
Theoretical Investigation of Rate Coefficients and Dynamical Mechanisms for N + N + N Three-Body Recombination Based on Full-Dimensional Potential Energy Surfaces

Abstract
1. Introduction
2. Computational Details
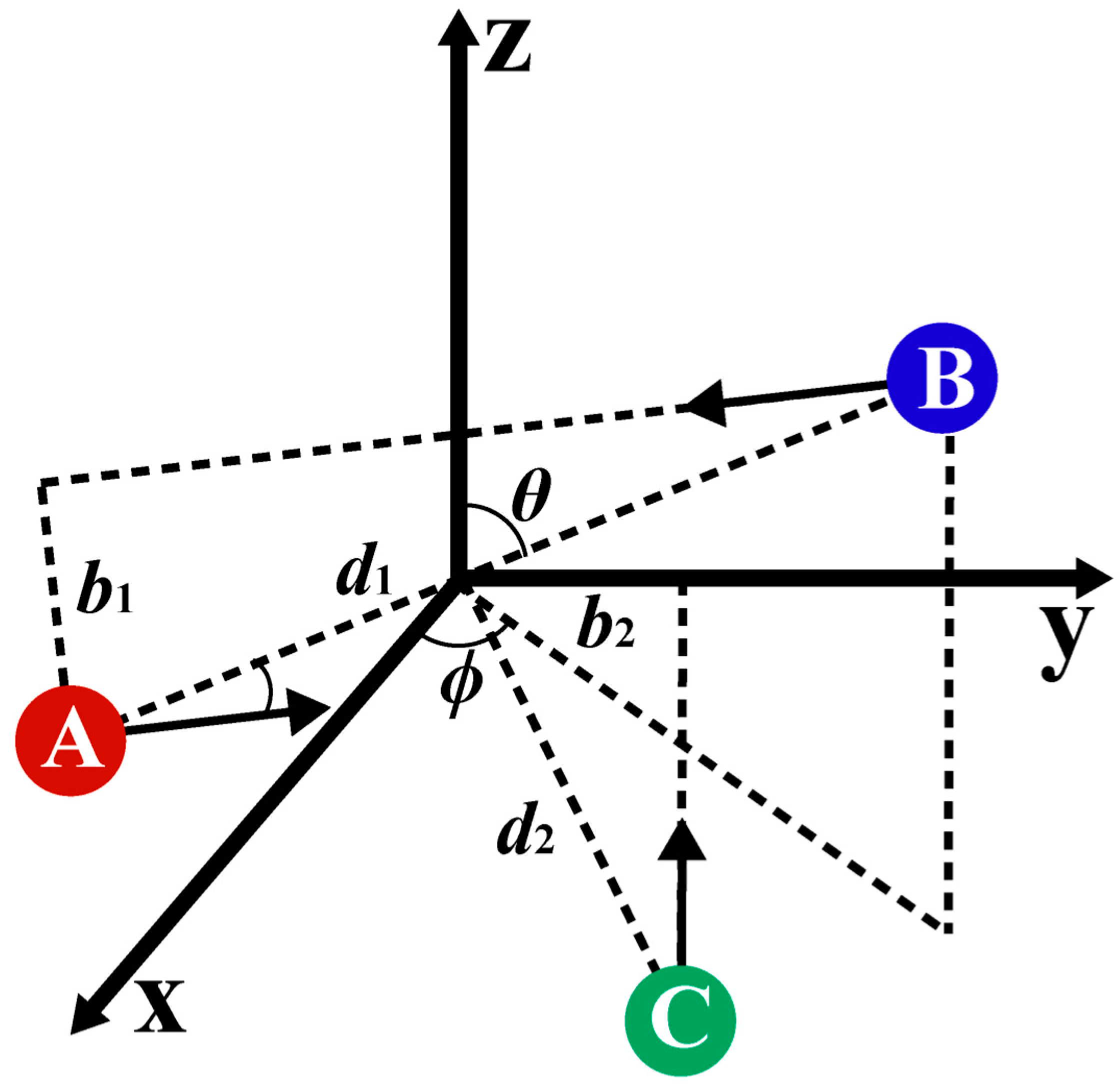
2.1. Binary Collision Parameters
2.2. Three-Body Collision Parameters
2.3. Rate Coefficients
2.4. Binning Method
3. Results and Discussion
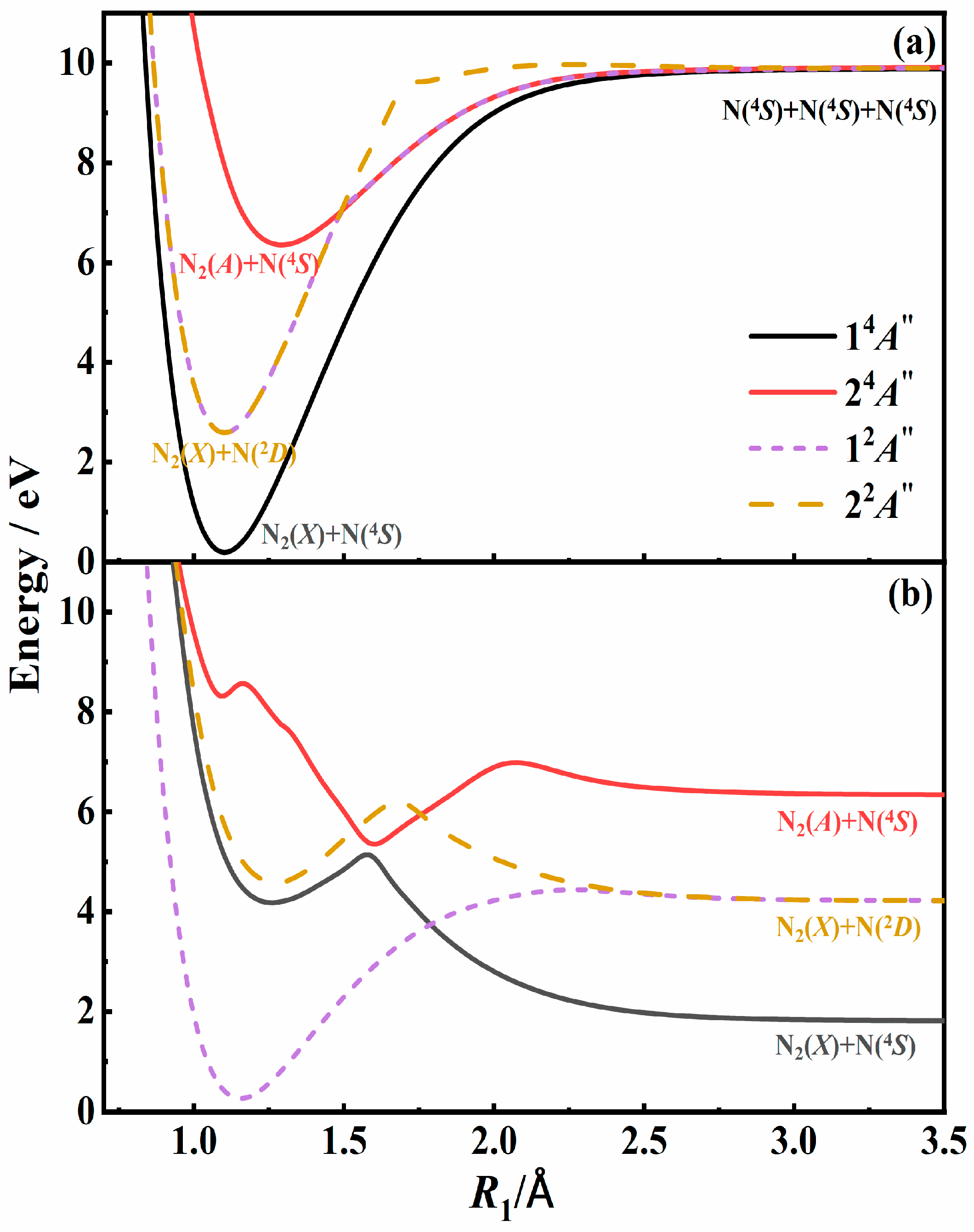
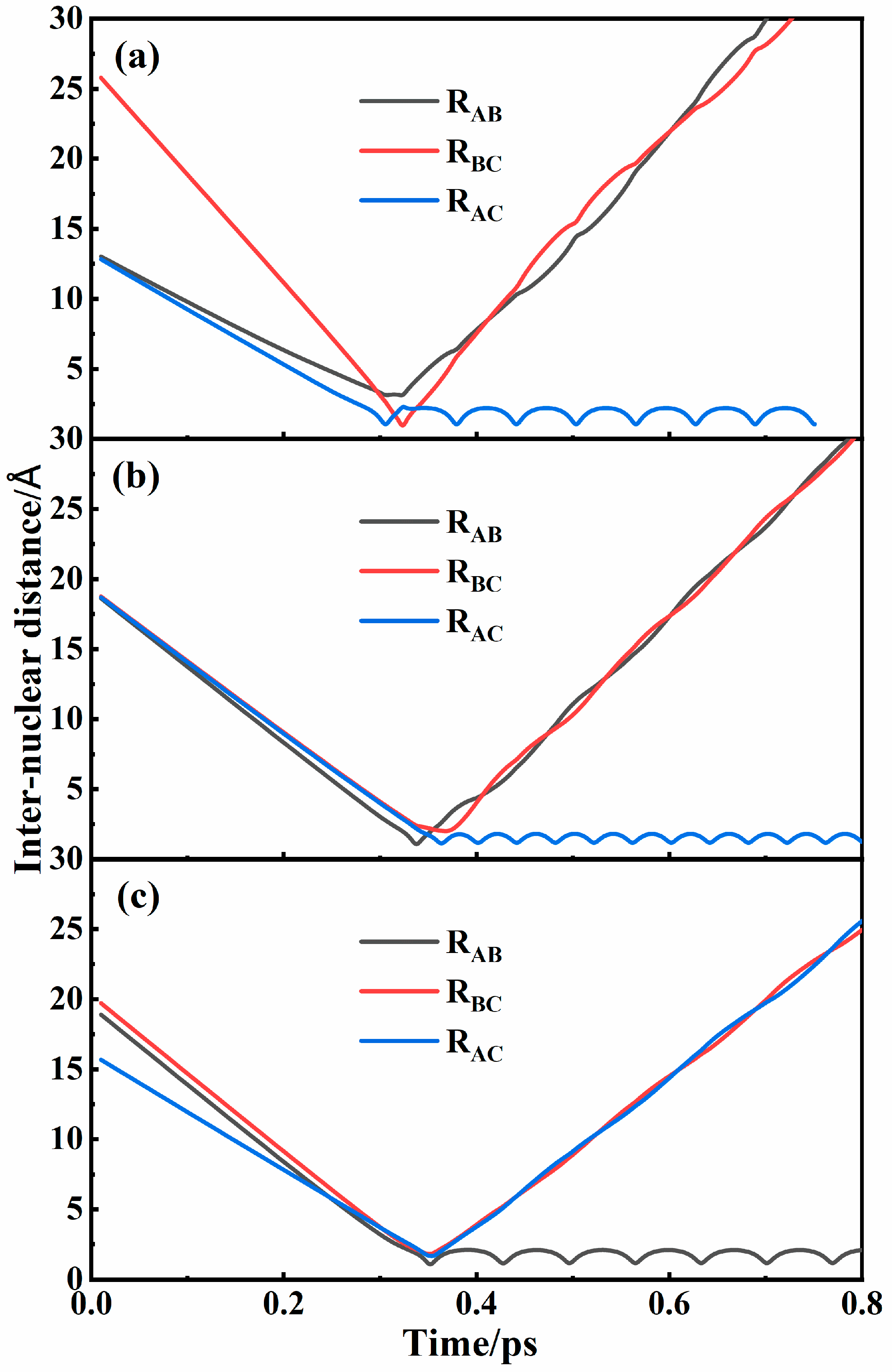
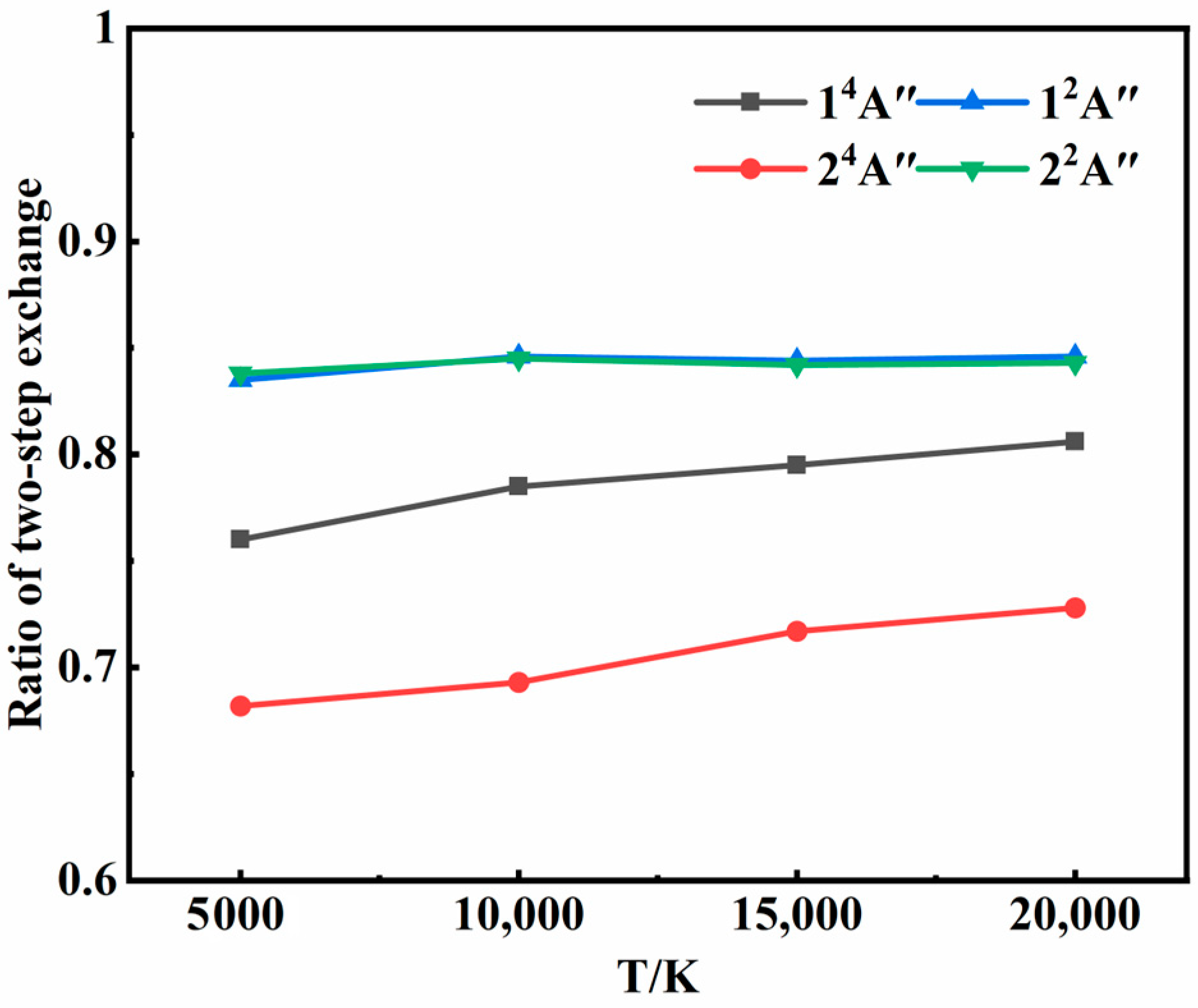
3.1. Recombination Mechanisms
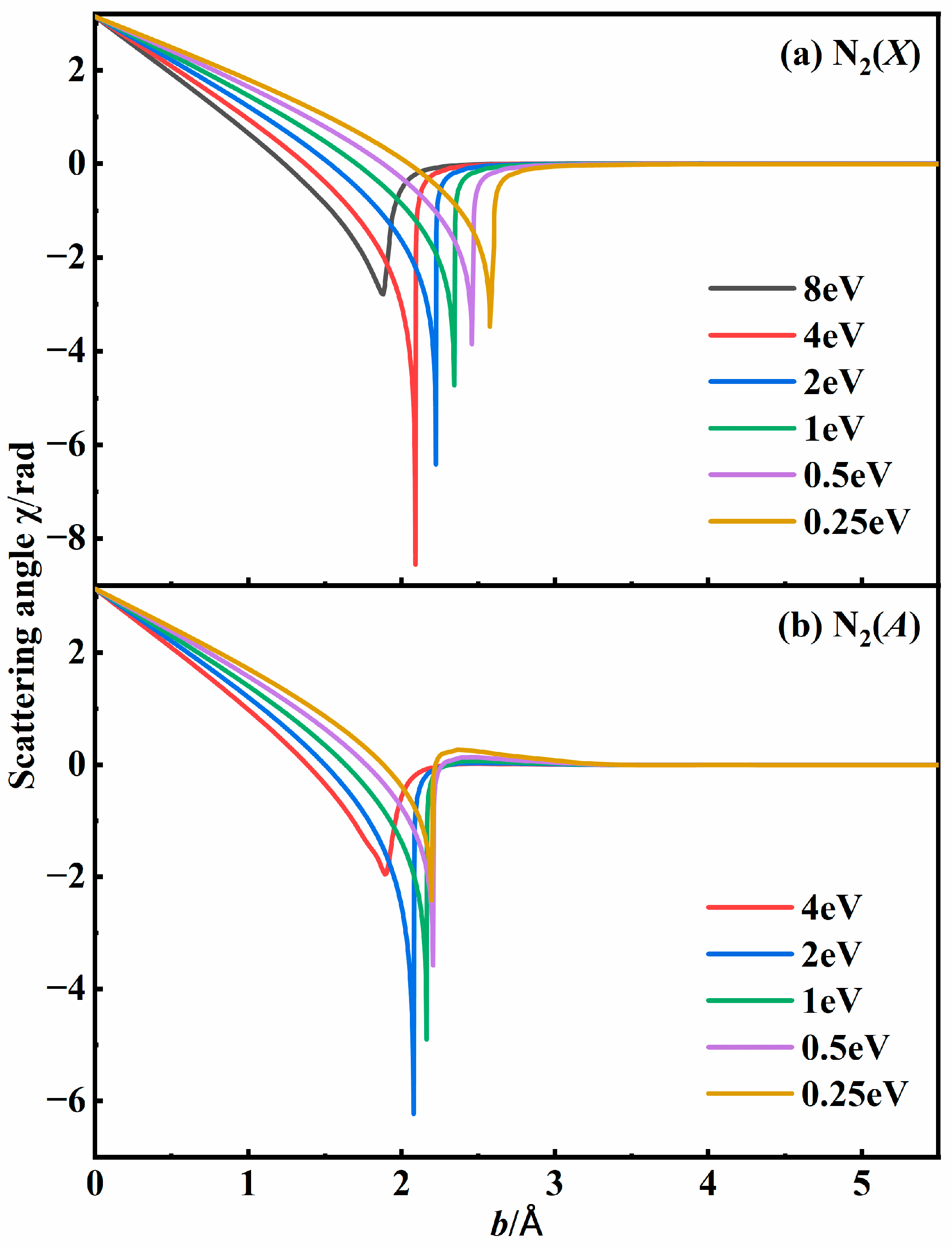
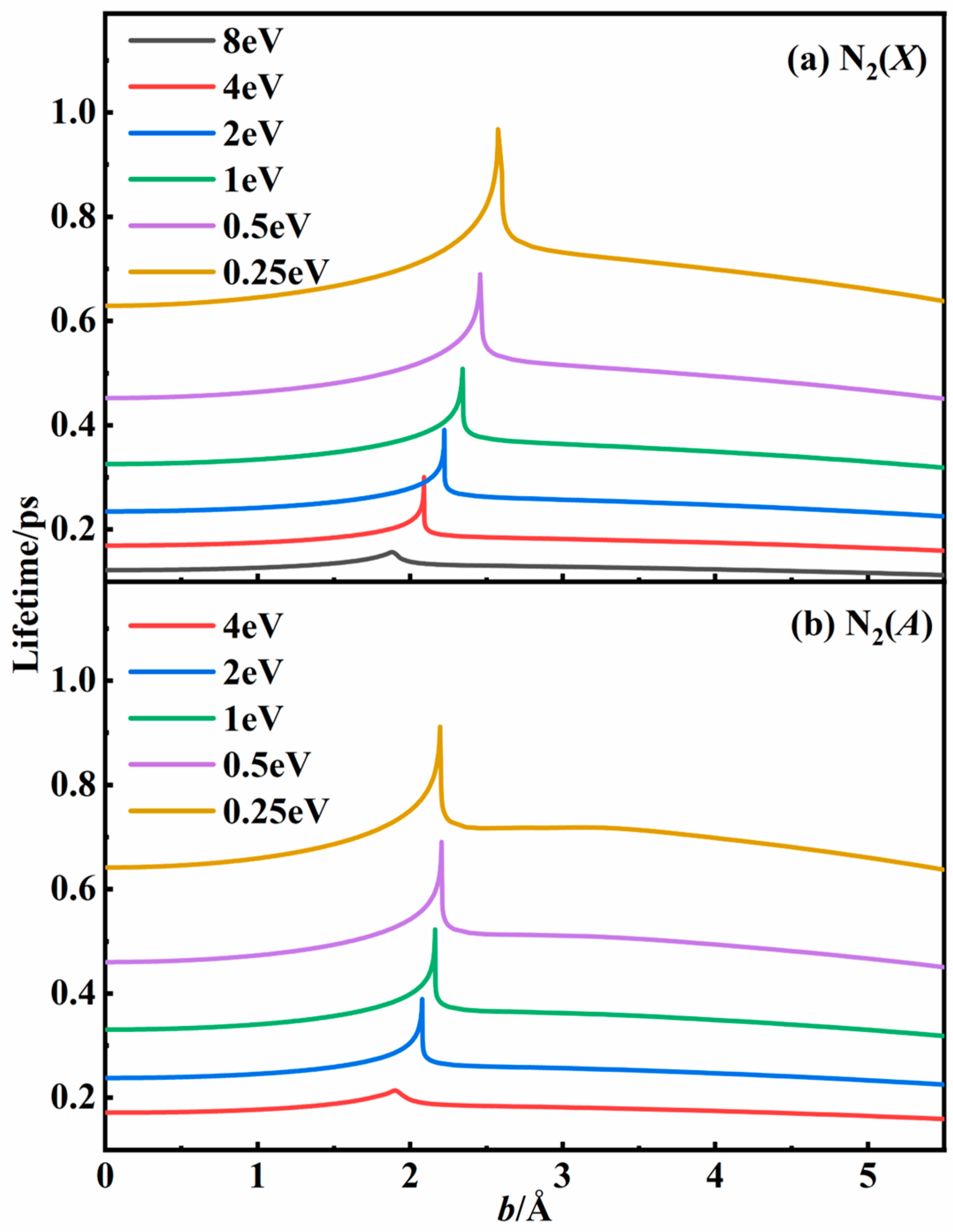
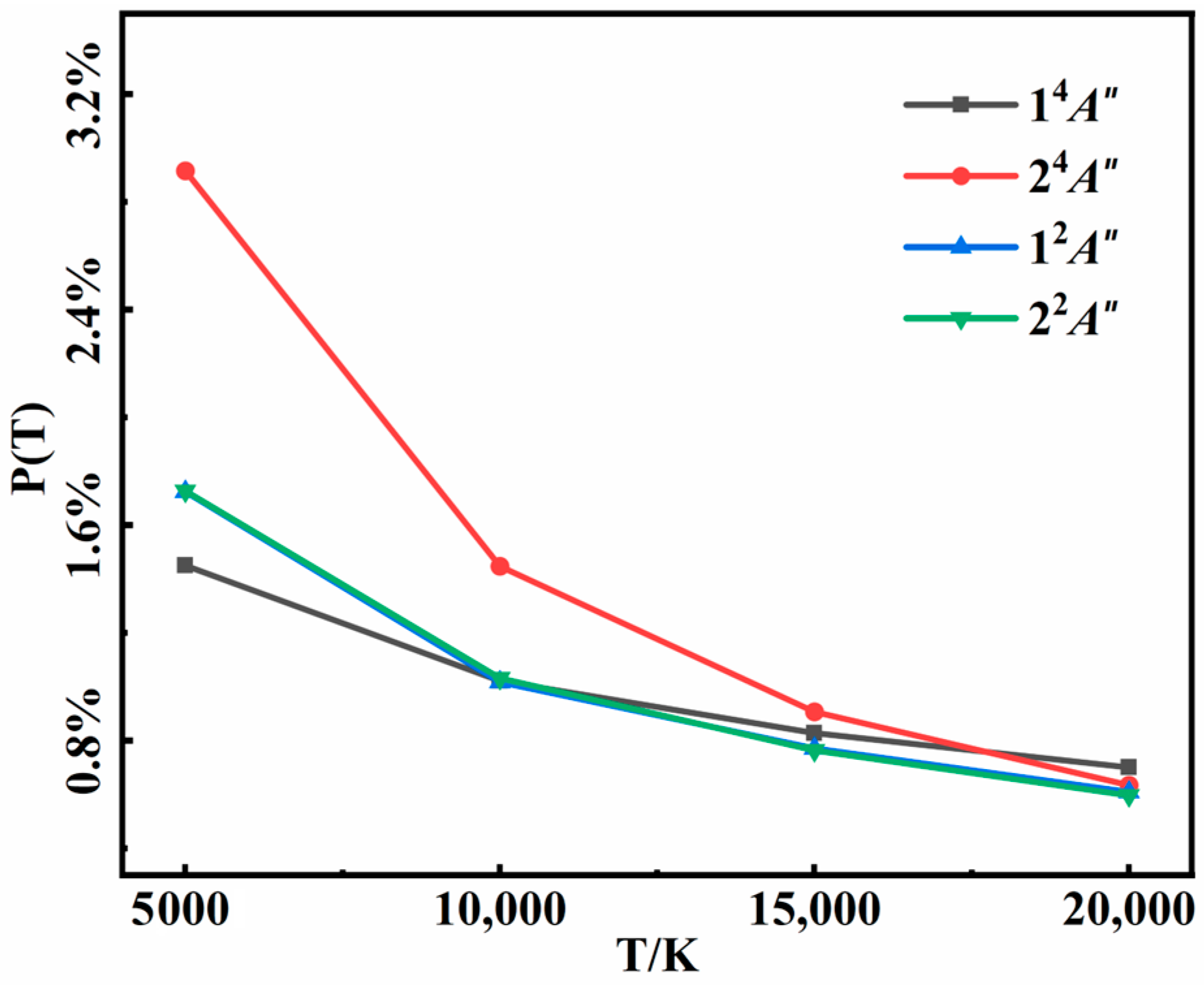
3.2. Recombination Probabilities
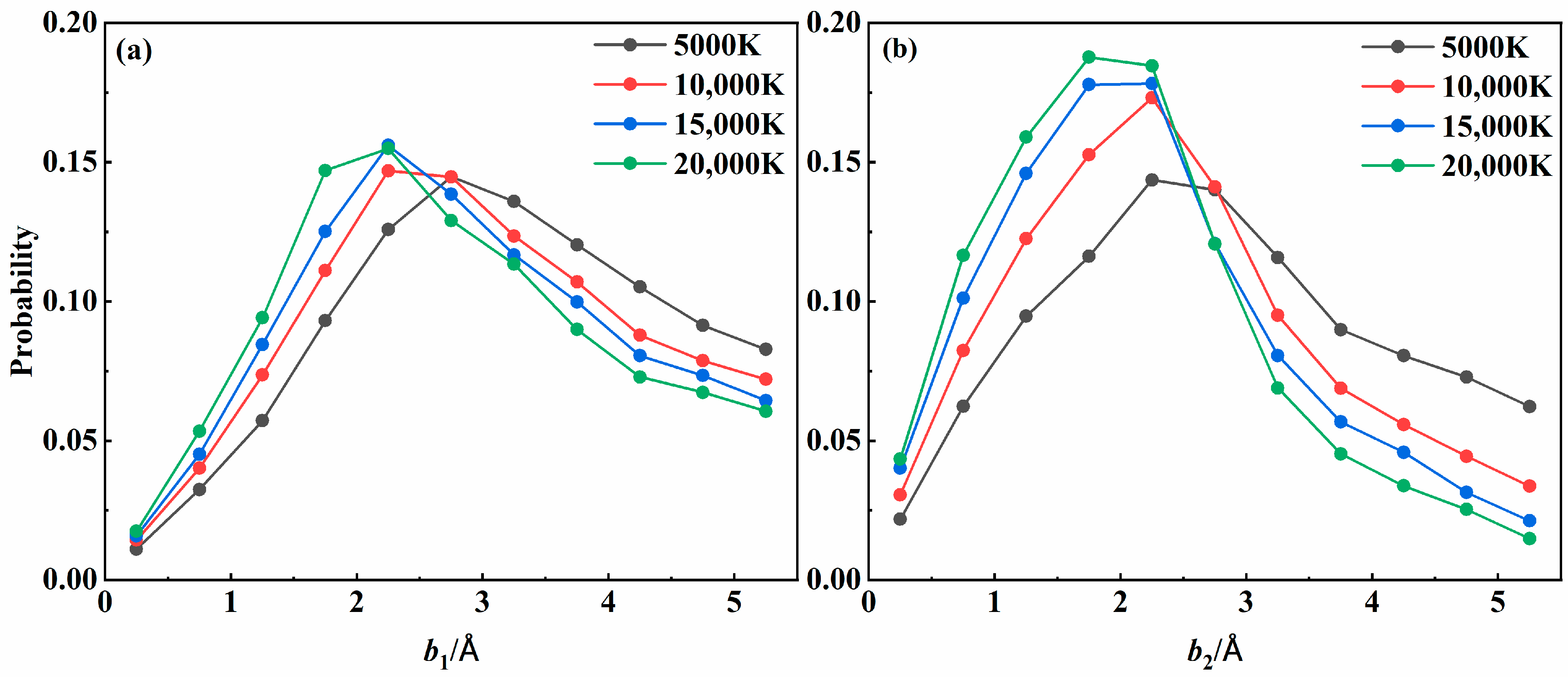
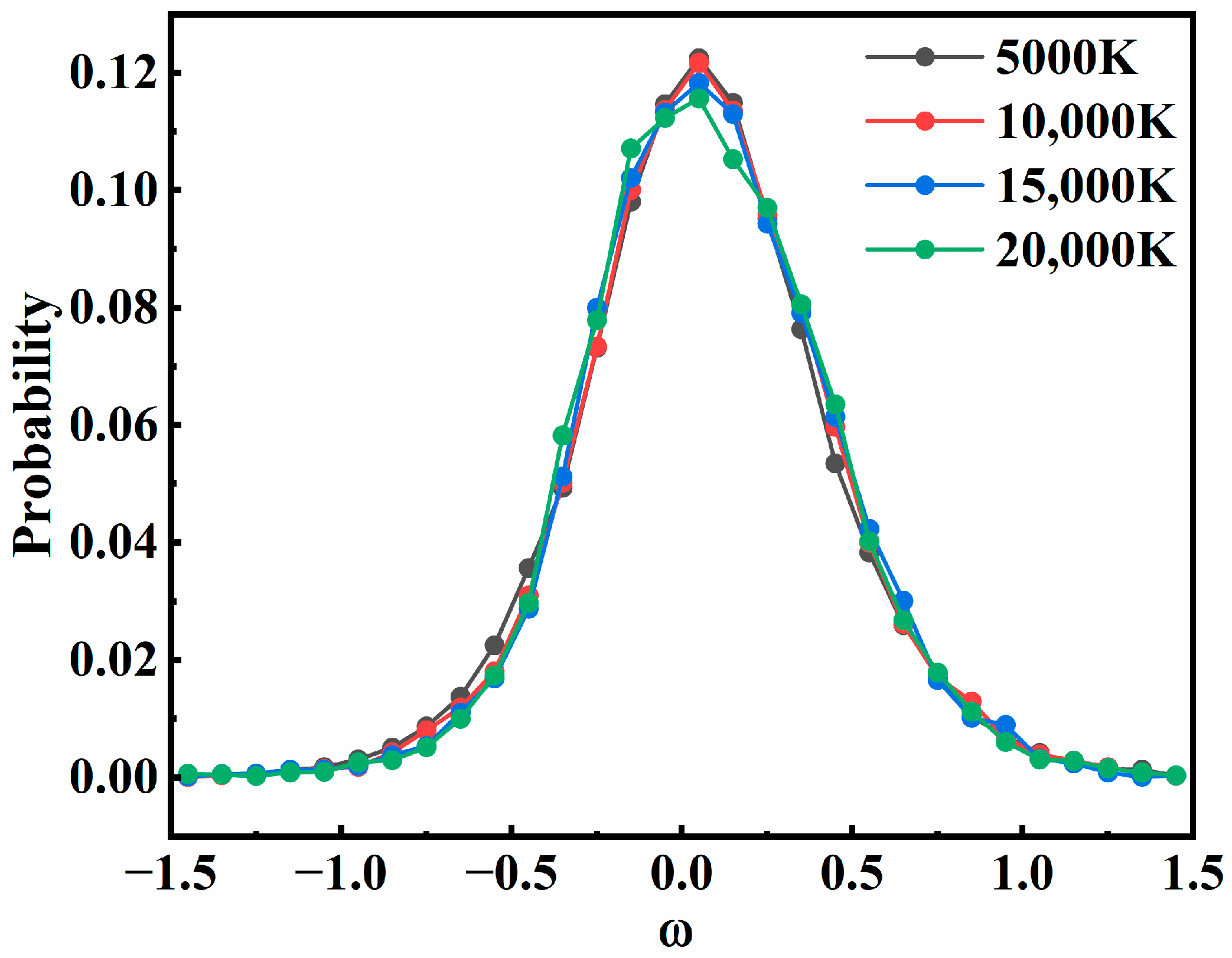
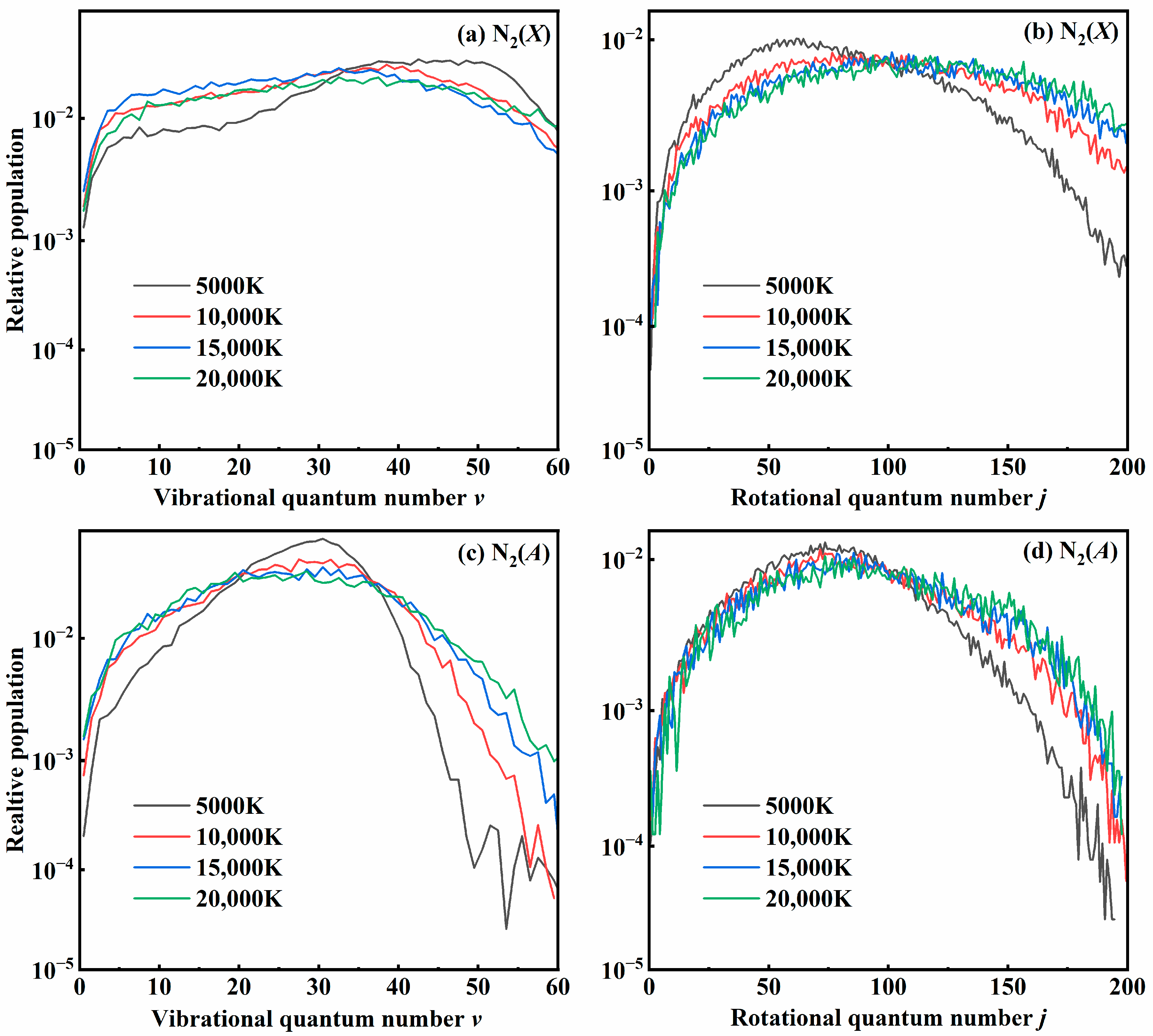
3.3. Quantum State Distribution
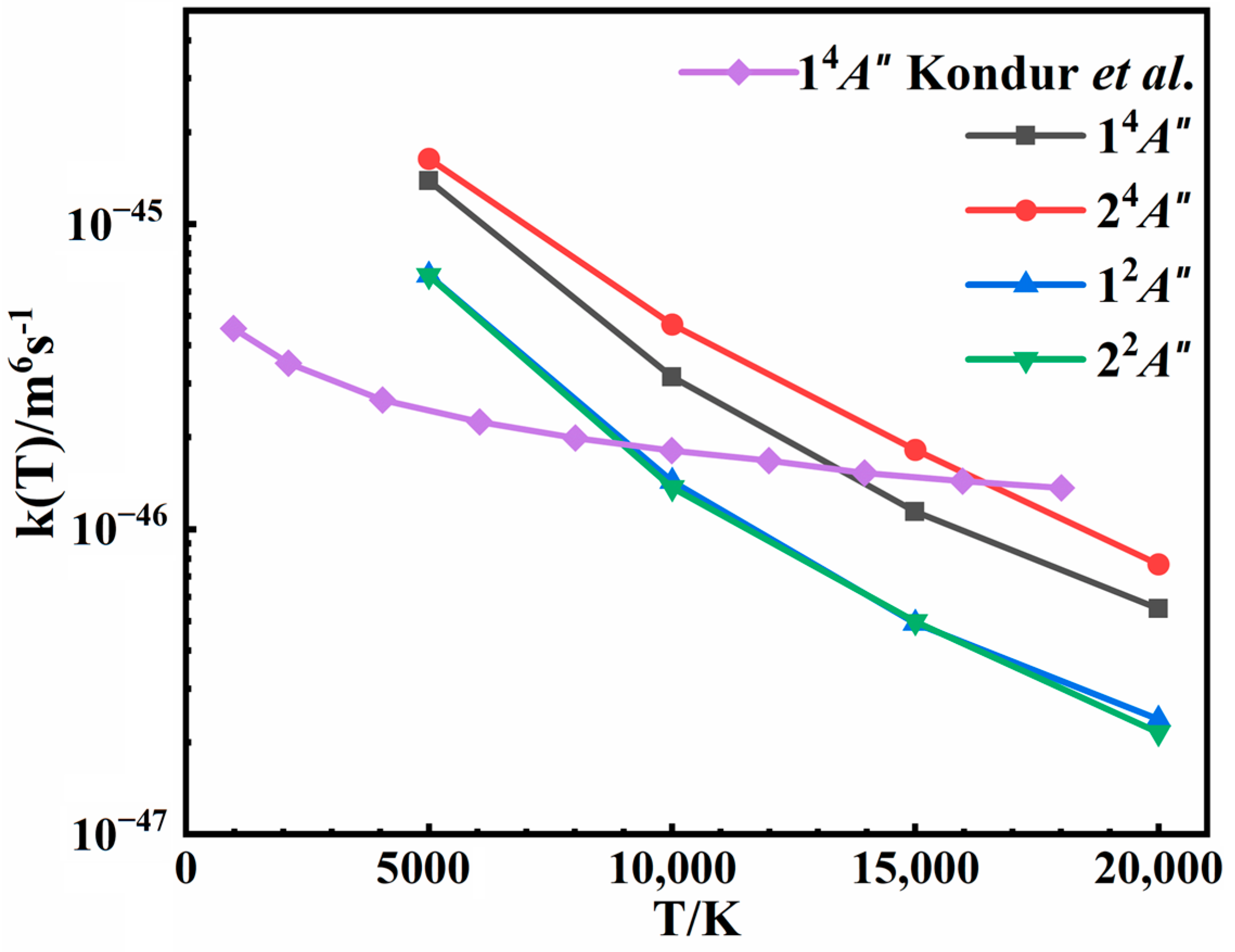
3.4. Rate Coefficients
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Magin, T.E.; Panesi, M.; Bourdon, A.; Jaffe, R.L.; Schwenke, D.W. Coarse-grain model for internal energy excitation and dissociation of molecular nitrogen. Chem. Phys. 2012, 398, 90–95. [Google Scholar] [CrossRef]
