
Electron Density and Molecular Orbital Analyses of the Nature of Bonding in the η3-CCH Agostic Rhodium Complexes Preceding the C–C and C–H Bond Cleavages


Abstract

1. Introduction
2. Results
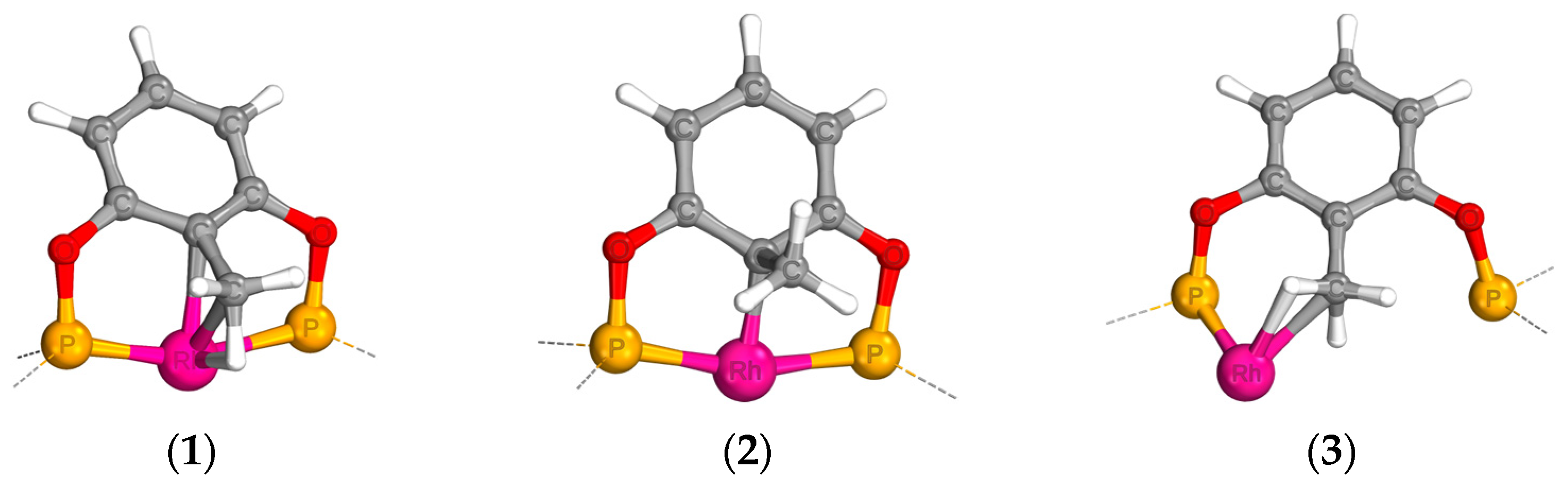
2.1. Geometries of the η3-Agostic Complexes
2.2. Electron Density Analysis
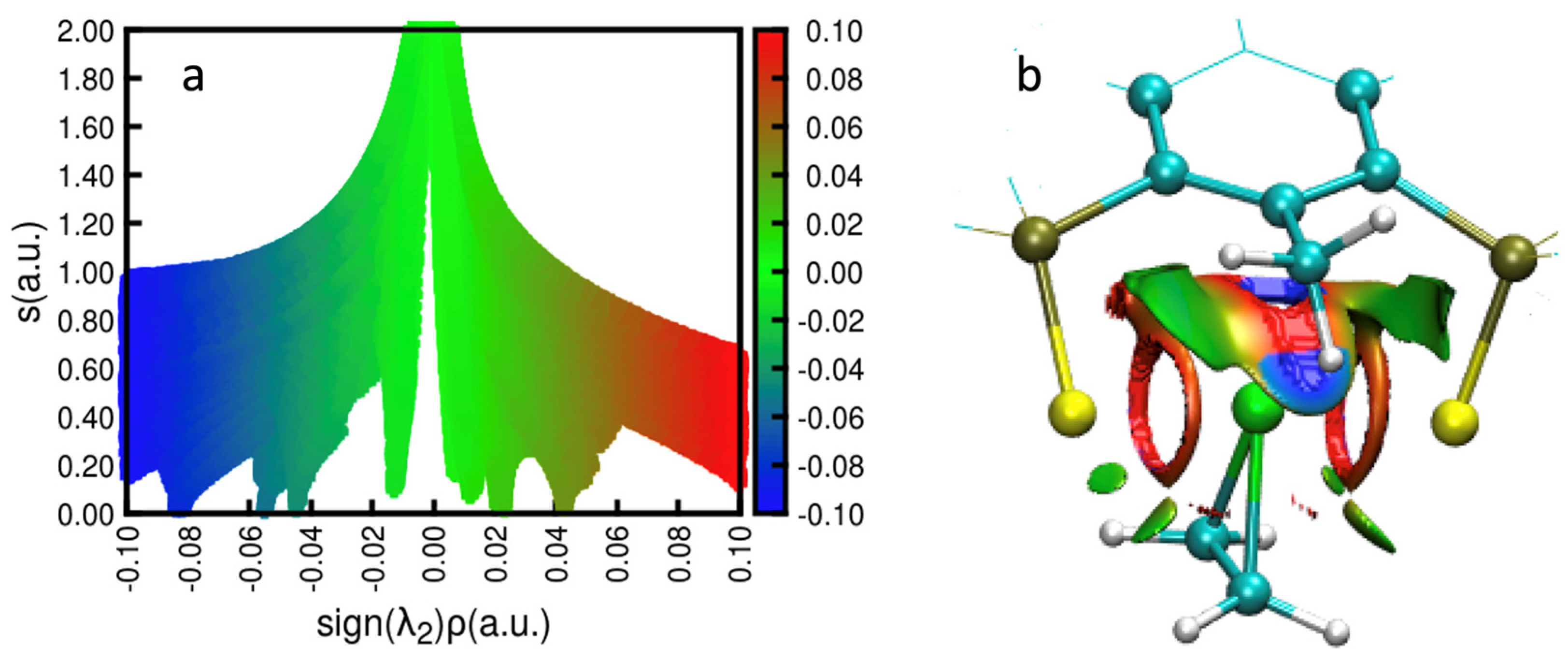
2.2.1. NCIPLOT
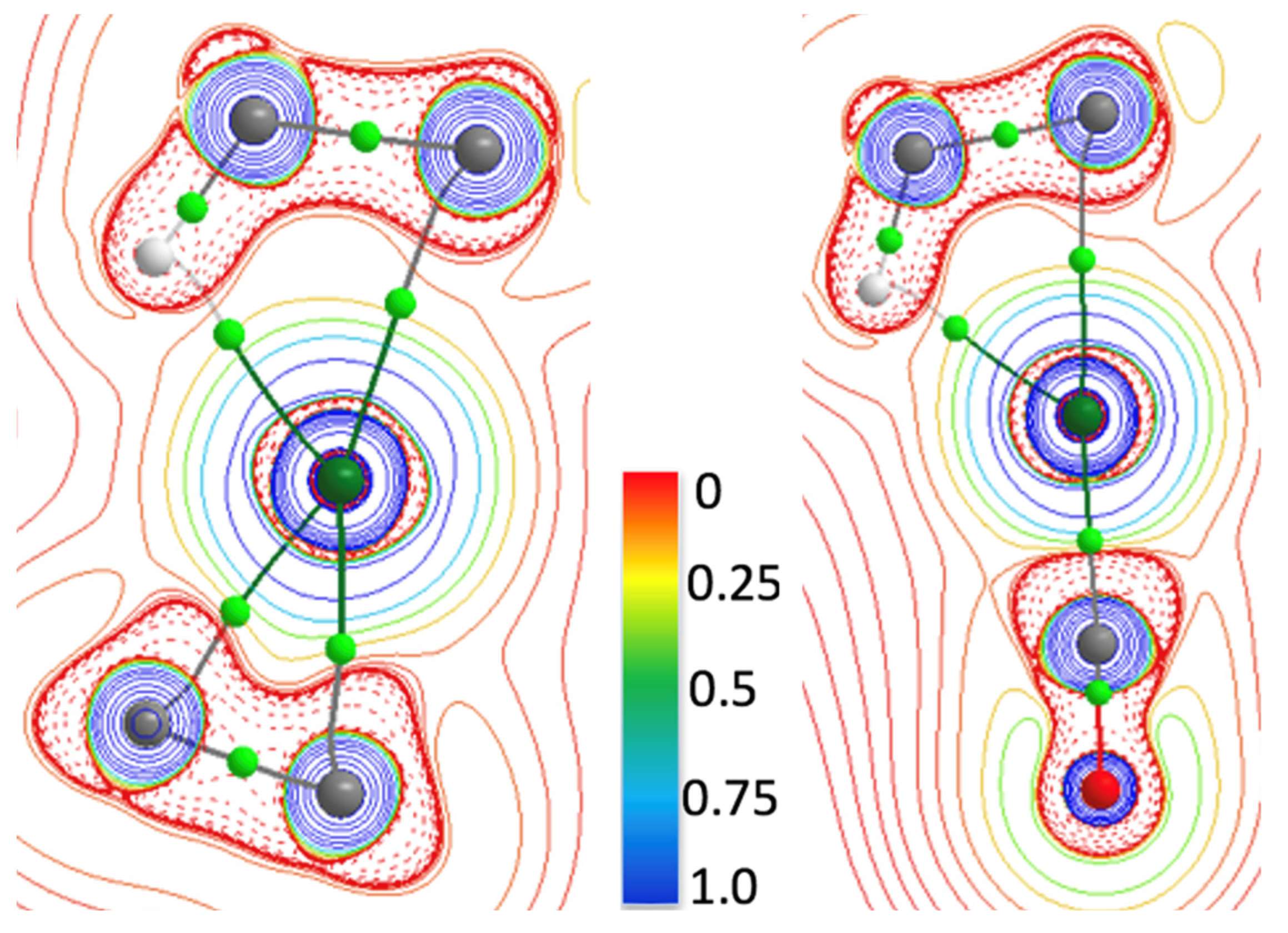
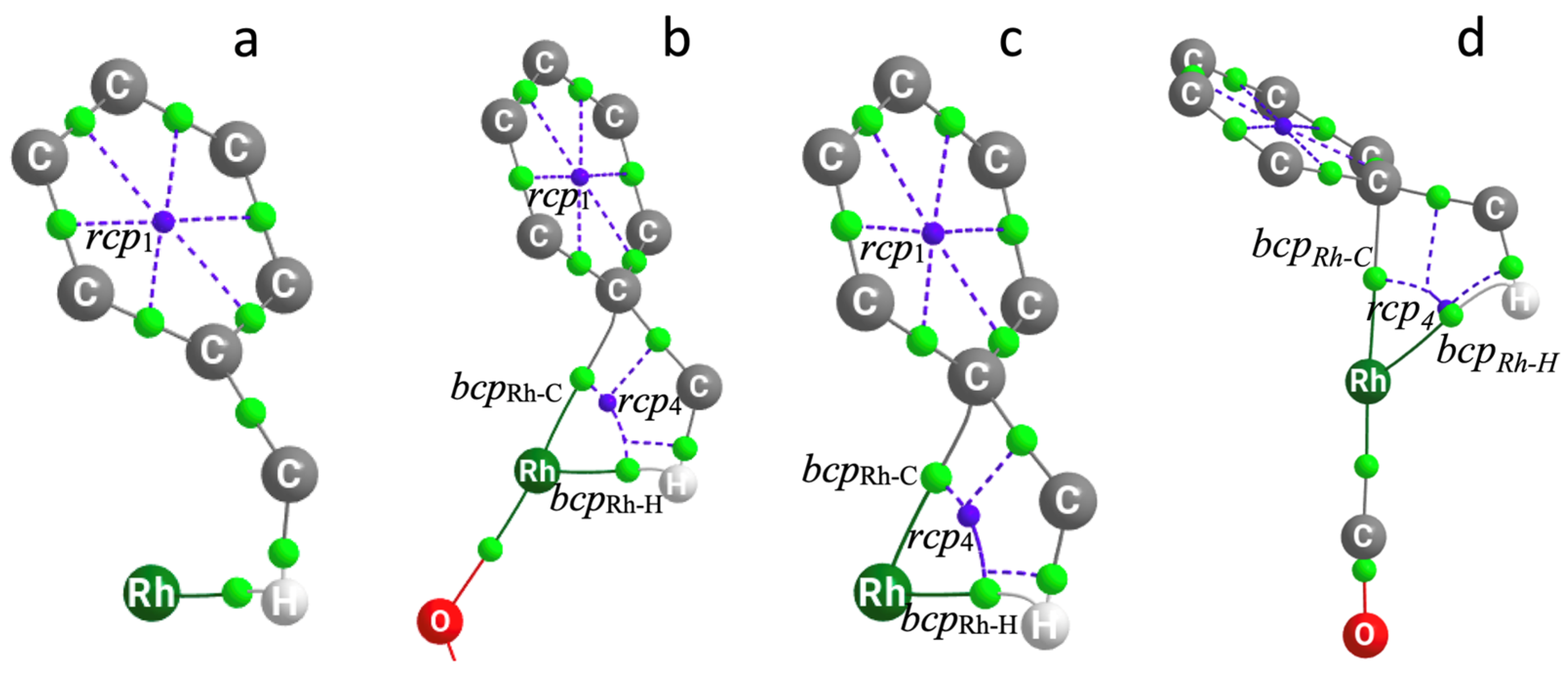
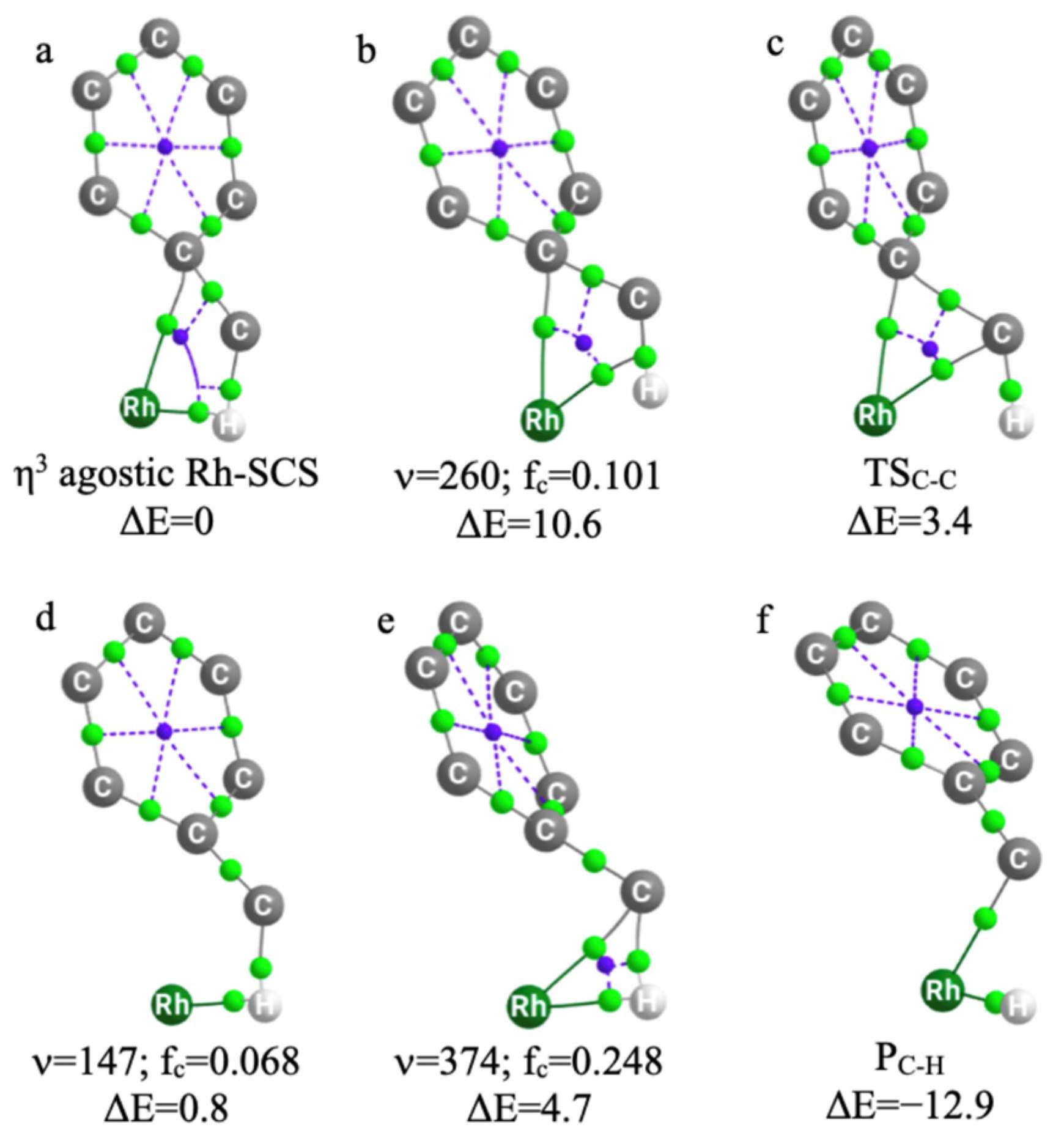
2.2.2. QTAIM
2.2.3. Electron Sharing Indices in 3D (ESI-3D)
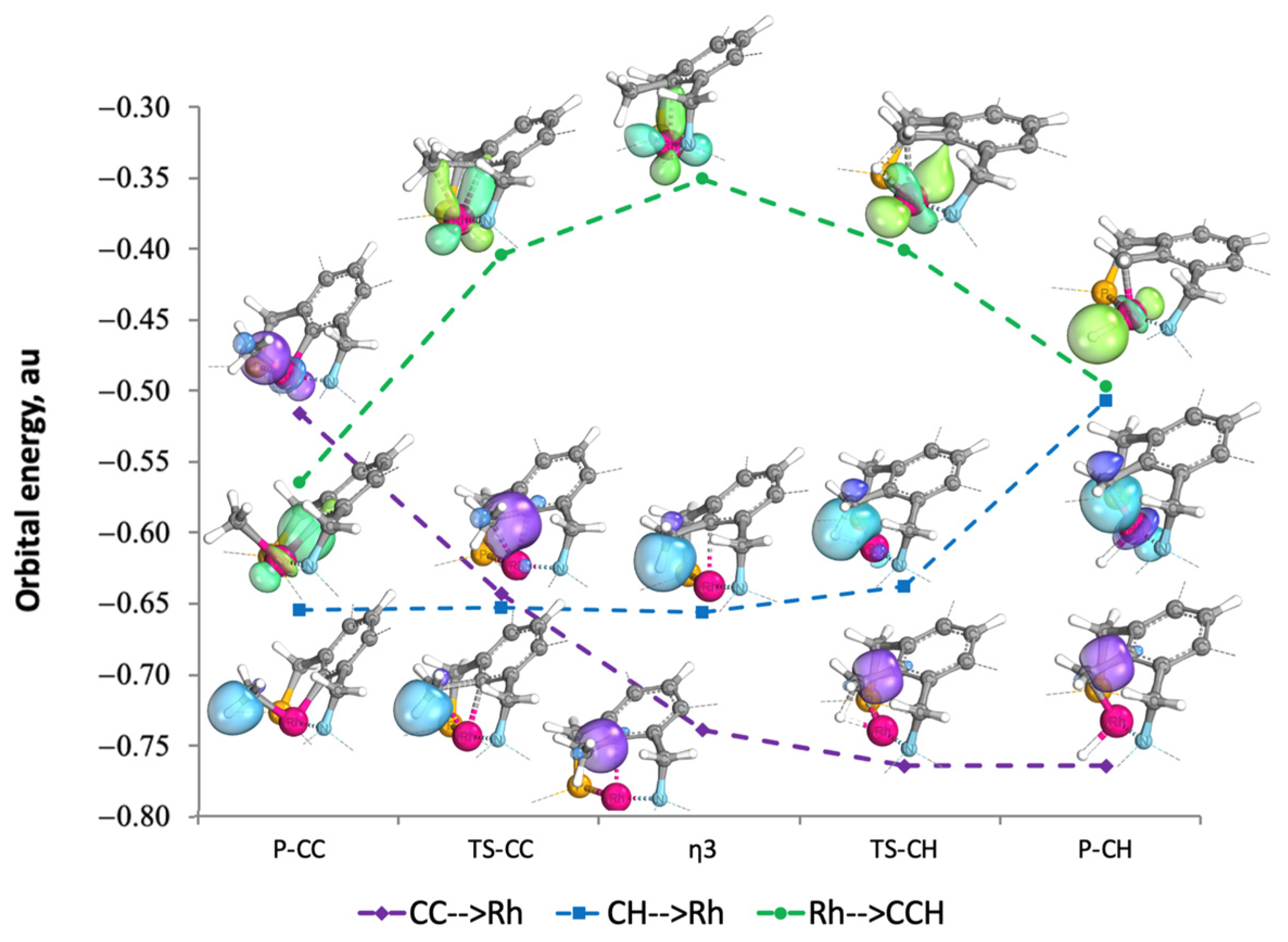
2.3. Molecular Orbital (MO) Analysis
2.3.1. NBO
2.3.2. IBO
3. Discussion
4. Computational Methodology
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murakami, M.; Ishida, N. Fundamental Reactions to Cleave Carbon–Carbon σ-Bonds with Transition Metal Complexes. In Cleavage of Carbon-Carbon Single Bonds by Transition Metals; Murakami, M., Murakami, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Brookhart, M.; Green, M.L.H. Carbon-hydrogen-transition metal bonds. J. Organomet. Chem. 1983, 250, 395–408. [Google Scholar] [CrossRef]
- Clot, E.; Eisenstein, O. Agostic Interactions from a Computational Perspective: One Name, Many Interpretations. In Principles and Applications of Density Functional Theory in Inorganic Chemistry II. Structure and Bonding; Mingos, D.M.P., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; Volume 113. [Google Scholar] [CrossRef]
- Scherer, W.; McGrady, G.S. Agostic Interactions in d0 Metal Alkyl Complexes. Angew. Chem. Int. Ed. 2004, 43, 1782–1806. [Google Scholar] [CrossRef] [PubMed]
- Brookhart, M.; Green, M.L.H.; Parkin, G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. USA 2007, 104, 6908–6914. [Google Scholar] [CrossRef] [PubMed]
- Komiya, N.; Hosokawa, T.; Adachi, J.; Inoue, R.; Kawamorita, S.; Naota, T. Regiospecific Remote Pt–H Interactions in Oligomethylene-Vaulted (N^C^N)-Pincer PtII Complexes. Eur. J. Inorg. Chem. 2018, 2018, 4771–4778. [Google Scholar] [CrossRef]
- Fang, R.-Q.; Yuan, H.-K.; Wang, X.-L.; Zhu, H.-L. Anagostic C-H⋅⋅⋅Ni interaction and DFT calculation of bis(μ-N,O-((E)-2-((cyclohexylmethylimino)methyl)phenol)–nickel(II) complex. Inorg. Nano-Met. Chem. 2017, 47, 756–760. [Google Scholar] [CrossRef]
- Lersch, M.; Tilset, M. Mechanistic Aspects of C−H Activation by Pt Complexes. Chem. Rev. 2005, 105, 2471–2526. [Google Scholar] [CrossRef]
- Sakaki, S. Theoretical Studies of CH σ-Bond Activation and Related Reactions by Transition-Metal Complexes. In Theoretical Aspects of Transition Metal Catalysis Topics in Organometallic Chemistry; Frenking, G., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 12, pp. 31–78. [Google Scholar] [CrossRef]
- Crabtree, R.H.; Holt, E.M.; Lavin, M.; Morehouse, S.M. Inter- vs. Intramolecular C-H Activation: A C-H-Ir Bridge in [IrH2(Mq)L2]BF4 and a C-H + M → C-M-H Reaction Trajectory. Inorg. Chem. 1985, 24, 1986–1992. [Google Scholar] [CrossRef]
- Findlater, M.; Cartwright-Sykes, A.; White, P.S.; Schauer, C.K.; Brookhart, M. Role of Coordination Geometry in Dictating the Barrier to Hydride Migration in d6 Square-Pyramidal Iridium and Rhodium Pincer Complexes. J. Am. Chem. Soc. 2011, 133, 12274–12284. [Google Scholar] [CrossRef]
