

Soloxolone N-3-(Dimethylamino)propylamide Restores Drug Sensitivity of Tumor Cells with Multidrug-Resistant Phenotype via Inhibition of P-Glycoprotein Efflux Function
 , and
, and
Abstract
1. Introduction
2. Results and Discussion
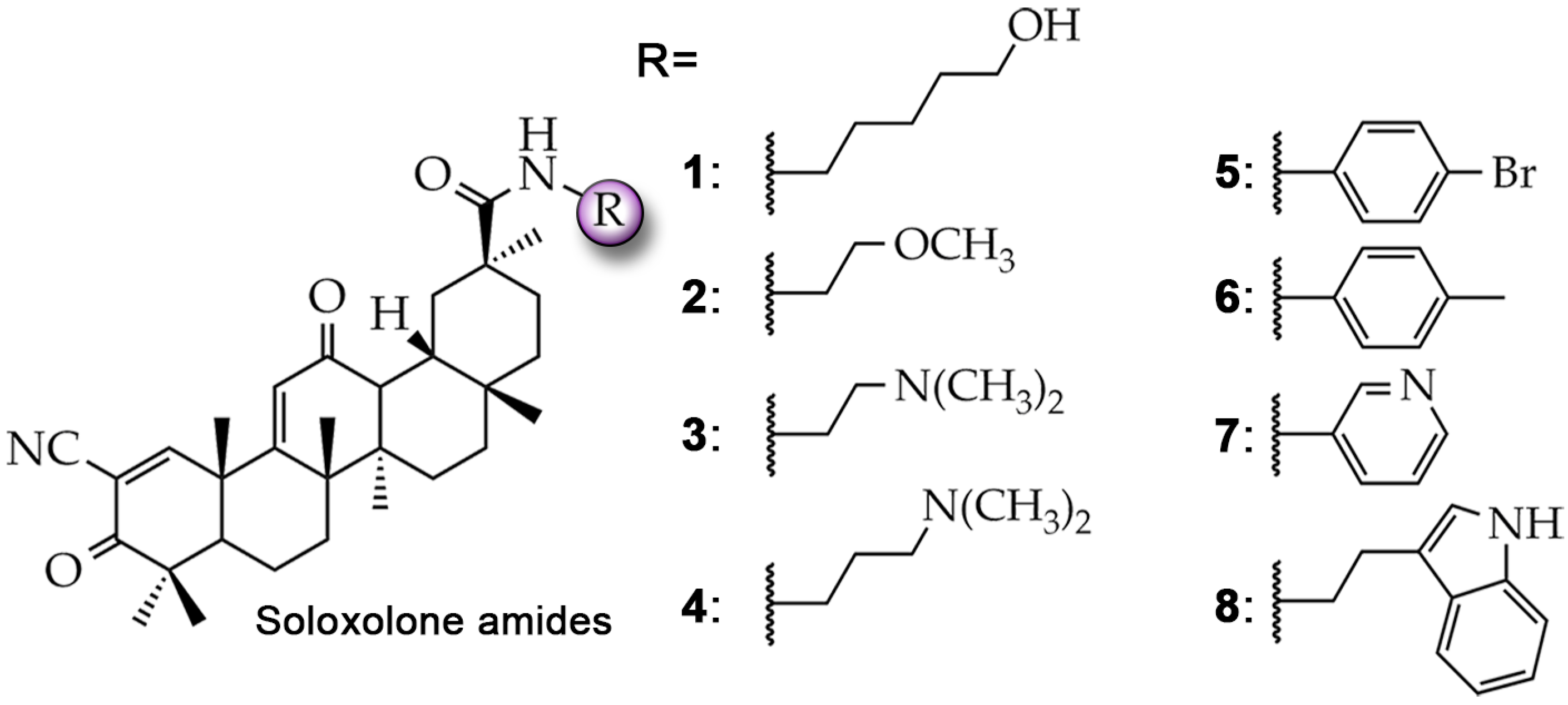
2.1. Soloxolone Amides 1–8 Can Interact with the Transmembrane Domain of P-gp
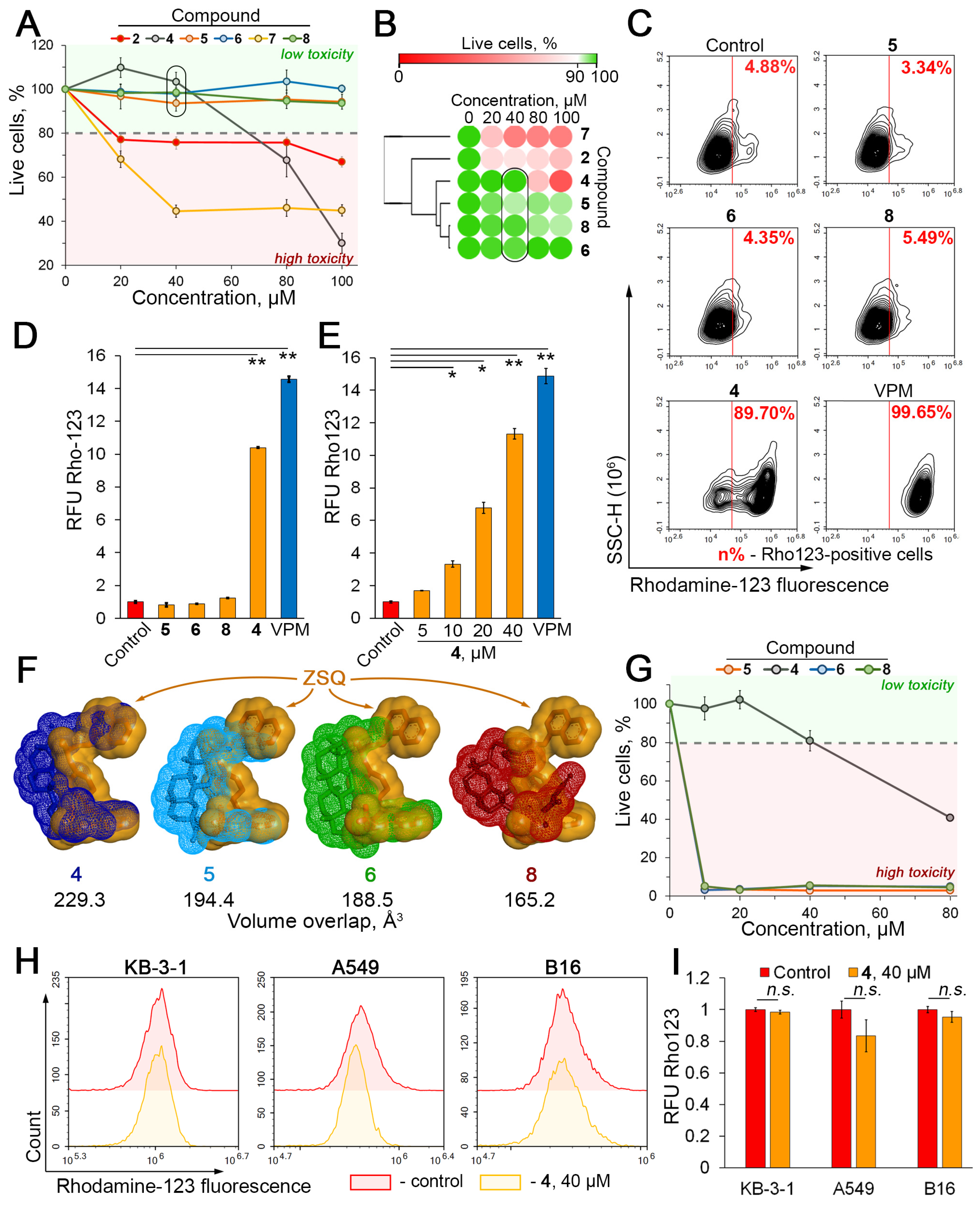
2.2. Compound 4 Enhances the Intracellular Accumulation of Rhodamine 123 in KB-8-5 Cells That Overexpress P-gp
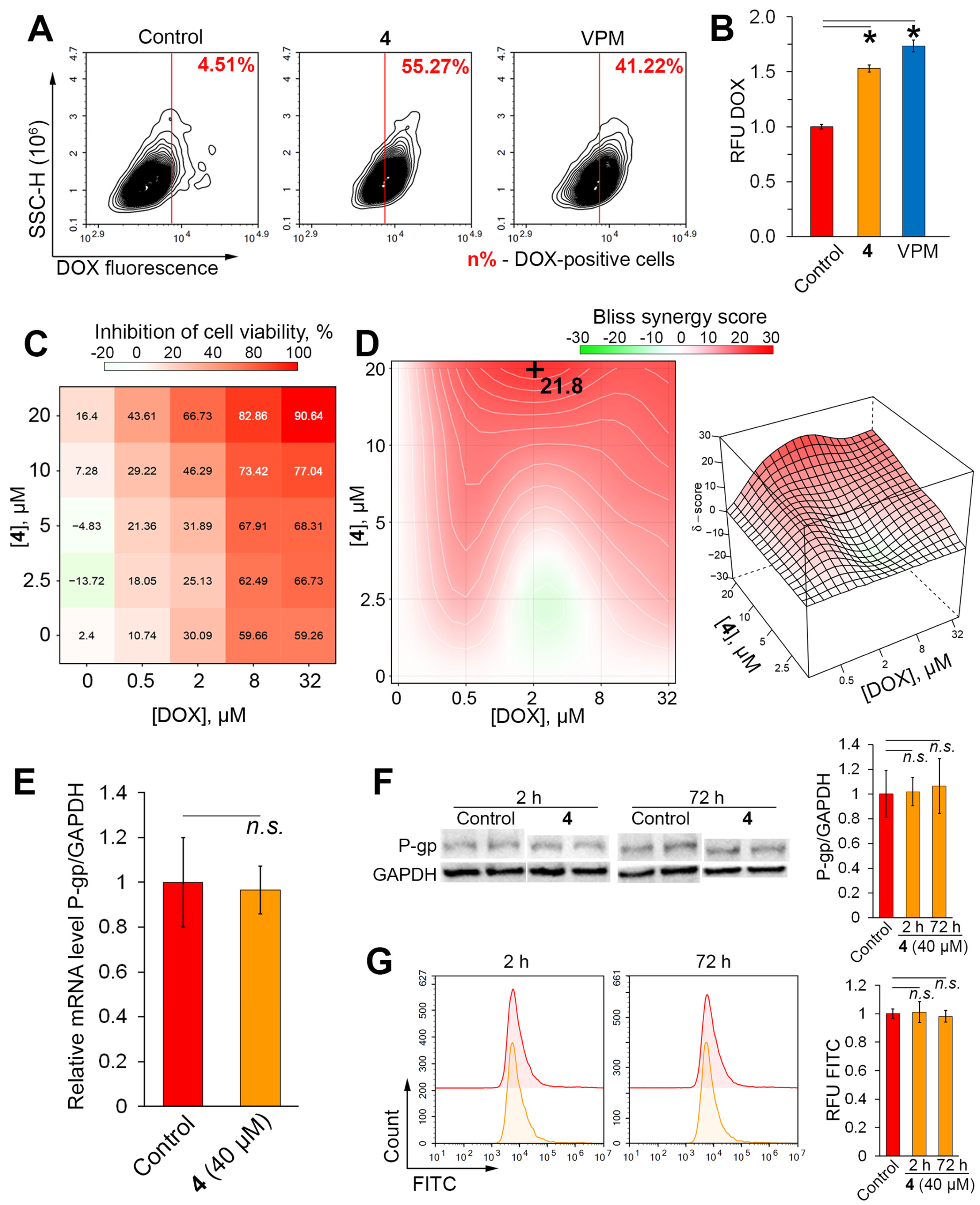
2.3. Compound 4 Increases Cytotoxicity and Intracellular Accumulation of Doxorubicin in KB-8-5 Cells
2.4. Compound 4 Binds to the Modulation Site of the P-gp Transmembrane Domain
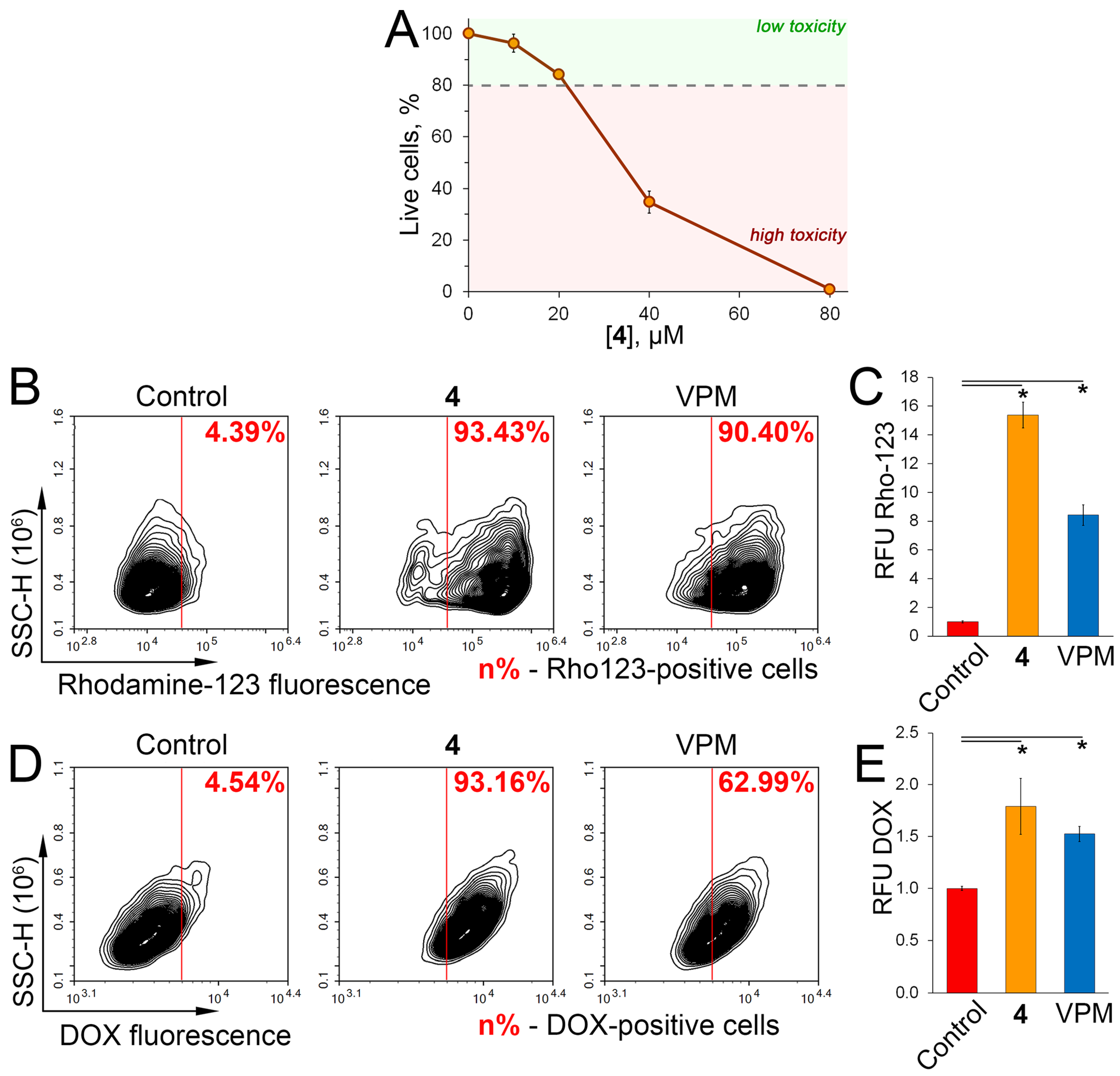
2.5. Verification of P-gp Inhibitory Activity of 4 in RLS40 Cells
2.6. Limitations of the Study
3. Materials and Methods
3.1. In Silico Prediction of P-glycoprotein Inhibitor Specificity of Soloxolone Amides
3.2. Cell Lines and Evaluated Compounds
3.3. Evaluation of Cytotoxicity by MTT Assay
3.4. Evaluation of Cytotoxicity by Water-Soluble Tetrazolium (WST) Test
3.5. Evaluation of the Combined Cytotoxicity of 4 and Doxorubicin
3.6. Rhodamine 123 and Doxorubicin Accumulation Assay
3.7. Kinetic Characterization for P-gp Inhibition
3.8. Assessment of ABCB1 Expression by Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
3.9. Western Blotting
3.10. Evaluation of P-gp Expression by Flow Cytometry
3.11. Molecular Docking
3.12. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, C.Y.; Shiranthika, C.; Wang, C.Y.; Chen, K.W.; Sumathipala, S. Reinforcement learning strategies in cancer chemotherapy treatments: A review. Comput. Methods Programs Biomed. 2023, 229, 107280. [Google Scholar] [CrossRef] [PubMed]
