
A Quantitative Re-Assessment of Microencapsulation in (Pre-Treated) Yeast

 , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
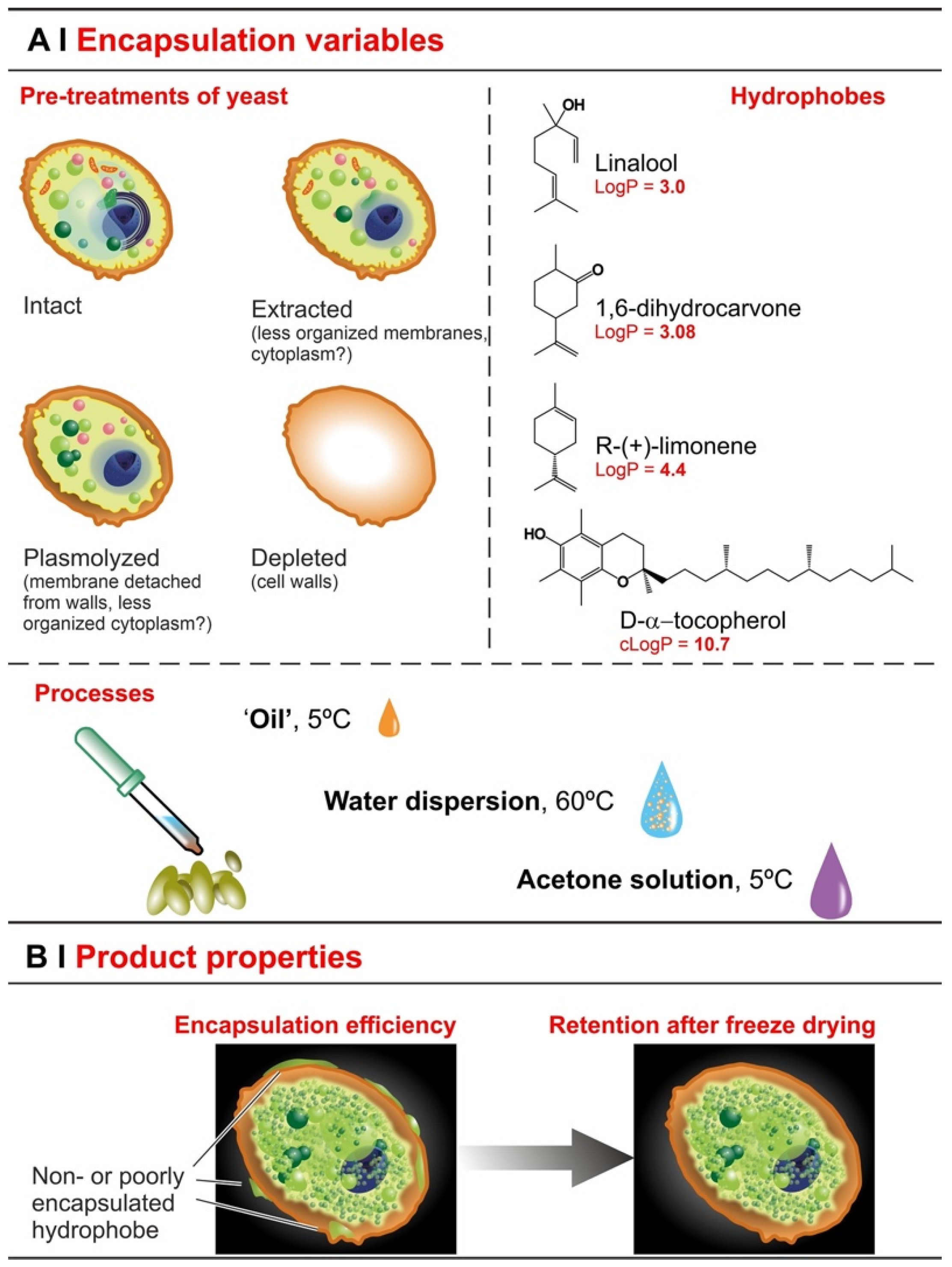
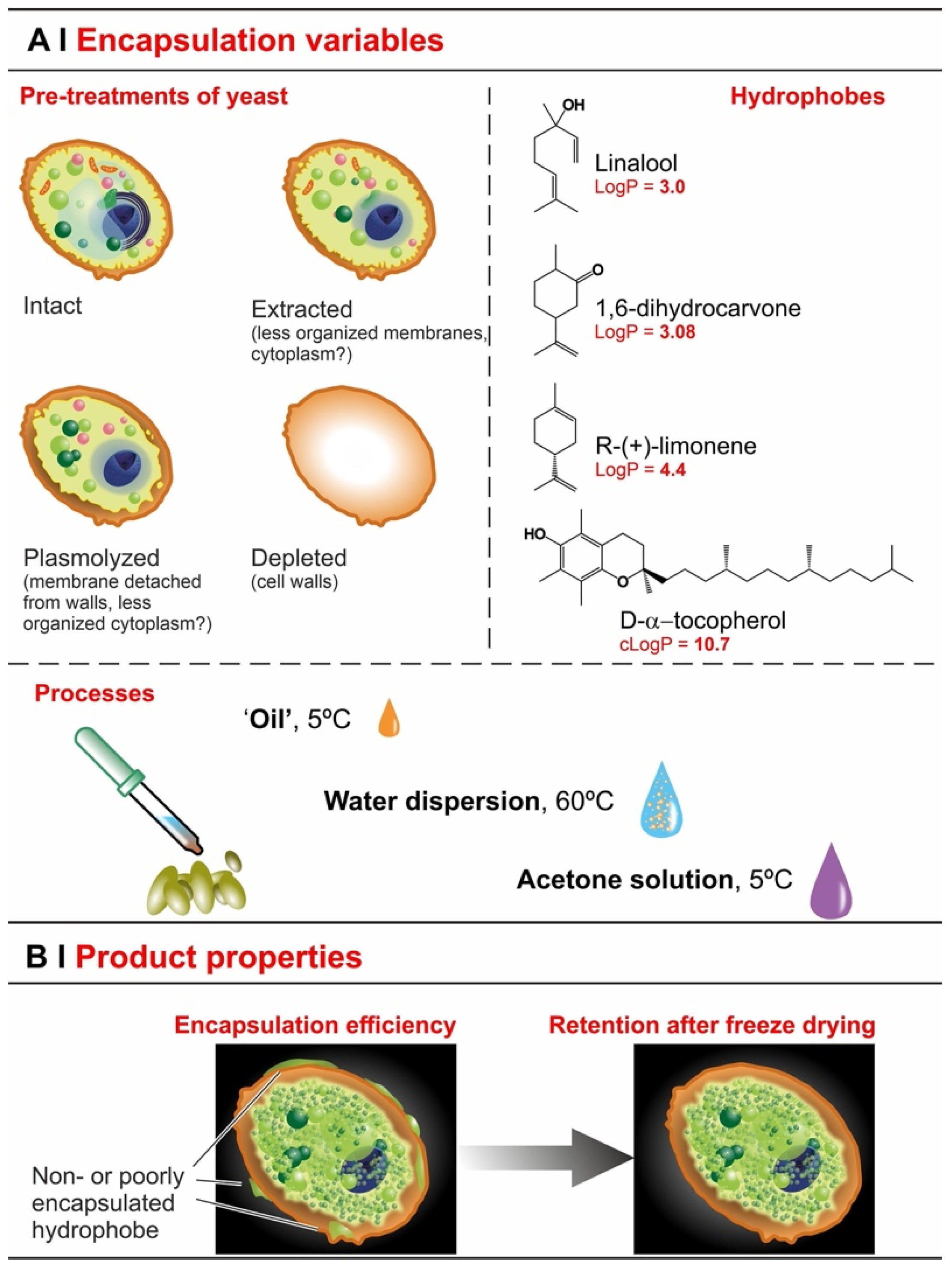
- Extraction with organic solvents (e.g., ethanol) partially solubilizes cell membranes [23], which should accelerate entrance, but—by removing intracellular lipids—it may also reduce encapsulation capacity.
- Plasmolysis via hypertonic shock produces invaginations on the plasma membrane [24] and increases the distance of the latter from the cell wall, separating the two irreversibly [19]. It may remove some cytoplasmic content [25], but is unlikely to seriously affect intracellular lipid content, and therefore hydrophobe encapsulation may not be seriously affected (lipid content is its thermodynamic driving force [26,27]). Plasmolysis may therefore affect the kinetics rather than the overall efficiency of the process; actually, there are reports that efficiency was actually increased by plasmolysis [11,13], as well as others claiming that it was not [3,12].
- Depletion leaves only one barrier in place (cell walls) and removes intracellular lipids (via saponification) [7], thus it seems reasonable to expect an even more accelerated permeation and a very reduced loading.