- Walter, M.D.; White, P.S.; Schauer, C.K.; Brookhart, M. Stability and Dynamic Processes in 16VE Iridium(III) Ethyl Hydride and Rhodium(I) σ-Ethane Complexes: Experimental and Computational Studies. J. Am. Chem. Soc. 2013, 135, 15933–15947. [Google Scholar] [CrossRef]
- Lein, M. Characterization of agostic interactions in theory and computation. Coord. Chem. Rev. 2009, 253, 625–634. [Google Scholar] [CrossRef]
- Suresh, C.H. Nature of α,β-CCC agostic bonding in metallacyclobutanes. J. Organomet. Chem. 2006, 691, 5366–5374. [Google Scholar] [CrossRef]
- Vigalok, A.; Milstein, D. Advances in metal chemistry of quinonoid compounds: New types of interactions between metals and aromatics. Acc. Chem. Res. 2001, 34, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Jaffart, J.; Etienne, M.; Reinhold, M.; McGrady, J.E.; Maseras, F. An unprecedented α-C–C agostic interaction in a cyclopropyl tris(pyrazolyl)boratoniobium complex. Chem. Commun. 2003, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Jaffart, J.; Cole, M.L.; Etienne, M.; Reinhold, M.; McGrady, J.E.; Maseras, F. C–H and C–C agostic interactions in cycloalkyl tris(pyrazolyl)boratoniobium complexes. Dalton Trans. 2003, 21, 4057–4064. [Google Scholar] [CrossRef]
- Gandelman, M.; Shimon, L.J.W.; Milstein, D. C-C versus C-H Activation and versus Agostic C-C Interaction Controlled by Electron Density at the Metal Center. Chem. Eur. J. 2003, 9, 4295–4300. [Google Scholar] [CrossRef]
- Harvey, B.G.; Ernst, R.D. Transition-Metal Complexes with (C–C)→M Agostic Interactions. Eur. J. Inorg. Chem. 2017, 2017, 1205–1226. [Google Scholar] [CrossRef]
- Etienne, M.; Weller, A.S. Intramolecular C–C agostic complexes: C–C sigma interactions by another name. Chem. Soc. Rev. 2014, 43, 242–259. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Czabaniuk, L.C. Structure and Reactivity of Late Transition Metal η3-Benzyl Complexes. Angew. Chem. Int. Ed. 2014, 53, 2826–2851. [Google Scholar] [CrossRef]
- Mulliken, R.S. Molecular scientists and molecular science: Some reminiscences. J. Chem. Phys. 1965, 43, S2–S11. [Google Scholar] [CrossRef]
- Ruedenberg, K. Atoms and Bonds in Molecules as Synergisms of Interactions between Electrons and Nuclei. J. Chem. Phys. 2022, 157, 210901. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence indices: A personal account. J. Comput. Chem. 2007, 28, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Frenking, G.; Krapp, A. Unicorns in the world of chemical bonding models. J. Comput. Chem. 2007, 28, 15–24. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Dapprich, S.; Frenking, G. Investigation of Donor—Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Popelier, P.; Logothetis, G. Characterization of an agostic bond on the basis of the electron density. J. Organomet. Chem. 1998, 555, 101–111. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Silvi, B.; Fourré, I.; Alikhani, M. The Topological Analysis of the Electron Localization Function. A Key for a Position Space Representation of Chemical Bonds. Monatshefte Für Chem./Chem. Mon. 2005, 136, 855–879. [Google Scholar] [CrossRef]
- Zins, E.-L.; Silvi, B.; Alikhani, M.E. Activation of C–H and B–H bonds through agostic bonding: An ELF/QTAIM insight. Phys. Chem. Chem. Phys. 2015, 17, 9258–9281. [Google Scholar] [CrossRef]