- Panesi, M.; Lani, A. Collisional radiative coarse-grain model for ionization in air. Phys. Fluids 2013, 25, 057101. [Google Scholar] [CrossRef]
- Munafò, A.; Panesi, M.; Magin, T.E. Boltzmann rovibrational collisional coarse-grained model for internal energy excitation and dissociation in hypersonic flows. Phys. Rev. E 2014, 89, 023001. [Google Scholar] [CrossRef] [PubMed]
- Sahai, A.; Lopez, B.; Johnston, C.O.; Panesi, M. Adaptive coarse graining method for energy transfer and dissociation kinetics of polyatomic species. J. Chem. Phys. 2017, 147, 054107. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Jaffe, R.L.; Schwenke, D.W.; Panesi, M. Construction of a coarse-grain quasi-classical trajectory method. I. Theory and application to N2–N2 system. J. Chem. Phys. 2018, 148, 054309. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Grover, M.S.; Schwartzentruber, T.E.; Panesi, M. Construction of a coarse-grain quasi-classical trajectory method. II. Comparison against the direct molecular simulation method. J. Chem. Phys. 2018, 148, 054310. [Google Scholar] [CrossRef]
- Panesi, M.; Munafò, A.; Magin, T.E.; Jaffe, R.L. Nonequilibrium shock-heated nitrogen flows using a rovibrational state-to-state method. Phys. Rev. E 2014, 90, 013009. [Google Scholar] [CrossRef]
- Pan, T.-J.; Wilson, T.J.; Stephani, K.A. Vibrational state-specific model for dissociation and recombination of the O2(3Σg−)+O(3P) system in DSMC. J. Chem. Phys. 2019, 150, 074305. [Google Scholar] [CrossRef]
- Valentini, P.; Schwartzentruber, T.E.; Bender, J.D.; Nompelis, I.; Candler, G.V. Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface. Phys. Fluids 2015, 27, 086102. [Google Scholar] [CrossRef]
- Schwartzentruber, T.E.; Grover, M.S.; Valentini, P. Direct molecular simulation of nonequilibrium dilute gases. J. Thermophys. Heat Transf. 2018, 32, 892–903. [Google Scholar] [CrossRef]
- Grover, M.S.; Torres, E.; Schwartzentruber, T.E. Direct molecular simulation of internal energy relaxation and dissociation in oxygen. Phys. Fluids 2019, 31, 076107. [Google Scholar] [CrossRef]
- Grover, M.S.; Schwartzentruber, T.E.; Varga, Z.; Truhlar, D.G. Vibrational energy transfer and collision-Induced dissociation in O+O2 collisions. J. Thermophys. Heat Transf. 2019, 33, 797–807. [Google Scholar] [CrossRef]