2. Results
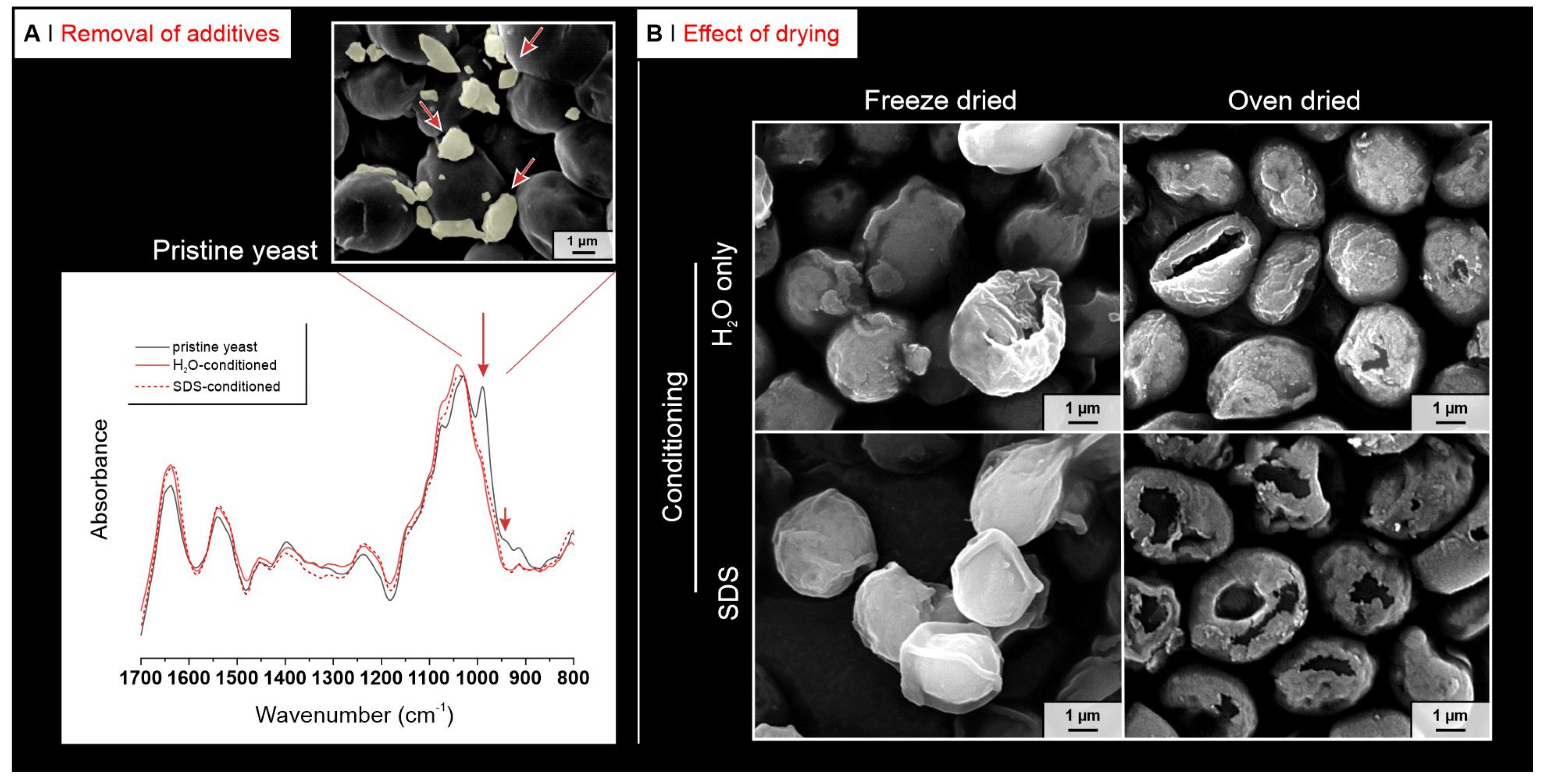
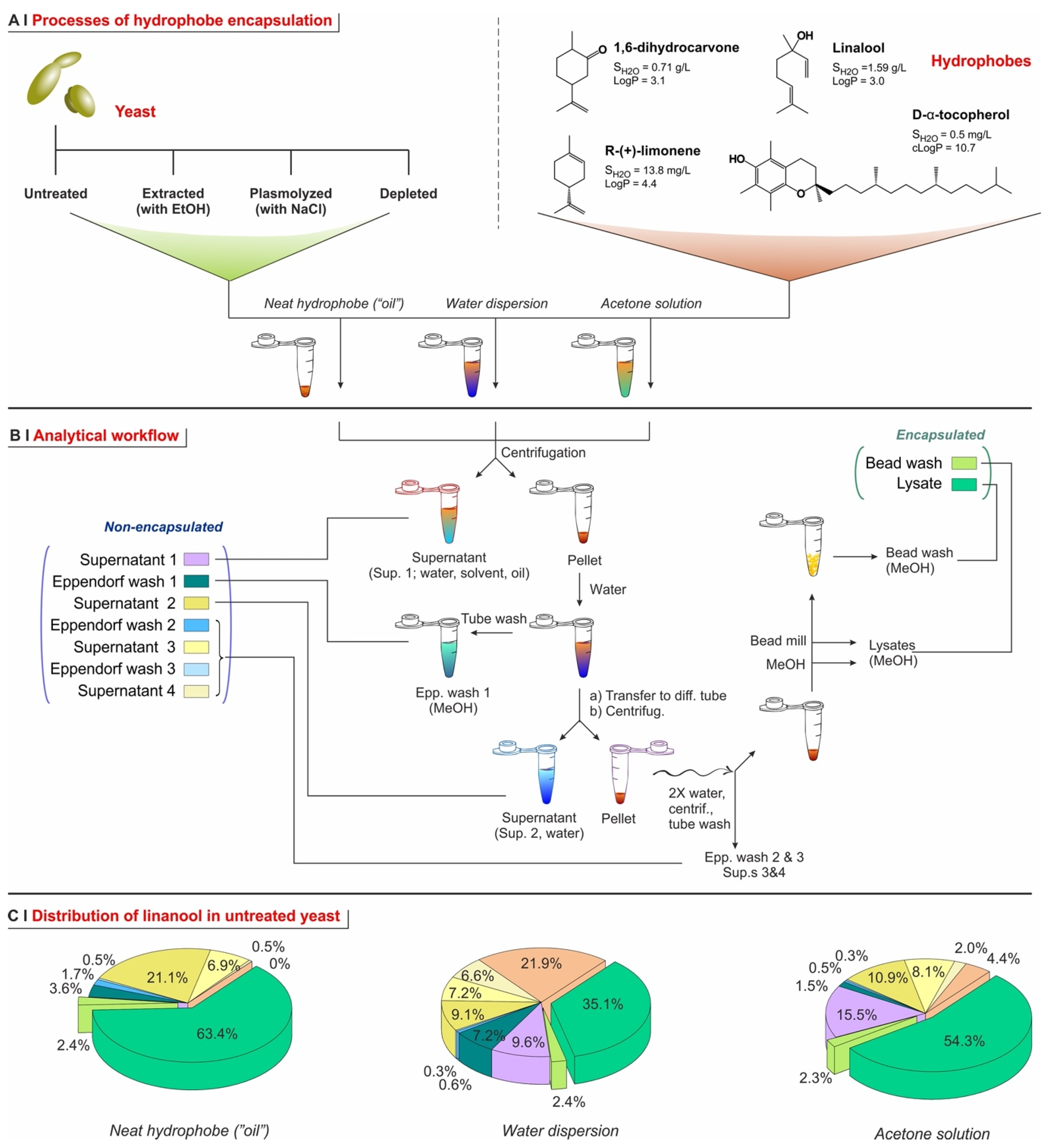
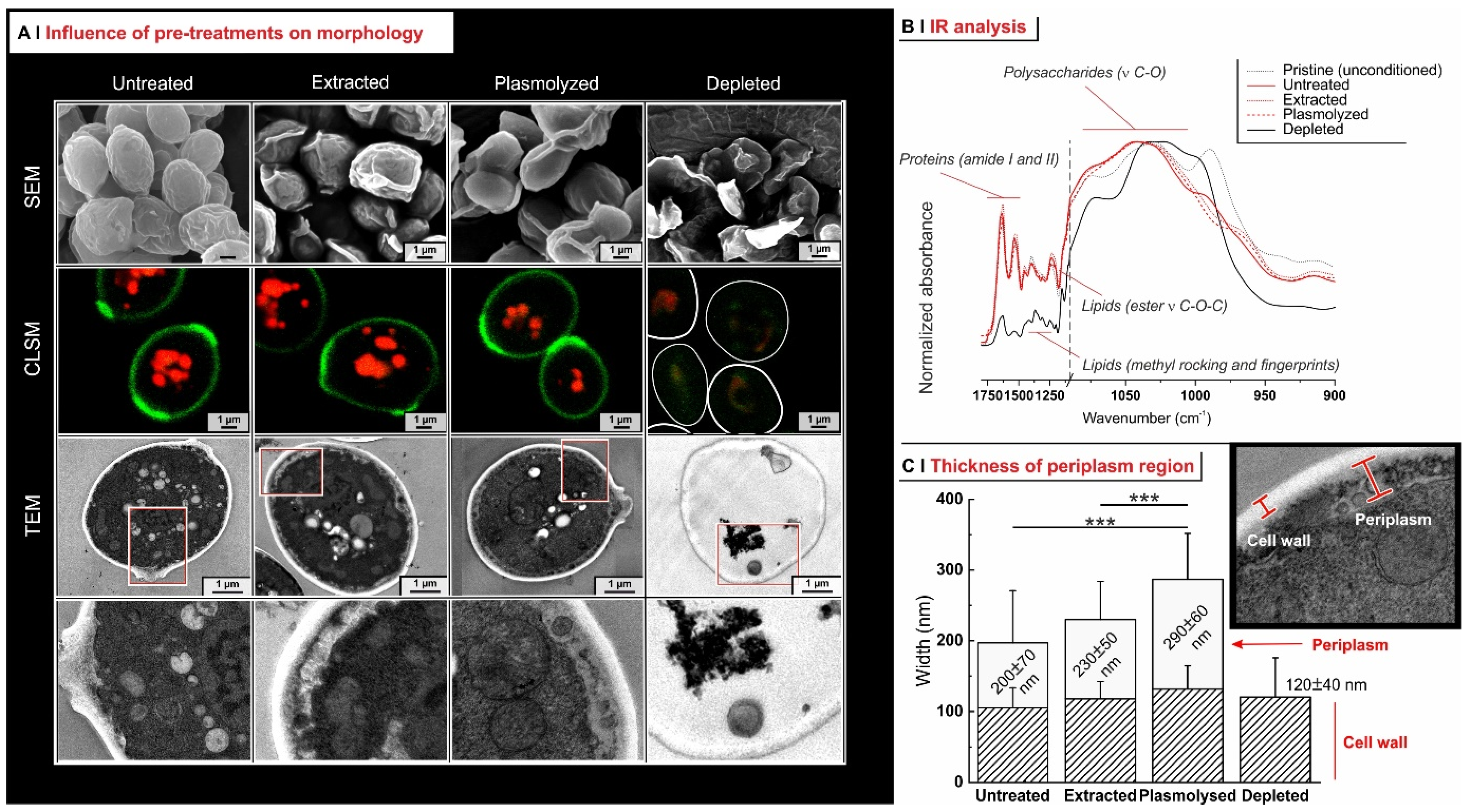
2.1. Yeast Conditioning
2.2. Yeast Pre-Treatment
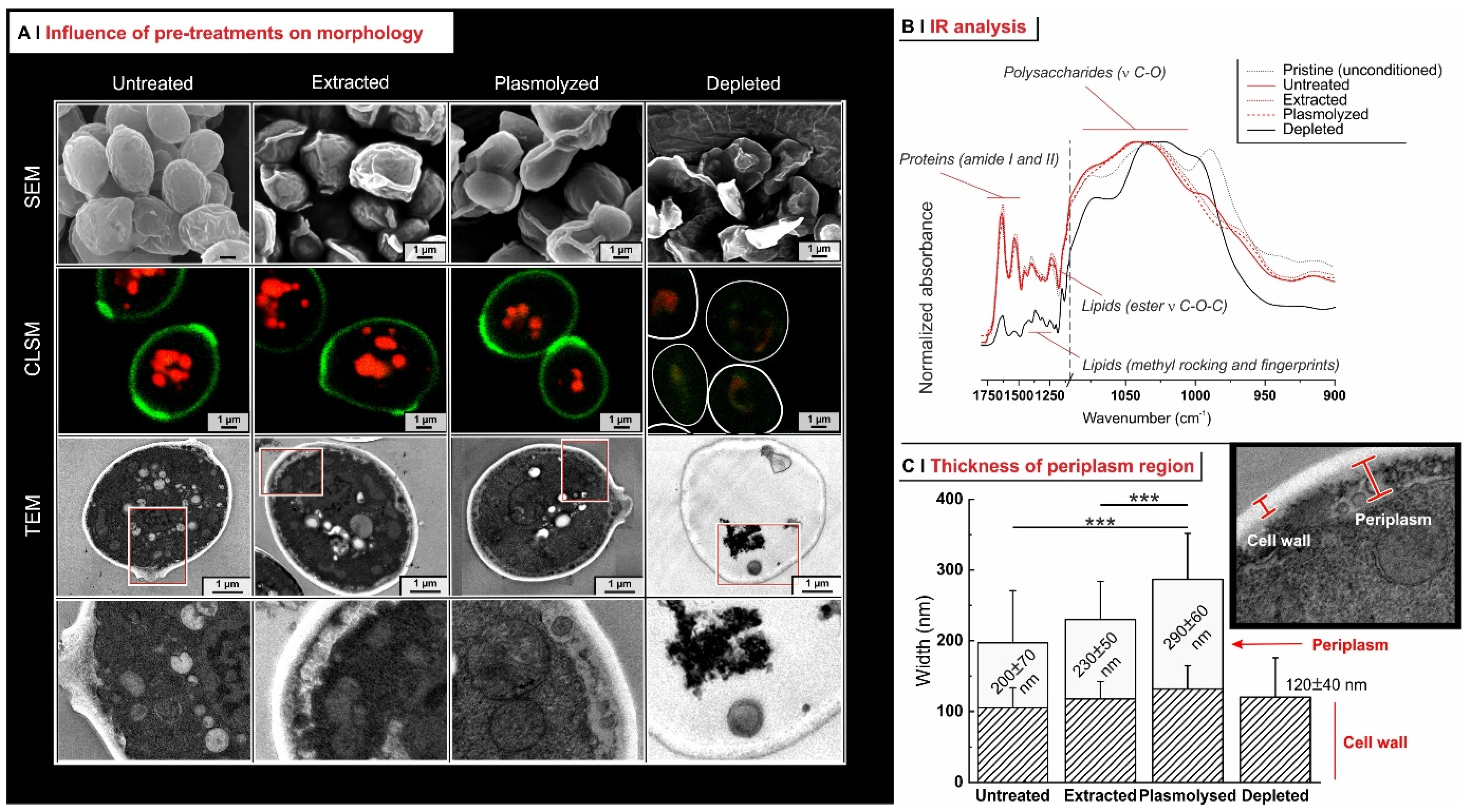
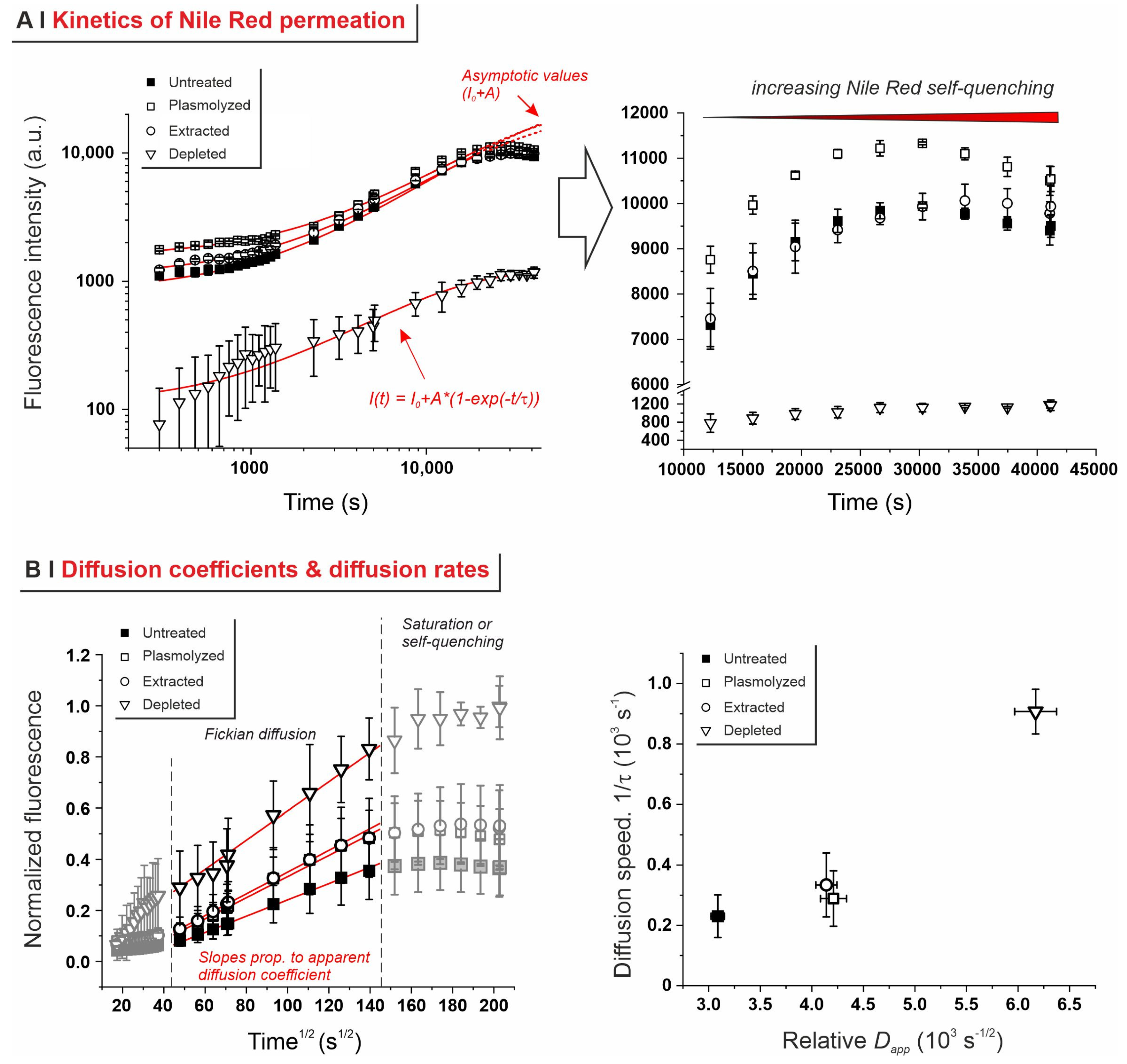
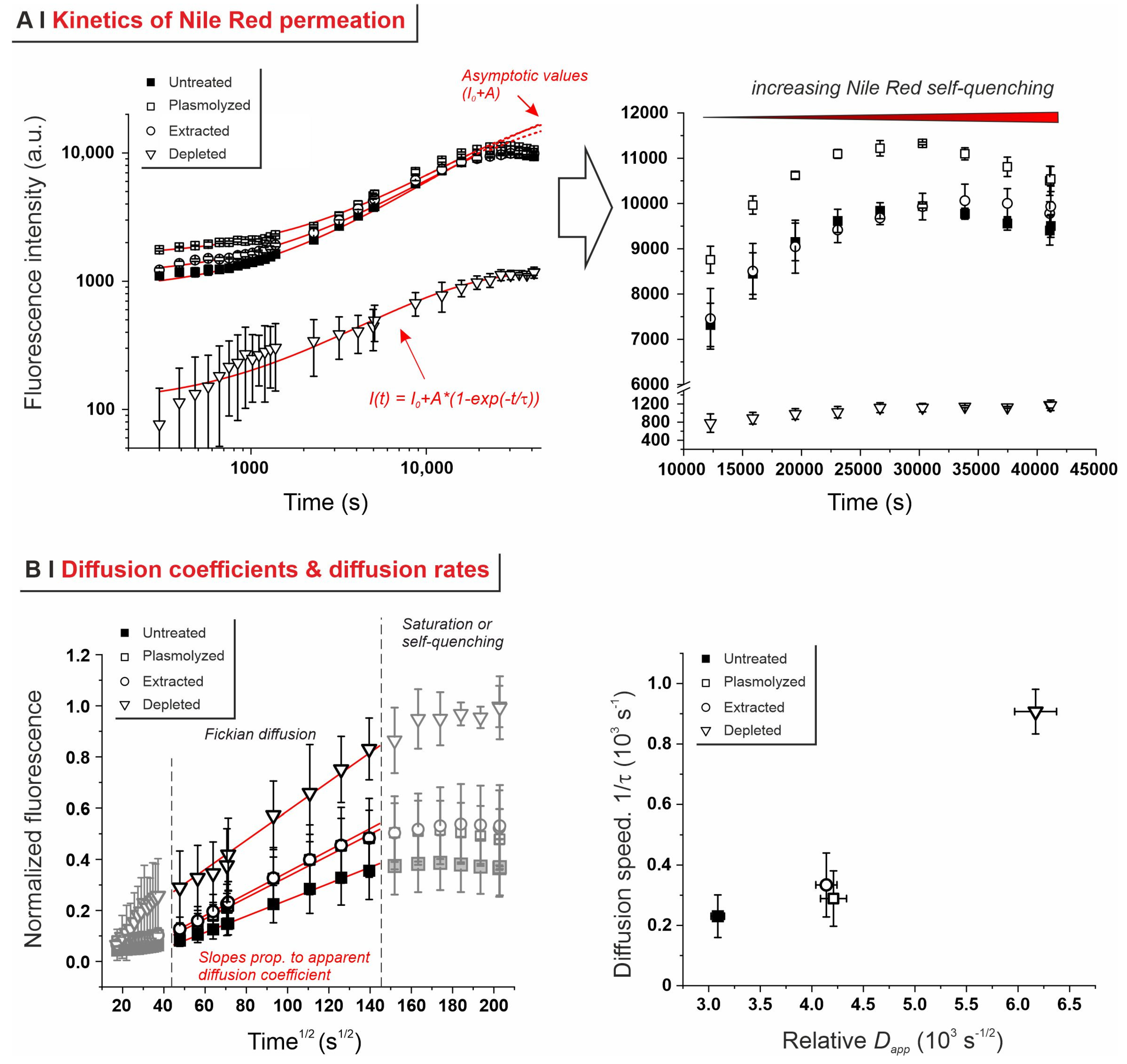
2.3. Effects of Pre-Treatments on Yeast Permeability
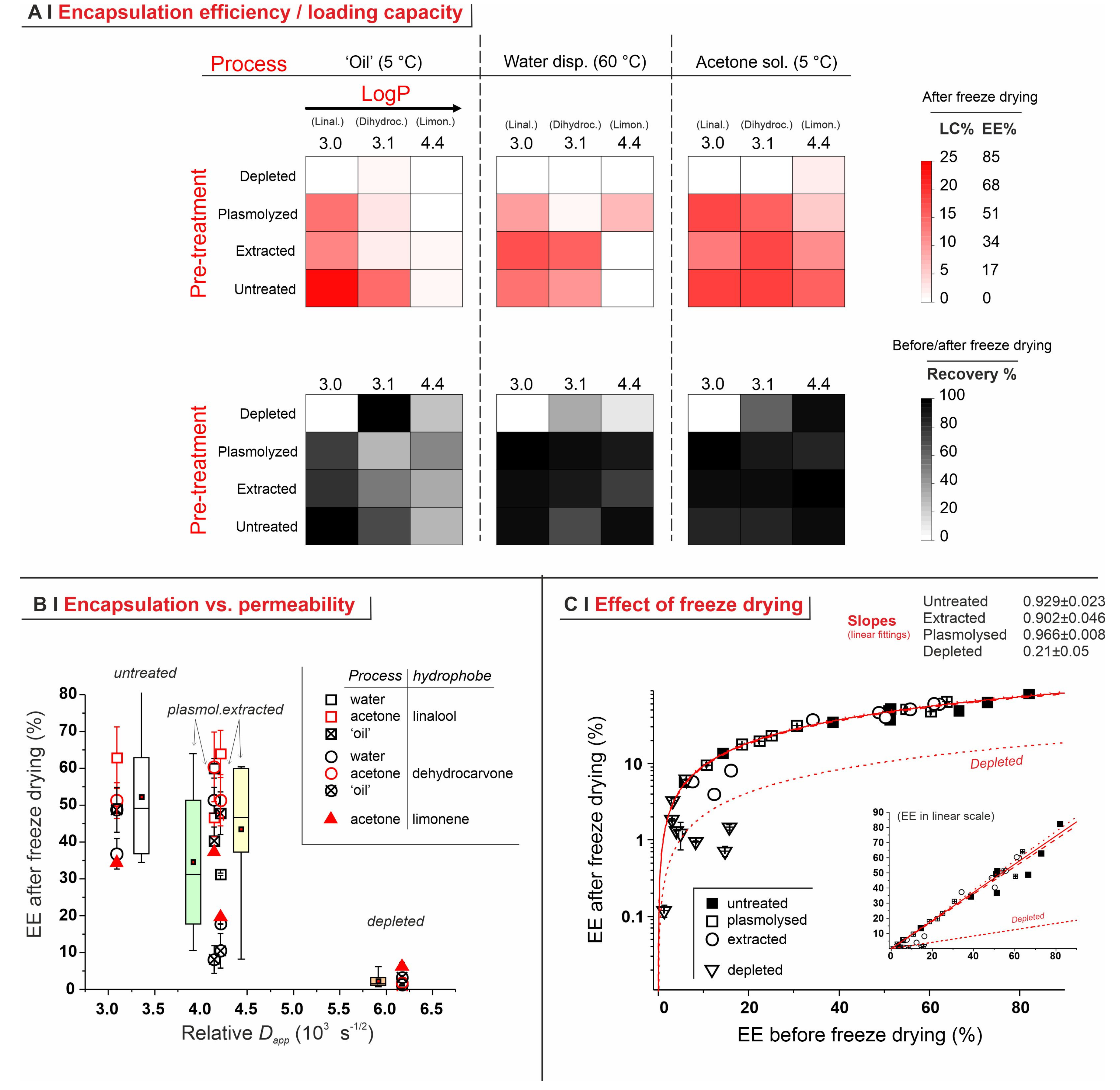
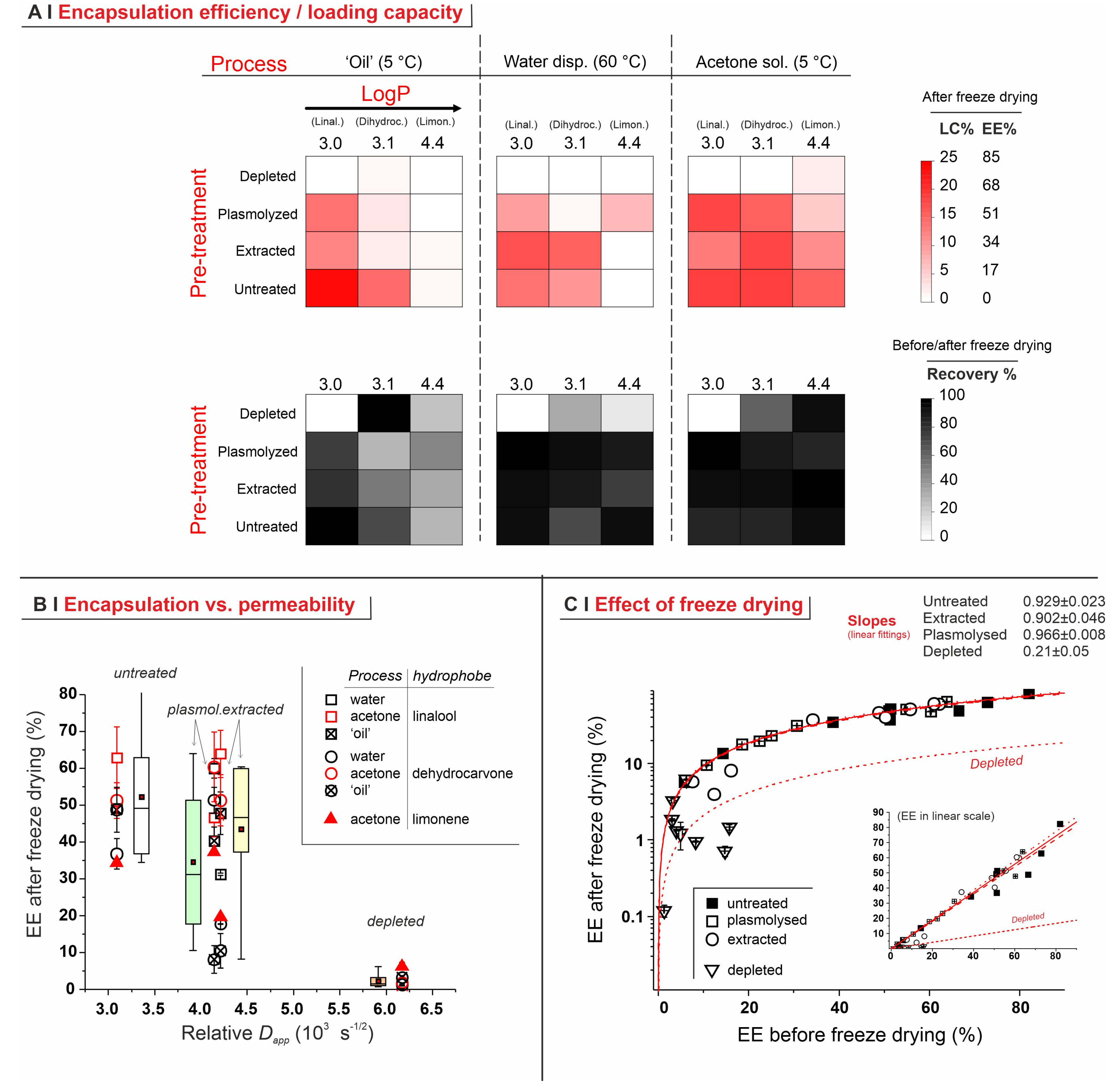
2.4. Encapsulation Processes for Single Hydrophobes
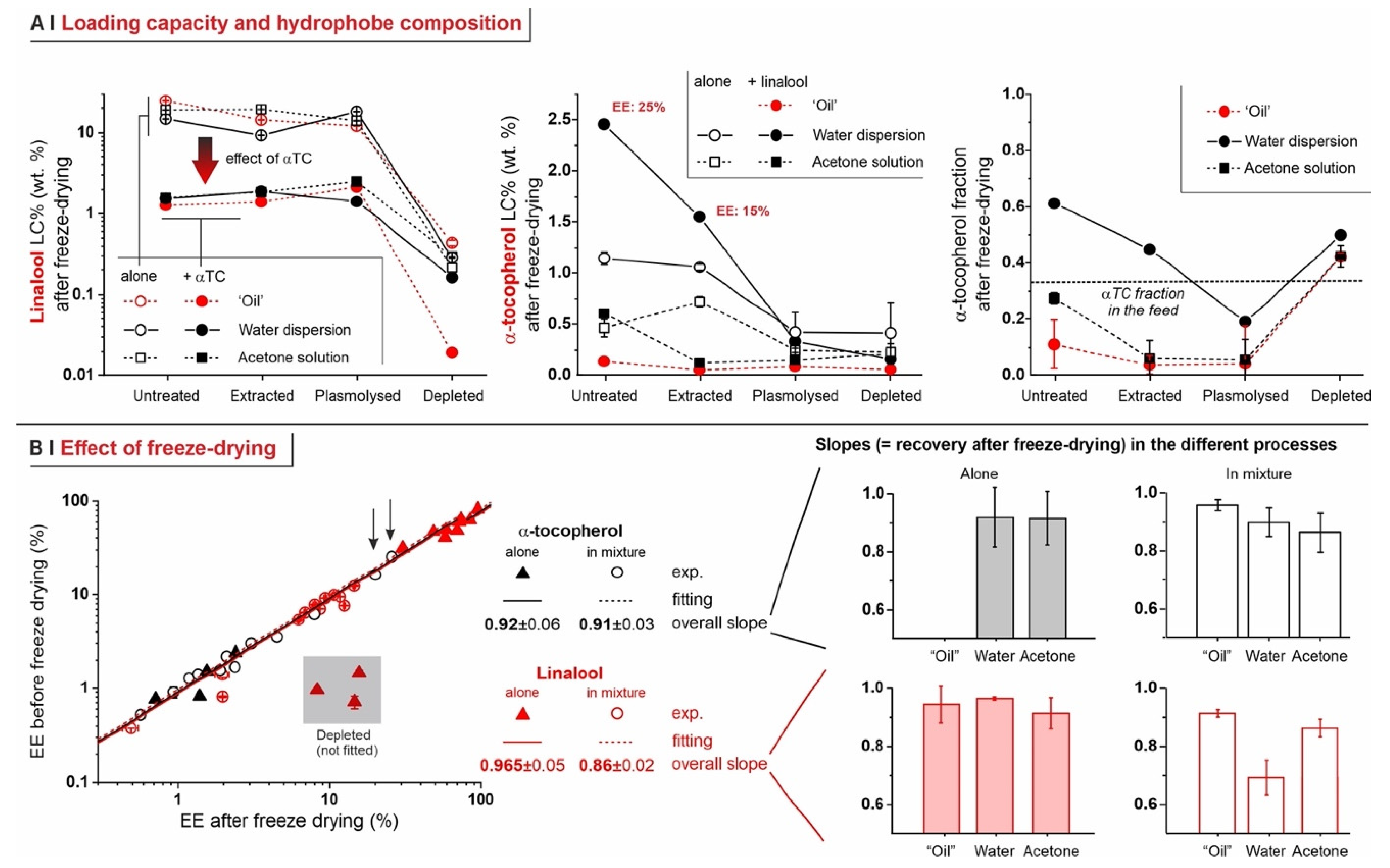
- Temperature. The encapsulation from water dispersion was performed at 60 °C. Such relatively high temperatures are routinely used in these processes [14,23] because the lower viscosity and higher diffusion coefficients of both water and hydrophobes, as well as the higher membrane fluidity [32], are believed to allow for a better/more rapid encapsulation; losses due to hydrophobe evaporation are sizeable but rather limited (up to around 10–15% in weight, see Figure 4C). On the other hand, when in pure ‘oil’ or acetone, a high temperature would entail excessive evaporation, thus these processes were operated at 5 °C, a temperature also used elsewhere for similar encapsulation protocols [7].