- Pendás, Á.M.; Contreras-García, J. Topological Approaches to the Chemical Bond; Spinger: Geneva, Switzerland, 2023. [Google Scholar] [CrossRef]
- Frenking, G.; Nikolaus Fröhlich, N. The Nature of the Bonding in Transition-Metal Compounds. Chem. Rev. 2000, 100, 717–774. [Google Scholar] [CrossRef]
- Kubas, G.J. Metal Dihydrogen and σ-Bond Complexes: Structure, Theory and Reactivity; Kluwer Academic Publishers: New York, NY, USA, 2001. [Google Scholar]
- García-Rodeja, Y.; Feixas, F.; Matito, E.; Solà, M. Three-Centre Electron Sharing Indices (3c-ESIs) as a Tool to Differentiate among (an)Agostic Interactions and Hydrogen Bonds in Transition Metal Complexes. Phys. Chem. Chem. Phys. 2022, 24, 29333–29337. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Jemmis, E.D. Origin of β-agostic interaction in d0 transition metal alkyl complexes: Influence of ligands. J. Organomet. Chem. 2018, 865, 37–44. [Google Scholar] [CrossRef]
- Xu, B.; Wang, Q.; Wang, X. Understanding the agostic bonding for group 4 metal methylidene complexes: A DFT approach. Comput. Theor. Chem. 2011, 976, 36–41. [Google Scholar] [CrossRef]
- Scherer, W.; Herz, V.; Brück, A.; Hauf, C.; Reiner, F.; Altmannshofer, S.; Leusser, D.; Stalke, D. The Nature of β-Agostic Bonding in Late-Transition-Metal Alkyl Complexes. Angew. Chem. Int. Ed. 2011, 50, 2845–2849. [Google Scholar] [CrossRef] [PubMed]
- Scherer, W.; Herz, V.; Hauf, C. On the Nature of β-Agostic Interactions: A Comparison Between the Molecular Orbital and Charge Density Picture. In Electron Density and Chemical Bonding, I. Structure and Bonding; Stalke, D., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 146. [Google Scholar] [CrossRef]
- Mebs, S.; Kalläne, S.I.; Braun, T. Hapticity of asymmetric rhodium-allyl compounds in the light of real-space bonding indicators. Z. Für Krist.—Cryst. Mater. 2018, 233, 615–626. [Google Scholar] [CrossRef]
- Montag, M.; Efremenko, I.; Cohen, R.; Shimon, L.J.W.; Leitus, G.; Diskin-Posner, Y.; Ben-David, Y.; Salem, H.; Martin, J.-M.L.; Milstein, D. Effect of CO on the Oxidative Addition of Arene C-H Bonds by Cationic Rhodium Complexes. Chem. Eur. J. 2010, 16, 328–353. [Google Scholar] [CrossRef]
- Hamdaoui, M.; Djukic, J.-P. Noncovalent Interactions in Key Metal-centred Catalytic Intermediates: Structure–Electronic Relationship. In Noncovalent Interactions in Catalysis; Mahmudov, Kamran, T., Eds.; RSC Catalysis Series; The Royal Society of Chemistry: London, UK, 2019; pp. 579–607. [Google Scholar]
- Escudero, J.; Besset, T. Non-Covalent Interactions in Transition Metal-Catalyzed Para-Selective CH Functionalization of Arenes. In Advances in Organometallic Chemistry; Pérez, P.J., Ed.; Academic Press: Cambridge, MA, USA, 2023; Volume 80, pp. 77–91. [Google Scholar] [CrossRef]
- Lu, Q.; Neese, F.; Bistoni, G. Formation of Agostic Structures Driven by London Dispersion. Angew. Chem. Int. Ed. 2018, 57, 4760–4764. [Google Scholar] [CrossRef]
- Lu, Q.; Neese, F.; Bistoni, G. London Dispersion Effects in the Coordination and Activation of Alkanes in σ-Complexes: A Local Energy Decomposition Study. Phys. Chem. Chem. Phys. 2019, 21, 11569–11577. [Google Scholar] [CrossRef]