- Esposito, F.; Armenise, I.; Capitelli, M. N–N2 state to state vibrational-relaxation and dissociation rates based on quasiclassical calculations. Chem. Phys. 2006, 331, 1–8. [Google Scholar] [CrossRef]
- Panesi, M.; Jaffe, R.L.; Schwenke, D.W.; Magin, T.E. Rovibrational internal energy transfer and dissociation of N2(1Σg+)-N(4Su) system in hypersonic flows. J. Chem. Phys. 2013, 138, 044312. [Google Scholar] [CrossRef] [PubMed]
- Korutla, S.; Koner, D.; Varandas, A.J.C.; Tammineni, R.R. Quantum and classical dynamics of the N(2D) + N2 reaction on its ground doublet state N3(12A″) potential energy surface. J. Phys. Chem. A 2021, 125, 5650–5660. [Google Scholar] [CrossRef]
- Galvão, B.R.L.; Varandas, A.J.C.; Braga, J.P.; Belchior, J.C. Electronic quenching of N(2D) by N2: Theoretical predictions, comparison with experimental rate constants, and impact on atmospheric modeling. J. Phys. Chem. Lett. 2013, 4, 2292–2297. [Google Scholar] [CrossRef]
- Bender, J.D.; Valentini, P.; Nompelis, I.; Paukku, Y.; Varga, Z.; Truhlar, D.G.; Schwartzentruber, T.; Candler, G.V. An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of N2 + N2 dissociation reactions. J. Chem. Phys. 2015, 143, 054304. [Google Scholar] [CrossRef]