- Alam, A. Chemotherapy Treatment and Strategy Schemes: A Review. Open Access J. Toxicol. 2018, 2, 555600. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.Y.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef] [PubMed]
- Morozova, N.G.; Shmendel, E.V.; Timofeev, G.A.; Ivanov, I.V.; Kubasova, T.S.; Plyavnik, N.V.; Markova, A.A.; Maslov, M.A.; Shtil, A.A. New design of cationic alkyl glycoglycerolipids toxic to tumor cells. Mendeleev Commun. 2019, 29, 166–168. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I.H. The Role of Multidrug Resistance Efflux Pumps in Cancer: Revisiting a JNCI Publication Exploring Expression of the MDR1 (P-glycoprotein) Gene. J. Natl. Cancer Inst. 2015, 107, djv222. [Google Scholar] [CrossRef]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Gupta, S.K.; Singh, P.; Ali, V.; Verma, M. Role of membrane-embedded drug efflux ABC transporters in the cancer chemotherapy. Oncol. Rev. 2020, 14, 144–151. [Google Scholar] [CrossRef]
- Volm, M.; Efferth, T. Role of P-Glycoprotein for Resistance of Tumors to Anticancer Drugs: From Bench to Bedside. In Resistance to Targeted Anti-Cancer Therapeutics; Efferth, T., Ed.; Department of Pharmaceutical Biology, Johannes Gutenberg University: Mainz, Germany, 2015; pp. 1–26. [Google Scholar]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Dong, J.; Qin, Z.; Zhang, W.D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R.; Chen, Z.S.; Cheng, X.D.; Qin, J.J. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: An update. Drug Resist. Updat. 2020, 49, 100681. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.S.; Liu, H.M. Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three Decades of P-gp Inhibitors: Skimming Through Several Generations and Scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Yuan, L.; Hu, C.; Cheng, X.; Qin, J.-J. Strategies to overcome cancer multidrug resistance (MDR) through targeting P-glycoprotein (ABCB1): An updated review. Pharmacol. Ther. 2023, 249, 108488. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; De Feo, V.; Zia-Ul-Haq, M. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Petrova, A.V.; Lobov, A.N.; Kukovinets, O.S.; Baev, D.S.; Kazakova, O.B. Synthesis of C17-[5-methyl-1,3]-oxazoles by N-propargylation of triterpenic acids and evaluation of their cytotoxic activity. Nat. Prod. Res. 2021, 35, 3850–3858. [Google Scholar] [CrossRef]
- Kazakova, O.; Smirnova, I.; Tret’yakova, E.; Csuk, R.; Hoenke, S.; Fischer, L. Cytotoxic Potential of a-Azepano- and 3-Amino-3,4-SeCo-Triterpenoids. Int. J. Mol. Sci. 2021, 22, 1714. [Google Scholar] [CrossRef]
- Krainova, G.; Beloglazova, Y.; Dmitriev, M.; Grishko, V. Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds. Molecules 2023, 28, 550. [Google Scholar] [CrossRef]
- Markov, A.V.; Odarenko, K.V.; Sen’kova, A.V.; Ilyina, A.A.; Zenkova, M.A. Evaluation of the Antitumor Potential of Soloxolone Tryptamide against Glioblastoma Multiforme Using in silico, in vitro, and in vivo Approaches. Biochemistry 2023, 88, 1008–1021. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Sen’kova, A.V.; Moralev, A.D.; Savin, I.A.; Komarova, N.I.; Salakhutdinov, N.F.; Zenkova, M.A.; Markov, A.V. Novel Epoxides of Soloxolone Methyl: An Effect of the Formation of Oxirane Ring and Stereoisomerism on Cytotoxic Profile, Anti-Metastatic and Anti-Inflammatory Activities In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 6214. [Google Scholar] [CrossRef]
- Odarenko, K.V.; Sen’kova, A.V.; Salomatina, O.V.; Markov, O.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Markov, A.V. Soloxolone para-methylanilide effectively suppresses aggressive phenotype of glioblastoma cells including TGF-β1-induced glial-mesenchymal transition in vitro and inhibits growth of U87 glioblastoma xenografts in mice. Front. Pharmacol. 2024, 15, 1428924. [Google Scholar] [CrossRef] [PubMed]