- Lin, X.; Wu, W.; Mo, Y. Agostic Interactions in Early Transition-Metal Complexes: Roles of Hyperconjugation, Dispersion, and Steric Effect. Chem. Eur. J. 2019, 25, 6591–6599. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, M.-H.; Huang, K.-W. Rhodium Pincer Complexes: Coordination, Reactivity and Catalysis. In Comprehensive Coordination Chemistry III; Constable, E.C., Parkin, G., Que, L., Jr., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 43–107. [Google Scholar] [CrossRef]
- Montag, M.; Efremenko, I.; Cohen, R.; Leitus, G.; Shimon, L.J.W.; Diskin-Posner, Y.; Ben-David, Y.; Martin, J.M.L.; Milstein, D. The impact of weak C-H⋯Rh interactions on the structure and reactivity of trans-[Rh(CO)2(phosphine)2]+: An experimental and theoretical examination. Chem. Eur. J. 2008, 14, 8183–8194. [Google Scholar] [CrossRef] [PubMed]
- Sajjad, M.A.; Harrison, J.A.; Nielson, A.J.; Schwerdtfeger, P. NBO Orbital Interaction Analysis for the Ambiphilic Metal–Ligand Activation/Concerted Metalation Deprotonation (AMLA/CMD) Mechanism Involved in the Cyclopalladation Reaction of N,N-Dimethylbenzylamine with Palladium Acetate. Organometallics 2018, 37, 3659–3669. [Google Scholar] [CrossRef]
- Lepetit, C.; Poater, J.; Alikhani, M.E.; Silvi, B.; Canac, Y.; Contreras-García, J.; Solà, M.; Chauvin, R. The Missing Entry in the Agostic–Anagostic Series: Rh(I)–η1-C Interactions in P(CH)P Pincer Complexes. Inorg. Chem. 2015, 54, 2960–2969. [Google Scholar] [CrossRef]
- Harrison, J.A.; Nielson, A.J.; Sajjad, M.A.; Saunders, G.C.; Schwerdtfeger, P. Steric and Electronic Manipulation of the Anagostic Interaction in 1-Tetralone Oxime and Imine Complexes of Rhodium(I). Eur. J. Inorg. Chem. 2016, 2016, 64–77. [Google Scholar] [CrossRef]
- Nielson, A.J.; Harrison, J.A.; Sajjad, M.A.; Schwerdtfeger, P. Electronic and Steric Manipulation of the Preagostic Interaction in Isoquinoline Complexes of Rh(I). Eur. J. Inorg. Chem. 2017, 2017, 2255–2264. [Google Scholar] [CrossRef]
- Arif Sajjad, M.A.; Harrison, J.A.; Nielson, A.J.; Schwerdtfeger, P. Interplay of Steric and Electronic Effects on the Bonding Components in Aromatic Ring Agostic Interactions. Organometallics 2017, 36, 4231–4237. [Google Scholar] [CrossRef]
- Poverenov, E.; Milstein, D. Noninnocent Behavior of PCP and PCN Pincer Ligands of Late Metal Complexes. In Organometallic Pincer Chemistry; van Koten, G., Milstein, D., Eds.; Springer Verlag: Berlin/Heidelberg, Germany, 2013; Volume 40, pp. 21–47. [Google Scholar] [CrossRef]
- Montag, M.; Efremenko, I.; Diskin-Posner, Y.; Ben-David, Y.; Martin, J.M.L.; Milstein, D. Exclusive C–C Oxidative Addition in a Rhodium Thiophosphoryl Pincer Complex and Computational Evidence for an η3-C–C–H Agostic Intermediate. Organometallics 2012, 31, 505–512. [Google Scholar] [CrossRef]
- Efremenko, I.; Montag, M. Revisiting C–C and C–H Bond Activation in Rhodium Pincer Complexes: Thermodynamics and Kinetics Involving a Common Agostic Intermediate. Organometallics 2022, 41, 2379–2393. [Google Scholar] [CrossRef]
- Cortés-Guzmán, F.; Bader, R.F.W. Complementarity of QTAIM and MO theory in the study of bonding in donor–acceptor complexes. Coord. Chem. Rev. 2005, 249, 633–662. [Google Scholar] [CrossRef]
- Finger, M.; Reinhold, J. Energy Density Distribution in Bridged Cobalt Complexes. Inorg. Chem. 2003, 42, 8128–8130. [Google Scholar] [CrossRef]