- Kondur, C.; Stephani, K.A. Effects of the third body (O and N) on the recombination of molecular nitrogen using quasi-classical trajectory methods. Chem. Phys. 2024, 576, 112092. [Google Scholar] [CrossRef]
- Varga, Z.; Truhlar, D.G. Potential energy surface for high-energy N+N2 collisions. Phys. Chem. Chem. Phys. 2021, 23, 26273–26284. [Google Scholar] [CrossRef]
- Geistfeld, E.C.; Torres, E.; Schwartzentruber, T. Quasi-classical trajectory analysis of three-body collision induced recombination in neutral nitrogen and oxygen. J. Chem. Phys. 2023, 159, 154111. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, S.; Zan, X.; Hu, H.; Xie, D.; Hu, X. Formation mechanisms of electronically excited nitrogen molecules from N+N2 and N+N+N collisions revealed by full-dimensional potential energy surfaces. J. Phys. Chem. A 2024, 128, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kondur, C.; Stephani, K.A. Molecular recombination dynamics of nitrogen from quasi-classical trajectory simulations of the N3 system. In Proceedings of the AIAA Scitech 2022 Forum, San Diego, CA, USA, 3–7 January 2022; p. 1906. [Google Scholar]
- Geistfeld, E.C.; Schwartzentruber, T.E. Three-body collision based recombination rate constants from quasi classical trajectory calculationsIn Proceedings of the AIAA Scitech 2021 Forum, Virtual, 11–15 and 19–21 January 2021; p. 0808. [Google Scholar]
- Kondur, C.; Stephani, K.A. Molecular recombination pathways of oxygen from quasiclassical trajectory calculations of the O3 system. In Proceedings of the AIAA Scitech 2020 Forum, Orlando, FL, USA, 6–10 January 2020; p. 1939. [Google Scholar]
- Azriel, V.M.; Kolesnikova, E.V.; Rusin, L.Y.; Sevryuk, M.B. Dynamical mechanisms of direct three-body recombination. J. Phys. Chem. A 2011, 115, 7055–7064. [Google Scholar] [CrossRef] [PubMed]
- Azriel, V.M.; Rusin, L.Y. The dynamics of direct three-body recombination of ions. Russ. J. Phys. Chem. A 2008, 2, 499–511. [Google Scholar] [CrossRef]
- Azriel, V.M.; Rusin, L.Y.; Sevryuk, M.B. Dynamics of two-stage direct three-body recombination of ions. Chem. Phys. 2013, 411, 26–34. [Google Scholar] [CrossRef]
- Kondur, C.; Stephani, K.A. Rate constants and molecular recombination pathways of oxygen from quasi-classical trajectory simulations of the O3 system. Chem. Phys. 2022, 552, 111357. [Google Scholar] [CrossRef]