- Identity of the solvent (only for acetone encapsulation). The use of an organic solvent has two potential advantages: it may increase the solubility of the hydrophobes in the yeast cell walls (as we have demonstrated for the non-volatile DMSO [5], which requires a more cumbersome removal), but it also reduces the viscosity and increases the volume of the hydrophobic phase, which allows for more homogeneous mixing with the yeast pellets. In the literature, ethanol is often used for this purpose, either pure [12] or in a 50% water mixture [23]. Here we have shown that, in linalool-based pilot experiments, acetone alone determines a higher encapsulation (see Supporting Information, Figure S6) and was thus used in all the encapsulation experiments from an organic solution. It is noteworthy that, as encapsulation from acetone was significantly higher than from ethanol, and the latter only slightly better than water slurry, this order qualitatively agrees with the dielectric constant of the carriers (66.7 [33], 29.1 [34], <19.45 [35], respectively, for water, ethanol, and acetone).
- Hydration of the yeast pellet. Cell wall hydration has been reported to be essential for permeation through yeast cell barriers (cell wall and plasma membrane) [15,26]. Using acetone solutions (5 °C), linalool encapsulation was indeed somehow more efficient on pre-wetted yeast (see Supporting Information, Figure S6). When using water dispersion (60 °C), there was no significant difference between dry and wet yeast, which is not surprising because its cells would be hydrated in situ by the dispersing medium. Using a neat hydrophobe (‘oil’), encapsulation efficiency was high for both wet and dry yeast, marginally better for the latter, possibly because of the more difficult mixing of an oil with a wet ‘cake’ of yeast. Therefore, further water dispersion and acetone solution experiments were performed on hydrated yeast, while those using oil were performed on dry yeast.
- Amount of solvent (encapsulation from ethanol or acetone). We have used linalool with identical doses (15 mg per 50 mg of yeast) but different concentrations, i.e., varying only the volume of solvent (acetone or ethanol; either pure or 50% in water; see Supporting Information, Figure S6). With pure solvents (no water), encapsulation efficiency increased proportionally to linalool concentration, which suggests a simple partition. Conversely, for 50% aqueous solvents, efficiency was generally higher at lower linalool concentrations, which hints to the solubility of the hydrophobe in the cell walls—remaining low because of their hydration—being the controlling factor. For further experiments, we have chosen to be under partition (thermodynamic) control, and therefore employed acetone (performing better than ethanol) as a pure solvent. Of note, we have, however, employed the second (120 mg/mL, corresponding to 2.5 µL solution/mg yeast) and not the first highest concentration, due to its better ability to wet the yeast pellets.
- Repeated additions. It has been reported [7] that repeated encapsulation cycles on the same sample may increase the overall loading capacity. We have, however, observed the opposite. A single exposure at a dose of 15 mg of linalool per 50 mg of yeast, at different concentrations in pure acetone, had a much higher encapsulation efficiency than five successive cycles under the same conditions (see Supporting Information, Figure S6). Interestingly, the latter corresponded to an overall combined dose of 75 mg of linalool per 50 mg of yeast, yet they resulted in a lower loading capacity, i.e., a lower content of linalool per yeast cell. This might be due to the extraction of a previously retained hydrophobe every time the pellet is exposed to fresh organic solvent, probably favored by the increasingly damaged cellular barriers. Therefore, for further experiments, we have only used single-addition experiments.