- Kluge, O.; Finger, M.; Reinhold, J. Orbital Contributions to the Molecular Charge and Energy Density Distributions in Co2(CO)8. Inorg. Chem. 2005, 44, 6494–6496. [Google Scholar] [CrossRef]
- Reinhold, J.; Kluge, O.; Mealli, C. Integration of Electron Density and Molecular Orbital Techniques to Reveal Questionable Bonds: The Test Case of the Direct Fe−Fe Bond in Fe2(CO)9. Inorg. Chem. 2007, 46, 7142–7147. [Google Scholar] [CrossRef]
- Jiao, Y.; Evans, M.E.; Morris, J.; Brennessel, W.W.; Jones, W.D. Rhodium–Carbon Bond Energies in Tp′Rh(CNneopentyl)(CH2X)H: Quantifying Stabilization Effects in M–C Bonds. J. Am. Chem. Soc. 2013, 135, 6994–7004. [Google Scholar] [CrossRef]
- Brayshaw, S.K.; Sceats, E.L.; Green, J.C.; Weller, A.S. C–C σ complexes of rhodium. Proc. Nat. Acad. Sci. USA 2007, 104, 6921–6926. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W.J. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Laplaza, R.; Peccati, F.; Boto, R.A.; Quan, C.; Carbone, A.; Piquemal, P.; Maday, Y.; Contreras-García, J. NCIPLOT and the analysis of noncovalent interactions using the reduced density gradient. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1497. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. Hydrogen–Hydrogen Bonding in Planar Biphenyl, Predicted by Atoms-In-Molecules Theory, Does Not Exist. Chem. Eur. J. 2006, 12, 2889–2895. [Google Scholar] [CrossRef]
- Grimme, S.; Mück-Lichtenfeld, C.; Erker, G.; Kehr, G.; Wang, H.; Beckers, H.; Willner, H. When Do Interacting Atoms Form a Chemical Bond? Spectroscopic Measurements and Theoretical Analyses of Dideuteriophenanthrene. Angew. Chem. Int. Ed. 2009, 48, 2592–2595. [Google Scholar] [CrossRef] [PubMed]
- Hazrah, A.S.; Nanayakkara, S.; Seifert, N.A.; Kraka, E.; Jäger, W. Structural study of 1- and 2-naphthol: New insights into the non-covalent H–H interaction in cis-1-naphthol. Phys. Chem. Chem. Phys. 2022, 24, 3722–3732. [Google Scholar] [CrossRef]
- Jabłoński, M. Bader’s Topological Bond Path Does Not Necessarily Indicate Stabilizing Interaction—Proof Studies Based on the Ng@[3n]cyclophane Endohedral Complexes. Molecules 2023, 28, 6353. [Google Scholar] [CrossRef]
- Farrugia, L.J.; Evans, C.; Tegel, M. Chemical Bonds without “Chemical Bonding”? A Combined Experimental and Theoretical Charge Density Study on an Iron Trimethylenemethane Complex. J. Phys. Chem. A 2006, 110, 7952–7961. [Google Scholar] [CrossRef]
- Mousavi, M.; Frenking, G. Bonding Analysis of Trimethylenemethane (TMM) Complexes [(CO)3M–TMM] (M. = Fe, Ru, Os, Rh+). Absence of Expected Bond Paths. J. Organomet. Chem. 2013, 748, 2–7. [Google Scholar] [CrossRef]
- Lane, J.R.; Contreras-García, J.; Piquemal, J.P.; Miller, B.J.; Kjaergaard, H.G. Are Bond Critical Points Really Critical for Hydrogen Bonding? J. Chem. Theory Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, S. Why Bond Critical Points Are Not “Bond” Critical Points. Chem. Eur. J. 2018, 24, 5401–5405. [Google Scholar] [CrossRef] [PubMed]
- Foroutan-Nejad, C.; Shahbazian, S.; Marek, R. Toward a Consistent Interpretation of the QTAIM: Tortuous Link between Chemical Bonds, Interactions, and Bond/Line Paths. Chem. Eur. J. 2014, 20, 10140–10152. [Google Scholar] [CrossRef]