- Moralev, A.D.; Salomatina, O.V.; Chernikov, I.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Markov, A.V. A Novel 3-meta-Pyridine-1,2,4-oxadiazole Derivative of Glycyrrhetinic Acid as a Safe and Promising Candidate for Overcoming P-Glycoprotein-Mediated Multidrug Resistance in Tumor Cells. ACS Omega 2023, 8, 48813–48824. [Google Scholar] [CrossRef] [PubMed]
- Laiolo, J.; Graikioti, D.G.; Barbieri, C.L.; Joray, M.B.; Antoniou, A.I.; Vera, D.M.A.; Athanassopoulos, C.M.; Carpinella, M.C. Novel betulin derivatives as multidrug reversal agents targeting P-glycoprotein. Sci. Rep. 2024, 14, 70. [Google Scholar] [CrossRef] [PubMed]
- Tolmacheva, I.; Beloglazova, Y.; Nazarov, M.; Gagarskikh, O.; Grishko, V. Synthesis and Anticancer Activity of A-Ring-Modified Derivatives of Dihydrobetulin. Int. J. Mol. Sci. 2023, 24, 9863. [Google Scholar] [CrossRef] [PubMed]
- Rybalkina, E.Y.; Moiseeva, N.I.; Karamysheva, A.F.; Eroshenko, D.V.; Konysheva, A.V.; Nazarov, A.V.; Grishko, V.V. Triterpenoids with modified A-ring as modulators of P-gp-dependent drug-resistance in cancer cells. Chem. Biol. Interact. 2021, 348, 109645. [Google Scholar] [CrossRef]
- Laiolo, J.; Barbieri, C.L.; Joray, M.B.; Lanza, P.A.; Palacios, S.M.; Vera, D.M.A.; Carpinella, M.C. Plant extracts and betulin from Ligaria cuneifolia inhibit P-glycoprotein function in leukemia cells. Food Chem. Toxicol. 2021, 147, 111922. [Google Scholar] [CrossRef]
- Moiseeva, N.; Eroshenko, D.; Laletina, L.; Rybalkina, E.; Susova, O.; Karamysheva, A.; Tolmacheva, I.; Nazarov, M.; Grishko, V. The Molecular Mechanisms of Oleanane Aldehyde-β-enone Cytotoxicity against Doxorubicin-Resistant Cancer Cells. Biology 2023, 12, 415. [Google Scholar] [CrossRef]