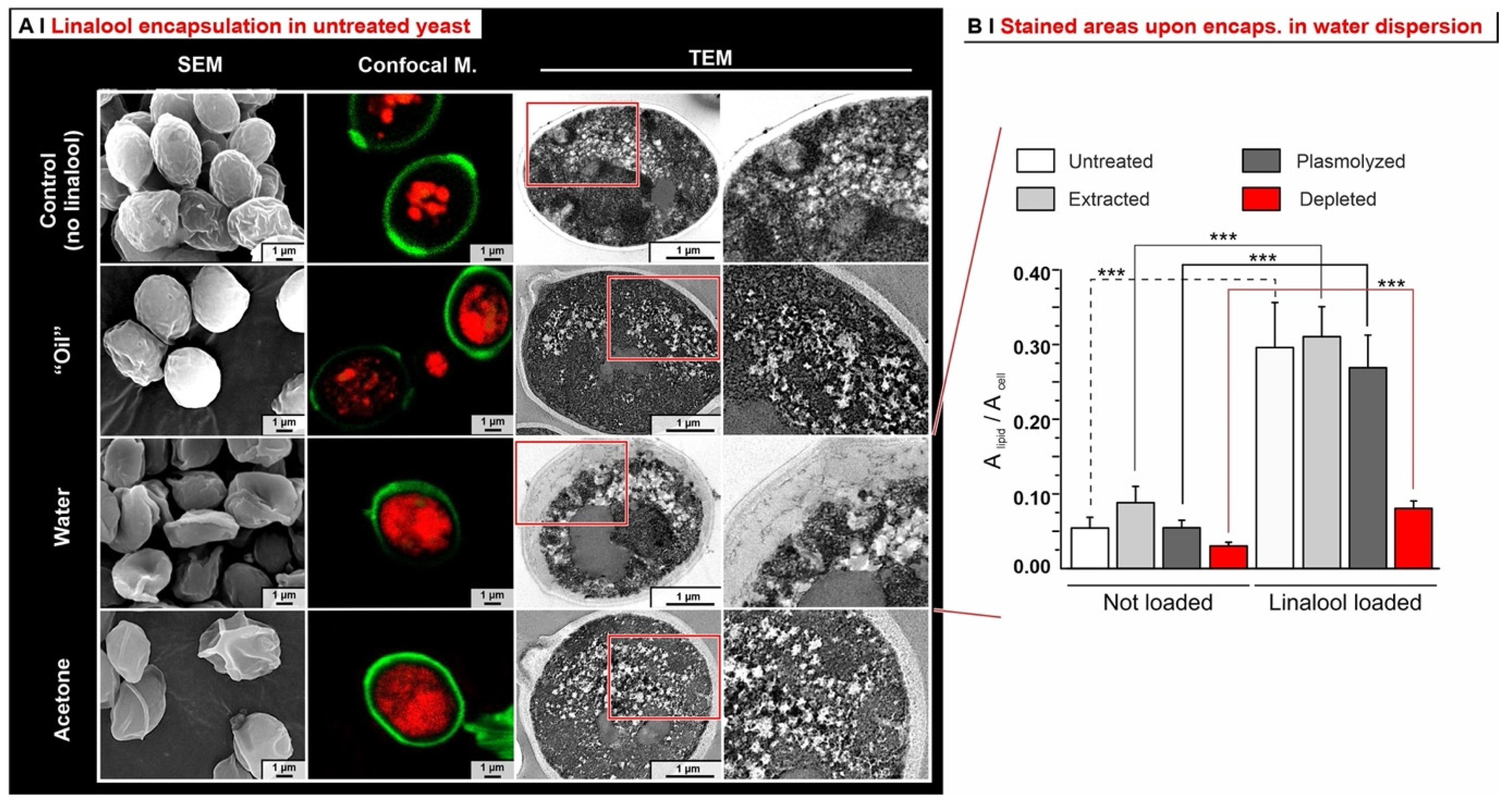
- Depleted cells: they are largely incapable of hosting any of the hydrophobes (see also Figure 5B), probably because of the depauperation of lipid components, as shown by Nile Red fluorescence.
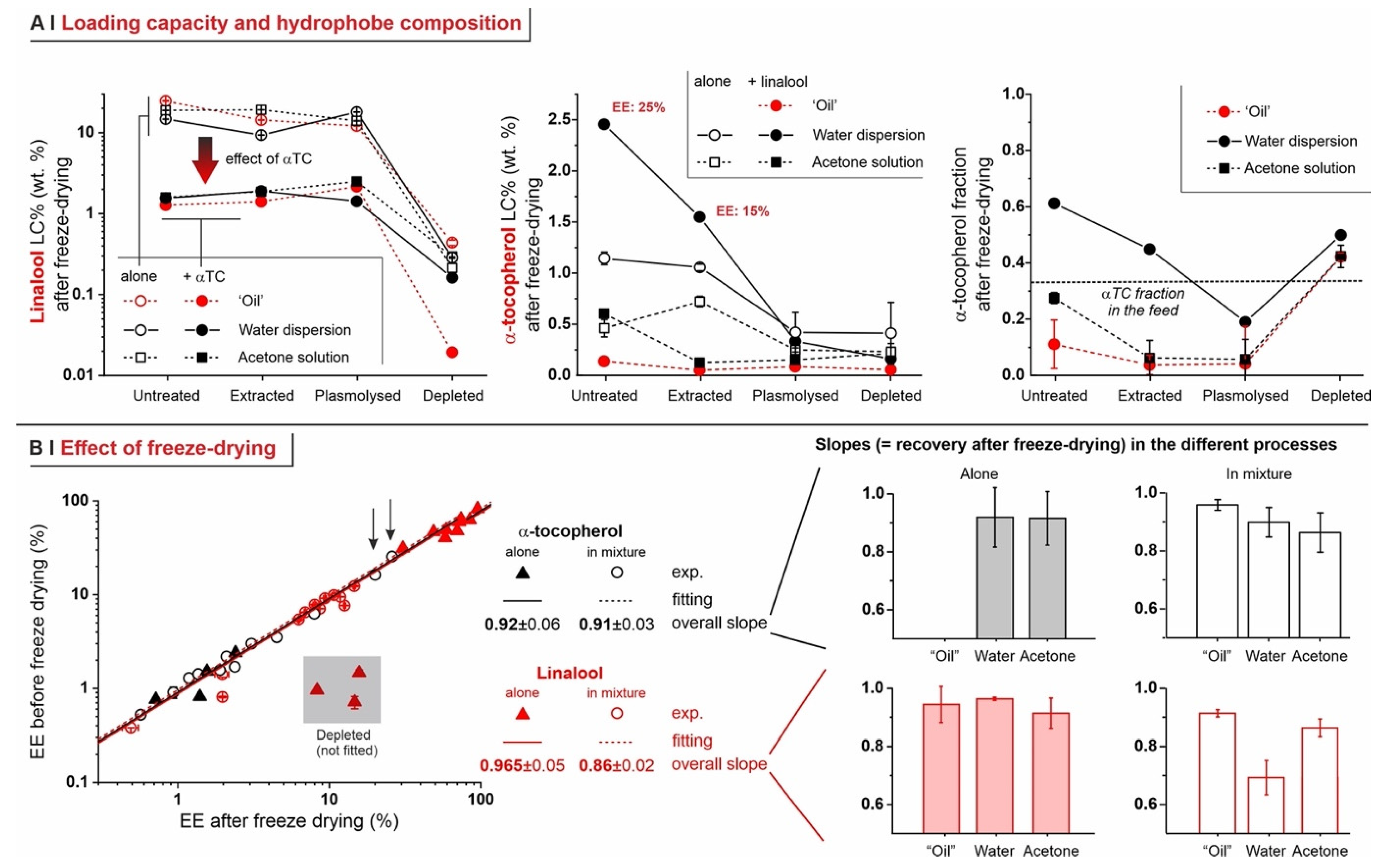
- Untreated cells: with a pure hydrophobe phase (‘oil’ or water dispersion), the encapsulation efficiency decreased in the order linalool (loading capacity up to 25% wt.) > 1,6-dihydrocarvone > limonene; the markedly better performance of linalool vs. 1,6-dihydrocarvone suggests that the linear/cyclic structural difference is possibly more important than the overall polarity, which would be an important correction to what was reported by Dardelle et al. [36] (i.e., logP being the key property). The trend was less apparent when the three hydrophobes were dissolved in acetone; it is indeed reasonable that a carrier capable of increasing solubility in cell walls and helping permeation through membranes would homogenize the performance of hydrophobes differing in structure and solubility.
- Plasmolyzed and extracted cells: in these two kinds of pre-treated yeast, generally linalool encapsulated best and limonene worst when using a pure hydrophobic phase, whereas no significant difference was recorded with acetone solutions. This is qualitatively similar to untreated cells, albeit mostly with a lower efficiency.