- Jabłoński, M. QTAIM-Based Comparison of Agostic Bonds and Intramolecular Charge-Inverted Hydrogen Bonds. J. Phys. Chem. A 2015, 119, 4993–5008. [Google Scholar] [CrossRef]
- Jabłoński, M. Geometry- and QTAIM-Based Comparison of Intramolecular Charge-Inverted Hydrogen Bonds, M···(H–Si) “Agostic Bond”, and M···(η2-SiH) σ Interactions. J. Phys. Chem. A 2015, 119, 11384–11396. [Google Scholar] [CrossRef]
- Jabłoński, M. Comparative study of geometric and QTAIM-based differences between XH⋯Y intramolecular charge-inverted hydrogen bonds, M1⋯(HX) agostic bonds and M2⋯(η2-XH) σ interactions (X=Si, Ge; Y=Al, Ga; M1=Ti, Co; M2=Mn, Fe, Cr). Comp. Theor. Chem. 2016, 1096, 54–65. [Google Scholar] [CrossRef]
- Castillo, N.; Matta, C.; Boyd, R. The first example of a cage critical point in a single ring: A novel twisted α-helical ring topology. Chem. Phys. Lett. 2005, 409, 265–269. [Google Scholar] [CrossRef]
- Rzepa, H.S.; Allan, C.S.M. Racemization of Isobornyl Chloride via Carbocations: A Nonclassical Look at a Classic Mechanism. J. Chem. Educ. 2010, 87, 221–228. [Google Scholar] [CrossRef]
- Coulson, C.A. The electronic structure of some polyenes and aromatic molecules. VII. Bonds of fractional order by the molecular orbital method. Proc. R. Soc. Lond. Ser. A. Math. Phys. Sci. 1939, 169, 413–428. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. J. Chem. Phys. 1955, 23, 2338–2343. [Google Scholar] [CrossRef]
- Mayer, I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence: Relations to Mulliken’s population analysis. Int. J. Quantum Chem. 1984, 26, 151–154. [Google Scholar] [CrossRef]
- Mayer, I. On bond orders and valences in the Ab initio quantum chemical theory. Int. J. Quantum Chem. 1986, 29, 73–84. [Google Scholar] [CrossRef]
- Mayer, I. Bond orders and valences from ab initio wave functions. Int. J. Quantum Chem. 1986, 29, 477–483. [Google Scholar] [CrossRef]
- Wiberg, K. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Stephens, M.E. Spatial localization of the electronic pair and number distributions in molecules. J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chem. Acc. 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Matito, E. ESI-3D: Electron Sharing Indices Program for 3D Molecular Space Partitioning; Institute of Computational chemistry and Catalysis (IQCC), University of Girona: Catalonia, Spain, 2006; Courtesy of Eduard Matito; Available online: https://quantchemdev.github.io/resources.html (accessed on 26 May 2023).
- Matito, E.; Duran, M.; Solá, M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization. J. Chem. Phys. 2005, 122, 014109. [Google Scholar] [CrossRef] [PubMed]
- Matito, E.; Solá, M.; Salvador, P.; Duran, M. Electron Sharing Indexes at the Correlated Level. Application to Aromaticity Measures. Faraday Discuss. 2007, 135, 325–345. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. Comment on “Natural Bond Orbitals and the Nature of the Hydrogen Bond”. J. Phys. Chem. A 2018, 122, 724–732. [Google Scholar] [CrossRef]
- Pudasaini, B.; Janesko, B.G. Agostic Interactions in Nickel(II) Complexes: Trans Influence of Ancillary Ligands on the Strength of the Bond. Organometallics 2014, 33, 84–93. [Google Scholar] [CrossRef]