- Gutzwiller, M.C.; José, J.V. Chaos in Classical and Quantum Mechanics; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Lahankar, S.A.; Zhang, J.; Minton, T.K.; Guo, H.; Lendvay, G. Dynamics of the O-atom exchange reaction 16O(3P)+18O18O(3Σg–)→16O18O(3Σg–)+18O(3P) at hyperthermal energies. J. Phys. Chem. A 2016, 120, 5348–5359. [Google Scholar] [CrossRef]
- Lendvay, G. Mechanism change in the dynamics of the O′+O2→O′O+O atom exchange reaction at high collision energies. J. Phys. Chem. A 2019, 123, 10230–10239. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T/K. | 5000 | 10,000 | 15,000 | 20,000 | |
|---|---|---|---|---|---|
| States | |||||
| 14A″ | 1.3876 × 10−45 | 3.1876 × 10−46 | 1.1423 × 10−46 | 5.4983 × 10−47 | |
| 24A″ | 1.6365 × 10−45 | 4.6891 × 10−46 | 1.8215 × 10−46 | 7.6794 × 10−47 | |
| 12A″ | 6.8119 × 10−46 | 1.4345 × 10−46 | 4.9191 × 10−47 | 2.3819 × 10−47 | |
| 22A″ | 6.7822 × 10−46 | 1.3682 × 10−46 | 4.9828 × 10−47 | 2.1555 × 10−47 | |