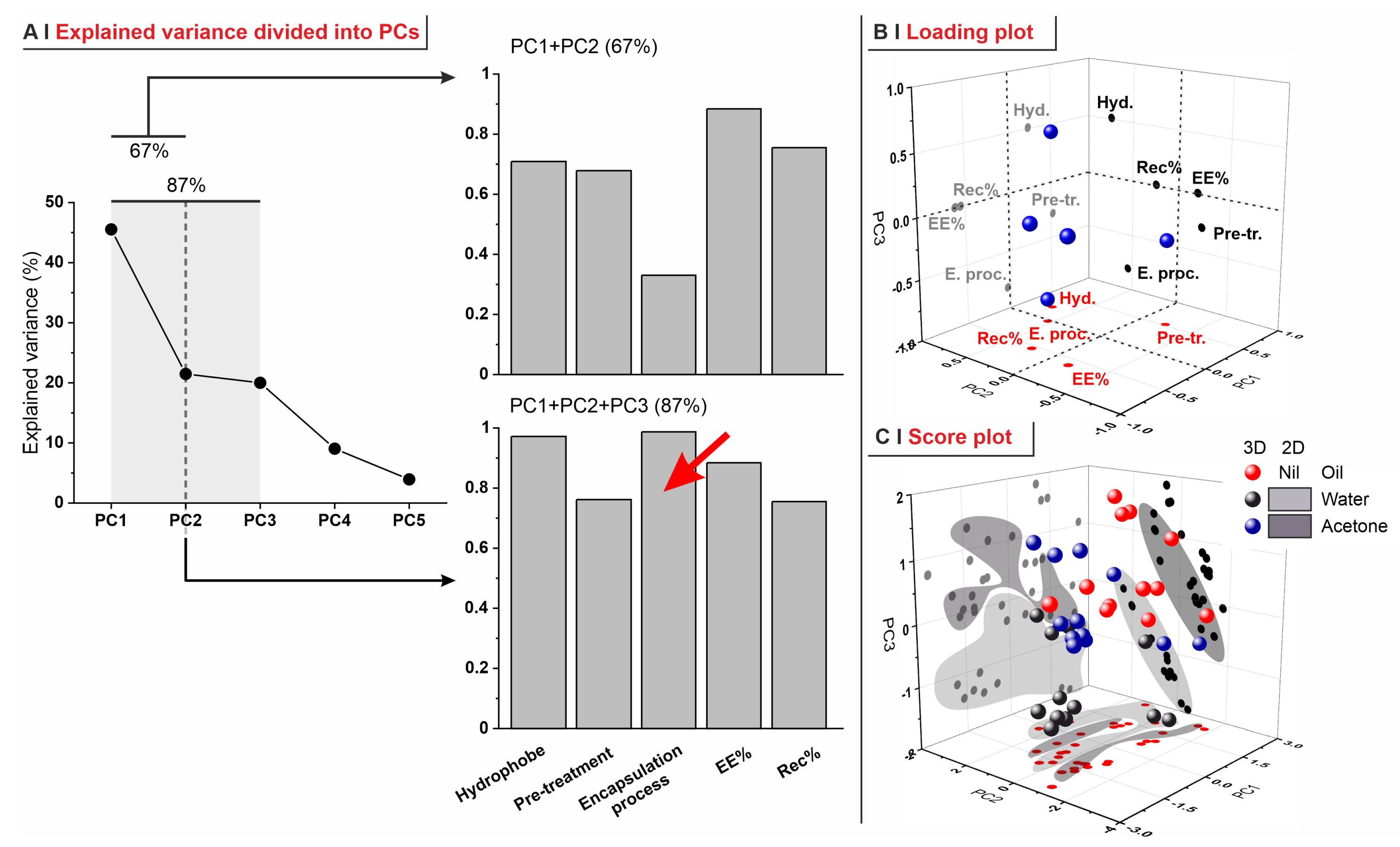
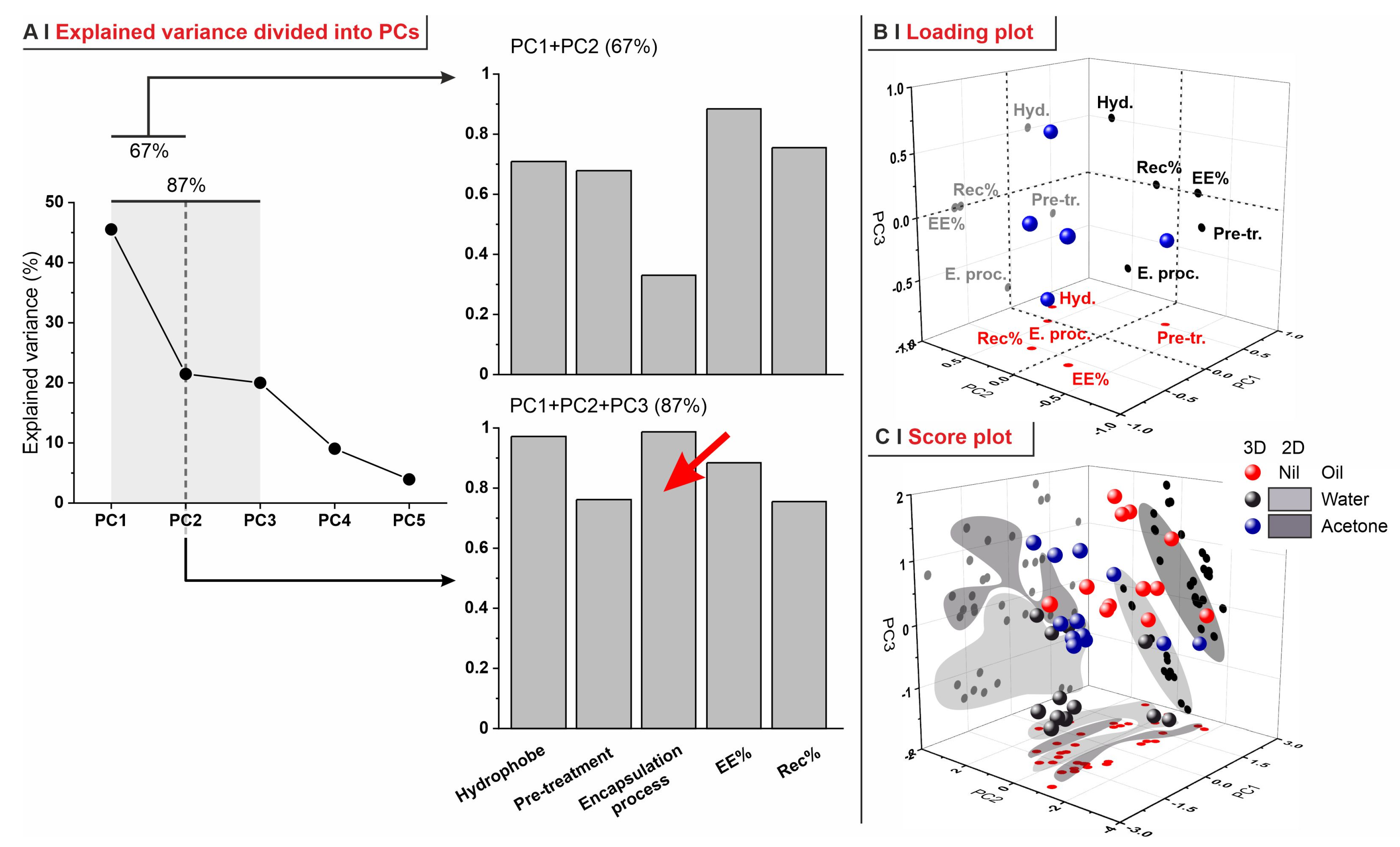
- (A)
- In PC1 vs. PC2, recovery was inversely correlated to pre-treatments and not correlated to the hydrophobe identity; this reflects the lower retention capacity of the most permeabilized yeasts (depleted).
- (B)
- In PC1 vs. PC3, EE% and recovery strongly correlated, independently of the encapsulation process; this is what was already observed in Figure 6C.
- (C)
- Although less relevant overall (only 41.5% of explained variance), the analysis of PC2 vs. PC3 confirmed that the nature of the encapsulation process, of the pre-treatment, and of the hydrophobe are completely independent of each other.
- (D)
- In none of these combinations of PCs do there seem to be correlations between EE% and LogP, although this is probably due to the small range of hydrophobes adopted in this study, as well as the different molecular structures (cyclic or linear).
2.5. Encapsulation of Hydrophobe Combinations
3. Conclusions
- (1)
- Analytics. We have developed methods that quantitatively account for fractions of material, such as those adsorbed on plasticware or on glassware, which are often overlooked in the literature, but are absolutely not negligible (see Figure 4C). This higher accuracy allows for a considerably more reliable assessment of the ‘real’ encapsulation (based on the loss during freeze-drying), as opposed to adsorption or physical entrapment. For example, we have demonstrated that maintaining the yeast cytoplasmic content of yeast cells is crucial to minimize the volatility of encapsulated hydrophobes; indeed, they easily evaporate or diffuse out of depleted cells. This is remarkable, since several studies have been based on the assumption that empty capsules could contain more hydrophobes [7].
- (2)
- Effects of yeast pre-treatments. A major point that we have addressed is whether pre-treating yeast may or may not lead to increased levels of encapsulated hydrophobes, a point where the literature reports are fragmented and occasionally contradictory. Plasmolyzed cells are sometimes reported as better performing [11], linking this to the extraction of proteins/nucleic acids at a temperature higher than the membrane transition temperature (45 °C > 37.5 °C) [38], but, all-in-all, the evidence is scant and the overall landscape unclear. Using conditioned yeast (washing and freeze-drying; removal of excipients without damaging cell structure; see Figure 1B), we have shown that yeast depletion—a saponification at high pH and temperature—was the most aggressive pre-treatment and modified both the morphology and composition of the yeast cells: they were reduced to their mere cell walls and the permeability through this last residual barrier was much larger than that through the original wall/membrane combination. The other two pre-treatments, i.e., plasmolysis and ethanol extraction, did not significantly alter cell morphology, nor lipid or protein levels (Figure 2A,B), but moderately increased permeability (increased Nile Red diffusion coefficient) and periplasmic thickness. Physico-chemically, these two pre-treatments are very different: one uses a hypertonic saline solution, the other a rather hydrophobic organic solvent; yet, they may have a similar, denaturing effect on proteins. Thus, it is tempting to ascribe the higher permeability and thickness to the denaturation of lipid-associated proteins causing alterations in the structure and anchoring of the cell membranes. If we add the encapsulation results to the above observations, it may be tempting to draft a direct relation between encapsulation efficiency, resistance to evaporation, and lipid (or protein) levels: they are all very low in depleted yeast, and rather similar in untreated, plasmolyzed, and extracted cells. However, the situation is more complex: analyzing together all processes and actives (Figure 6B), encapsulation seemed not only to massively decrease upon depletion, but to possibly already falter upon plasmolysis/ethanol extraction, which is a trend opposite to what is seen for permeability (Figure 2A,B and Figures S5). There appears, therefore, to be an inverse relation between the speed and capacity of encapsulation, which suggests the presence of key components—which we surmise to be membrane proteins—acting as gatekeepers of hydrophobe loading. Therefore, a tentative interpretation is that protein denaturation caused by plasmolysis/ethanol extraction mildly increases permeability and decreases encapsulation, possibly via an altered organization of lipid-based barrier structures (i.e., the cell membrane), while removal of lipids (and proteins) in the more aggressive conditions of yeast depletion, produces more drastic effects, including a much higher volatility.