- Debnath, T.; Ash, T.; Banu, T.; Das, A.K. Investigation of agostic interaction through NBO analysis and its impact on β-hydride elimination and dehydrogenation: A DFT approach. Theor. Chem. Acc. 2016, 135, 1–14. [Google Scholar] [CrossRef]
- Thakur, T.S.; Desiraju, G.R. Theoretical investigation of C–H⋯M interactions in organometallic complexes: A natural bond orbital (NBO) study. J. Mol. Struct. THEOCHEM 2007, 810, 143–154. [Google Scholar] [CrossRef]
- Barthes, C.; Lepetit, C.; Canac, Y.; Duhayon, C.; Zargarian, D.; Chauvin, R. P(CH)P Pincer Rhodium(I) Complexes: The Key Role of Electron-Poor Imidazoliophosphine Extremities. Inorg. Chem. 2013, 52, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chem. Educ. Res. Pract. Eur. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Knizia, G. Intrinsic atomic orbitals: An unbiased bridge between quantum theory and chemical concepts. J. Chem. Theory Comput. 2013, 9, 4834–4843. [Google Scholar] [CrossRef]
- Knizia, G.; Klein, J.E.M.N. Electron flow in reaction mechanisms—Revealed from first principles. Angew. Chem. Int. Ed. 2015, 54, 5518–5522. [Google Scholar] [CrossRef]
- Klein, J.E.M.N.; Havenith, R.W.A.; Knizia, G. The Pentagonal-Pyramidal Hexamethylbenzene Dication: Many Shades of Coordination Chemistry at Carbon. Chem. Eur. J. 2018, 24, 12340–12345. [Google Scholar] [CrossRef]
- Macchi, P.; Proserpio, D.M.; Sironi, A. Experimental Electron Density Studies for Investigating the Metal π-Ligand Bond: The Case of Bis(1,5-cyclooctadiene)nickel. J. Am. Chem. Soc. 1998, 120, 1447–1455. [Google Scholar] [CrossRef]
- Ball, P. Beyond the bond. Nature 2011, 469, 26–28. [Google Scholar] [CrossRef]
- Benítez, F.J.; Gutiérrez-Oliva, S.; Herrera, B.; Toro-Labbé, A. Basis Electronic Activity of Molecular Systems. A Theory of Bond Reactivity. J. Phys. Chem. A 2024, 128, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 122, 154101. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions. J. Chem. Phys. 2006, 123, 024101. [Google Scholar] [CrossRef]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Pantazis, D.A.; McGrady, J.E.; Maseras, F.; Etienne, M. Critical Role of the Correlation Functional in DFT Descriptions of an Agostic Niobium Complex. J. Chem. Theory Comput. 2007, 3, 1329–1336. [Google Scholar] [CrossRef]
- Tognetti, V.; Joubert, L. On the Influence of Density Functional Approximations on Some Local Bader’s Atoms-in-Molecules Properties. J. Phys. Chem. A 2011, 115, 5505–5515. [Google Scholar] [CrossRef]
- Pantazis, D.A.; McGrady, J.E.; Besora, M.; Maseras, F.; Etienne, M. On the Origin of α- and β-Agostic Distortions in Early-Transition-Metal Alkyl Complexes. Organometallics 2008, 27, 1128–1134. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Brémond, É.; Tognetti, V.; Chermette, H.; Sancho-García, J.C.; Joubert, L.; Adamo, C. Electronic Energy and Local Property Errors at QTAIM Critical Points While Climbing Perdew’s Ladder of Density-Functional Approximations. J. Chem. Theory Comput. 2022, 18, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Boto, R.A.; Peccati, F.; Laplaza, R.; Quan, C.; Carbone, A.; Piquemal, J.-P.; Maday, Y.; Contreras-Garcia, J. NCIPLOT4: A New Step towards a Fast Quantification of Noncovalent Interactions. Available online: https://github.com/juliacontrerasgarcia/nciplot. (accessed on 10 August 2024).