- Markov, A.V.; Ilyina, A.A.; Salomatina, O.V.; Sen’kova, A.V.; Okhina, A.A.; Rogachev, A.D.; Salakhutdinov, N.F.; Zenkova, M.A. Novel Soloxolone Amides as Potent Anti-Glioblastoma Candidates: Design, Synthesis, In Silico Analysis and Biological Activities In Vitro and In Vivo. Pharmaceuticals 2022, 15, 603. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Wang, P.-H.; Tu, Y.-S.; Tseng, Y.J. PgpRules: A decision tree based prediction server for P-glycoprotein substrates and inhibitors. Bioinformatics 2019, 35, 4193–4195. [Google Scholar] [CrossRef]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN Web Server for ADMET Predictions. Front. Pharmacol. 2017, 8, 00889. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.J.; Ferreira, M.J.U.; Dos Santos, D.J.V.A. Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Phondeth, L.; Kamaraj, R.; Nilles, J.; Weiss, J.; Haefeli, W.E.; Pávek, P.; Theile, D. Rifabutin but not rifampicin can partly out-balance P-glycoprotein induction by concurrent P-glycoprotein inhibition through high affinity binding to the inhibitory site. Arch. Toxicol. 2024, 98, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.M.D.; Almazni, I.A.; Moawadh, M.S.; Alharbi, Z.M.; Helmi, N.; Alqahtani, L.S.; Hussain, T.; Alafnan, A.; Moin, A.; Elkhalifa, A.O.; et al. Targeting NF-κB signaling cascades of glioblastoma by a natural benzophenone, garcinol, via in vitro and molecular docking approaches. Front. Chem. 2024, 12, 1352009. [Google Scholar] [CrossRef]
- Shah, D.; Ajazuddin; Bhattacharya, S. Role of natural P-gp inhibitor in the effective delivery for chemotherapeutic agents. J. Cancer Res. Clin. Oncol. 2023, 149, 367–391. [Google Scholar] [CrossRef]
- Singh, S.; Prasad, N.R.; Chufan, E.E.; Patel, B.A.; Wang, Y.J.; Chen, Z.S.; Ambudkar, S.V.; Talele, T.T. Design and synthesis of human ABCB1 (P-glycoprotein) inhibitors by peptide coupling of diverse chemical scaffolds on carboxyl and amino termini of (S)-valine-derived thiazole amino acid. J. Med. Chem. 2014, 57, 4058–4072. [Google Scholar] [CrossRef]
