, 5050. https://doi.org/10.3390/molecules29215050
Xue H, Xi Y, Kishimoto N. Quantum Chemical Model Calculations of Adhesion and Dissociation between Epoxy Resin and Si-Containing Molecules. Molecules. 2024; 29(21):5050. https://doi.org/10.3390/molecules29215050
Chicago/Turabian StyleXue, Hao, Yingxiao Xi, and Naoki Kishimoto. 2024. "Quantum Chemical Model Calculations of Adhesion and Dissociation between Epoxy Resin and Si-Containing Molecules" Molecules 29, no. 21: 5050. https://doi.org/10.3390/molecules29215050
APA StyleXue, H., Xi, Y., & Kishimoto, N. (2024). Quantum Chemical Model Calculations of Adhesion and Dissociation between Epoxy Resin and Si-Containing Molecules. Molecules, 29(21), 5050. https://doi.org/10.3390/molecules29215050