- Keith, T.A. AIMAll version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018; Available online: https://nbo7.chem.wisc.edu (accessed on 23 January 2022).
- Knizia, G. IboView v.20211019-RevA. Available online: http://www.iboview.org/ (accessed on 23 January 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rcp and bcp | No Ancillary Ligand | MeOH | C2H4 | CO | |
|---|---|---|---|---|---|
| PCP | rcp4-bcpRh–C | - | 0.430 | 0.522 | 0.486 |
| rcp4-bcpRh–H | - | 0.814 | 0.684 | 0.713 | |
| POCOP | rcp4-bcpRh–C | 0.505 | - | 0.936 | 0.931 |
| rcp4-bcpRh–H | 0.727 | - | 0.217 | 0.212 | |
| PCN | rcp4-bcpRh–C | 0.807 | 0.507 | 0.760 | 0.873 |
| rcp4-bcpRh–H | 0.354 | 0.772 | 0.478 | 0.292 | |
| PCO | rcp4-bcpRh–C | - | - | 0.734 | 0.981 |
| rcp4-bcpRh–H | - | - | 0.459 | 0.098 | |
| SCS | rcp4-bcpRh–C | 0.310 | - | 0.749 | 0.266 |
| rcp4-bcpRh–H | 0.817 | - | 0.414 | 0.892 |
| P/S→Rh | Rh | P/S | P/N/O/S→Rh | Rh | P/N/O/S | |||
| PCP |  | 0.50 | 1.44 |  | 0.33 | 1.63 a | ||
| POCOP | 0.52 | 1.44 | 0.31 | 1.66 a | ||||
| PCN | 0.52 | 1.45 | 0.22 | 1.70 b | ||||
| PCO | 0.70 | 1.28 | 0.09 | 1.88 b | ||||
| SCS | 0.38 | 1.59 a,b | 0.37 | 1.59 b | ||||
| Rh→P/S | Rh→P/N/O/S | |||||||
| PCP |  | 1.90 | 0.04 | 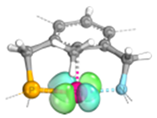 | 1.90 | - | ||
| POCOP | 1.91 | 0.02 | 1.87 | 0.08 a | ||||
| PCN | 1.94 | 0.02 | 1.92 | - b | ||||
| PCO | 1.91 | 0.05 | 1.94 | - | ||||
| SCS | 1.99 | - | 1.95 | - | ||||
| C–C→Rh | Rh | Cipso | CMe | C–H→Rh | Rh | CMe | HMe | |
| PCP | 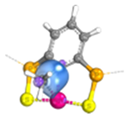 | 0.02 | 0.95 | 1.01 | 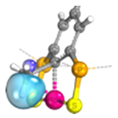 | 0.19 | 0.98 | 0.79 |
| POCOP | 0.05 | 0.96 | 0.97 | 0.11 | 1.03 | 0.83 | ||
| PCN | 0.05 | 0.96 | 0.98 | 0.08 | 1.07 | 0.83 | ||
| PCO | 0.03 | 0.95 | 1.01 | 0.17 | 0.99 d | 0.79 | ||
| SCS | 0.15 | 0.91 | 0.92 | 0.08 | 1.11 | 0.78 | ||
| Rh→CCH | Rh→CH | |||||||
| PCP |  | 1.93 | 0.03 | - | 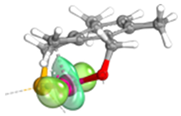 | 1.86 | 0.03 | 0.06 |
| POCOP | 1.85 | 0.12 c | - | 1.93 | 0.02 | 0.03 | ||
| PCN | 1.82 | 0.09 | - | 1.96 | - | - | ||
| PCO | 1.83 | 0.06 e | - | 1.92 | 0.03 | 0.03 | ||
| SCS | 1.54 | 0.33 | 0.07 | 1.96 | - | 0.02 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Efremenko, I. Electron Density and Molecular Orbital Analyses of the Nature of Bonding in the η3-CCH Agostic Rhodium Complexes Preceding the C–C and C–H Bond Cleavages. Molecules 2024, 29, 4788. https://doi.org/10.3390/molecules29204788
Efremenko I. Electron Density and Molecular Orbital Analyses of the Nature of Bonding in the η3-CCH Agostic Rhodium Complexes Preceding the C–C and C–H Bond Cleavages. Molecules. 2024; 29(20):4788. https://doi.org/10.3390/molecules29204788
Chicago/Turabian StyleEfremenko, Irena. 2024. "Electron Density and Molecular Orbital Analyses of the Nature of Bonding in the η3-CCH Agostic Rhodium Complexes Preceding the C–C and C–H Bond Cleavages" Molecules 29, no. 20: 4788. https://doi.org/10.3390/molecules29204788
APA StyleEfremenko, I. (2024). Electron Density and Molecular Orbital Analyses of the Nature of Bonding in the η3-CCH Agostic Rhodium Complexes Preceding the C–C and C–H Bond Cleavages. Molecules, 29(20), 4788. https://doi.org/10.3390/molecules29204788