- Zeng, R.; Yang, X.M.; Li, H.W.; Li, X.; Guan, Y.; Yu, T.; Yan, P.; Yuan, W.; Niu, S.L.; Gu, J.; et al. Simplified Derivatives of Tetrandrine as Potent and Specific P-gp Inhibitors to Reverse Multidrug Resistance in Cancer Chemotherapy. J. Med. Chem. 2023, 66, 4086–4105. [Google Scholar] [CrossRef]
- Ibba, R.; Sestito, S.; Ambrosio, F.A.; Marchese, E.; Costa, G.; Fiorentino, F.P.; Fusi, F.; Marchesi, I.; Polini, B.; Chiellini, G.; et al. Discovery of pyridoquinoxaline-based new P-gp inhibitors as coadjutant against Multi Drug Resistance in cancer. Eur. J. Med. Chem. 2024, 276, 116647. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, B.; Dai, Q.Q.; Zhang, X.N.; Zhang, J.Y.; Zuo, J.H.; Liu, H.; Chen, Z.S.; Li, G.B.; Wang, S.; et al. Discovery of New 4-Indolyl Quinazoline Derivatives as Highly Potent and Orally Bioavailable P-Glycoprotein Inhibitors. J. Med. Chem. 2021, 64, 14895–14911. [Google Scholar] [CrossRef]
- Błauż, A.; Rychlik, B. Drug-selected cell line panels for evaluation of the pharmacokinetic consequences of multidrug resistance proteins. J. Pharmacol. Toxicol. Methods 2017, 84, 57–65. [Google Scholar] [CrossRef]
- Ye, L.-Y.; Hu, S.; Xu, H.-E.; Xu, R.-R.; Kong, H.; Zeng, X.-N.; Xie, W.-P.; Wang, H. The effect of tetrandrine combined with cisplatin on proliferation and apoptosis of A549/DDP cells and A549 cells. Cancer Cell Int. 2017, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Song, Y.; Li, C.; Zhang, Z.; Xue, Z.; Huang, Q.; Yu, L.; Zhu, D.; Cao, Z.; Lu, A.; et al. The naturally occurring flavonoid nobiletin reverses methotrexate resistance via inhibition of P-glycoprotein synthesis. J. Biol. Chem. 2022, 298, 101756. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.H.; Bao, Y.L.; Wu, Y.; Yu, C.L.; Meng, X.Y.; Li, Y.X. Cantharidin reverses multidrug resistance of human hepatoma HepG2/ADM cells via down-regulation of P-glycoprotein expression. Cancer Lett. 2008, 272, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lu, Q.; Hu, X. Down-regulation of P-glycoprotein expression in MDR breast cancer cell MCF-7/ADR by honokiol. Cancer Lett. 2006, 243, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Ling, V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997, 250, 130–137. [Google Scholar] [CrossRef]
- Rajaei, N.; Rahgouy, G.; Panahi, N.; Razzaghi-Asl, N. Bioinformatic analysis of highly consumed phytochemicals as P-gp binders to overcome drug-resistance. Res. Pharm. Sci. 2023, 18, 505–516. [Google Scholar] [CrossRef]
- Patutina, O.A.; Mironova, N.L.; Logashenko, E.B.; Popova, N.A.; Nikolin, V.P.; Vasil’ev, G.V.; Kaledin, V.I.; Zenkova, M.A.; Vlasov, V.V. Cyclophosphamide metabolite inducing apoptosis in RLS mouse lymphosarcoma cells is a substrate for P-glycoprotein. Bull. Exp. Biol. Med. 2012, 152, 348–352. [Google Scholar] [CrossRef]
- Chang, Y.T.; Lin, Y.C.; Sun, L.; Liao, W.C.; Wang, C.C.N.; Chou, C.Y.; Morris-Natschke, S.L.; Lee, K.H.; Hung, C.C. Wilforine resensitizes multidrug resistant cancer cells via competitive inhibition of P-glycoprotein. Phytomedicine 2020, 71, 153239. [Google Scholar] [CrossRef]
- Singh, D.V.; Godbole, M.M.; Misra, K. A plausible explanation for enhanced bioavailability of P-gp substrates in presence of piperine: Simulation for next generation of P-gp inhibitors. J. Mol. Model. 2013, 19, 227–238. [Google Scholar] [CrossRef]
- Shchulkin, A.V.; Abalenikhina, Y.V.; Kosmachevskaya, O.V.; Topunov, A.F.; Yakusheva, E.N. Regulation of P-Glycoprotein during Oxidative Stress. Antioxidants 2024, 13, 215. [Google Scholar] [CrossRef]
- Thomas, C.; Tampé, R. Structural and Mechanistic Principles of ABC Transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.S.E.; Sen’kova, A.V.; Nadyrova, A.I.; Savin, I.A.; Markov, A.V.; Mitkevich, V.A.; Makarov, A.A.; Ilinskaya, O.N.; Mironova, N.L.; Zenkova, M.A. Antitumour activity of the ribonuclease binase from Bacillus pumilus in the RLS40 tumour model is associated with the reorganisation of the miRNA network and reversion of cancer-related cascades to normal functioning. Biomolecules 2020, 10, 1509. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Type | Title 3 |
|---|---|---|
| ABCB1 | Forward | 5′-AATGGCTACATGAGAGCGGAG-3′ |
| Reverse | 5′-AATGTTCTGGCTTCCGTTGC-3′ | |
| GAPDH | Forward | 5′-ACCCCCAATGTGTCCGTCGT-3′ |
| Reverse | 5′-TACTCCTTGGAGGCCATGTA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moralev, A.D.; Salomatina, O.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Markov, A.V. Soloxolone N-3-(Dimethylamino)propylamide Restores Drug Sensitivity of Tumor Cells with Multidrug-Resistant Phenotype via Inhibition of P-Glycoprotein Efflux Function. Molecules 2024, 29, 4939. https://doi.org/10.3390/molecules29204939
Moralev AD, Salomatina OV, Salakhutdinov NF, Zenkova MA, Markov AV. Soloxolone N-3-(Dimethylamino)propylamide Restores Drug Sensitivity of Tumor Cells with Multidrug-Resistant Phenotype via Inhibition of P-Glycoprotein Efflux Function. Molecules. 2024; 29(20):4939. https://doi.org/10.3390/molecules29204939
Chicago/Turabian StyleMoralev, Arseny D., Oksana V. Salomatina, Nariman F. Salakhutdinov, Marina A. Zenkova, and Andrey V. Markov. 2024. "Soloxolone N-3-(Dimethylamino)propylamide Restores Drug Sensitivity of Tumor Cells with Multidrug-Resistant Phenotype via Inhibition of P-Glycoprotein Efflux Function" Molecules 29, no. 20: 4939. https://doi.org/10.3390/molecules29204939
APA StyleMoralev, A. D., Salomatina, O. V., Salakhutdinov, N. F., Zenkova, M. A., & Markov, A. V. (2024). Soloxolone N-3-(Dimethylamino)propylamide Restores Drug Sensitivity of Tumor Cells with Multidrug-Resistant Phenotype via Inhibition of P-Glycoprotein Efflux Function. Molecules, 29(20), 4939. https://doi.org/10.3390/molecules29204939