| Total | 4.3835 × 10−45 | 1.0650 × 10−45 | 3.9540 × 10−46 | 1.7716 × 10−46 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Wei, Z.; Hu, H.; Hu, X.; Xie, D. Theoretical Investigation of Rate Coefficients and Dynamical Mechanisms for N + N + N Three-Body Recombination Based on Full-Dimensional Potential Energy Surfaces. Molecules 2024, 29, 4933. https://doi.org/10.3390/molecules29204933
Xu C, Wei Z, Hu H, Hu X, Xie D. Theoretical Investigation of Rate Coefficients and Dynamical Mechanisms for N + N + N Three-Body Recombination Based on Full-Dimensional Potential Energy Surfaces. Molecules. 2024; 29(20):4933. https://doi.org/10.3390/molecules29204933
Chicago/Turabian StyleXu, Chong, Zhenxuan Wei, Huayu Hu, Xixi Hu, and Daiqian Xie. 2024. "Theoretical Investigation of Rate Coefficients and Dynamical Mechanisms for N + N + N Three-Body Recombination Based on Full-Dimensional Potential Energy Surfaces" Molecules 29, no. 20: 4933. https://doi.org/10.3390/molecules29204933
APA StyleXu, C., Wei, Z., Hu, H., Hu, X., & Xie, D. (2024). Theoretical Investigation of Rate Coefficients and Dynamical Mechanisms for N + N + N Three-Body Recombination Based on Full-Dimensional Potential Energy Surfaces. Molecules, 29(20), 4933. https://doi.org/10.3390/molecules29204933