- (3)
- Effects of the process. In single-compound encapsulation experiments performed without a carrier (=no solvent, i.e., ‘oil’ or water dispersion), logP appeared to negatively correlate with EE%. It seems reasonable to link this effect to the hydrophobe’s solubility in the cell walls, which we have previously described as a major determinant of encapsulation [37] and would indeed decrease with increasing logP. With acetone solutions, perhaps not surprisingly, the encapsulation efficiency was not significantly affected by the nature of the hydrophobe. This prompted us to try double-compound encapsulation, where a first hydrophobe would act as a ‘carrier’ for one more difficult to encapsulate on its own. Using a linalool/α-tocopherol mixture, the former is safe and perfumed, and diffuses easily into yeast; the second is usable as a ‘drug’. However, this approach does not appear to hold a general validity and should be assessed on a case-by-case basis, since results are difficult to predict: in our case, α-tocopherol benefitted from the double-compound process only under those conditions (water dispersion; untreated yeast or extracted with ethanol) where it appreciably encapsulated on its own. Further, this effect was, in part, counterbalanced by α-tocopherol hindering the linalool’s entrance.
- (4)
- Additional noteworthy observations:
- (A)
- Cell wall pre-hydration appears to be beneficial to achieve high EE%, but only when encapsulation is not performed in a water environment.
- (B)
- The practice of repeating encapsulation cycles has often been used in the literature, based on the belief that it would increase the final loading of the yeast [7]. In our study, however, even when evaporation was completely avoided (5 °C, sealed Eppendorf tubes), we have not observed any significant improvement in loading, whereas, clearly, the overall efficiency of the process was much decreased. Therefore, it seems to us that a strong caveat should be issued in relation to this practice.
4. Materials and Methods
4.1. Materials
4.2. Preparative Procedures
- (A)
- Cells untreated (control): wet pellets were dispersed in Milli-Q water and centrifuged, discarding the supernatant, three more times.
- (B)
- Cells extracted with ethanol: wet pellets were transferred into a 250 mL round-bottom flask, re-suspended in 80 mL of 50% v. ethanol/water and left at room temperature under stirring at 300 rpm for 2 h. Yeast cells were centrifuged and then washed thrice with Milli-Q water.
- (C)
- Cells plasmolyzed with hypertonic sodium chloride: wet pellets were suspended in 80 mL of 0.9% wt. sodium chloride and centrifuged discarding the supernatant three times, followed by a re-suspension in 80 mL of 20% wt. sodium chloride; the dispersion was transferred into a 250 mL round-bottom flask, left at 45 °C under 300 rpm magnetic stirring for 2 h, centrifuged, and washed once with 40 mL of 0.9% wt. sodium chloride and thrice with Milli-Q water each time after centrifugation.
- (D)
- Cells depleted and reduced to cell walls: wet pellets were resuspended in 80 mL of 1M sodium hydroxide, transferred into a 250 mL round-bottom flask equipped with a condenser, and left at 85 °C under 300 rpm stirring for 1 h. Yeast was centrifuged, discarding the supernatant, and re-suspended in 60 mL of Milli-Q water, transferred back to a clean 250 mL round-bottom flask equipped with a condenser; the pH was adjusted to 4.5 with a 0.5 M hydrochloric acid solution (final volume 80 mL) and the temperature was risen to 60 °C for 1 h, while keeping the dispersion under 300 rpm stirring. Yeast was centrifuged, washed once with Milli-Q water, twice with isopropanol, then with acetone and finally with Milli-Q water three times (always after centrifugation). It was then freeze-dried as reported in the Supporting Information, Section S1. The techniques used to characterize the differently pre-treated yeast cells are reported in the Supporting Information, Section S3.
- (A)
- Encapsulation from an ‘oil phase’ (=no solvent or dispersant). For a comparison between wet and dry yeast, 50 mg of conditioned but untreated yeast (control) was used either directly in a dry form, or as a wet pellet (suspended in 1 mL of Milli-Q Water and centrifuged discarding the supernatant, to yield roughly 210–220 mg of wet material) and exposed to linalool. In detail, yeast (wet or dry) was introduced into 2 mL Eppendorf tubes, and linalool was directly pipetted on top of it in an amount corresponding to 30% wt. of the yeast’s dry weight, vortexing immediately for 30 s; Eppendorf tubes were sealed with Parafilm and incubated in an orbital shaker (5 °C, 54 rpm, 2 h). Since dry yeast showed higher encapsulation (Figure S6, symbols in the bottom row), all further screening experiments of ‘oil’ encapsulation, e.g., with different hydrophobes, only used dry yeast.
- (B)
- Encapsulation from water dispersion. One g of freeze-dried pre-treated yeast was dispersed in 10 mL of Milli-Q water, gently vortexing to ensure homogeneity. An amount of 500 μL aliquots (corresponding to 50 mg of dry yeast) was transferred into 2 mL Eppendorf tubes, centrifuged, discarding the supernatant, diluted in 500-x μL of Milli-Q water, and finally topped up with x μL of hydrophobe (x is the volume corresponding to 30% wt./15 mg of hydrophobe through the density values reported above). The slurries were vortexed again, sealed with Parafilm and incubated in a Thermomixer C (Eppendorf AG, Hamburg, Germany) at 60 °C, 900 rpm for 4 h. In order to estimate the possible loss of hydrophobe due to evaporation, a 2 mL Eppendorf tube containing the same amounts of water and hydrophobes, but no yeast, was incubated as the yeast-containing samples and analyzed.
- (C)
- Encapsulation from organic solution. (1) Choice of the encapsulation conditions. These experiments were conducted with linalool as a probe at 30% wt. in respect to the mass of dry yeast. Fifty mg of conditioned but untreated yeast (control) was used directly in a dry form, or as a wet pellet (suspended in 1 mL of Milli-Q Water and centrifuged discarding the supernatant, to yield roughly 210–220 mg of wet material); the yeast suspension was treated with additional variable volumes (500, 250, 125, or 62.5 µL) of liquid phase (water, ethanol, acetone, 50% v/v water/ethanol, and 50% v/v water/acetone) always containing 15 mg of linalool and incubated in an orbital shaker (5 °C, 54 rpm) for 2 h. In experiments featuring repeated encapsulation cycles, the yeast was centrifuged, discarding the supernatant, air dried for 24 h, subjected to the same conditions of encapsulation for a total of four additional cycles of encapsulation (each cycle with 15 mg of linalool) and washing. (2) Encapsulation in acetone. As a result of the screening in point 1 (see Supporting Information, Figure S6), the optimized protocol for encapsulation in organic solution resulted in the following procedure. Fifty mg of freeze-dried yeast in the form of a wet pellet was dispersed into 125 µL of a 120 mg/mL solution of hydrophobe in acetone (barely enough to cover the whole pellet), quickly vortexed, sealed with Parafilm, and incubated in an orbital shaker (5 °C, 54 rpm, 2 h).
4.3. Characterization Techniques
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, S.; Tao, M.; Huang, K. Encapsulation of Manuka essential oil in yeast microcarriers for enhanced thermal stability and antimicrobial activity. Food Bioprocess Technol. 2021, 14, 2195–2206. [Google Scholar] [CrossRef]
- Shi, G.; Rao, L.; Yu, H.; Xiang, H.; Yang, H.; Ji, R. Stabilization and encapsulation of photosensitive resveratrol within yeast cell. Int. J. Pharm. 2008, 349, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Paramera, E.I.; Konteles, S.J.; Karathanos, V.T. Stability and release properties of curcumin encapsulated in Saccharomyces cerevisiae, β-cyclodextrin and modified starch. Food Chem. 2011, 125, 913–922. [Google Scholar] [CrossRef]
- Iassonova, D.R.; Hammond, E.G.; Beattie, S.E. Oxidative stability of polyunsaturated triacylglycerols encapsulated in oleaginous yeast. J. Am. Oil Chem. Soc. 2008, 85, 711–716. [Google Scholar] [CrossRef]
- Ciamponi, F.; Duckham, C.; Tirelli, N. Yeast cells as microcapsules. Analytical tools and process variables in the encapsulation of hydrophobes in S. cerevisiae. Appl. Microbiol. Biotechnol. 2012, 95, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Moon, N.J.; Hammond, E.G.; Glatz, B.A. Conversion of cheese whey and whey permeate to oil and single-cell protein. J. Dairy Sci. 1978, 61, 1537–1547. [Google Scholar] [CrossRef]
- Scariot, D.; Volpato, H.; Fernandes, N.; Soares, E.; Ueda-Nakamura, T.; Dias-Filho, B.; Din, Z.; Rodrigues-Filho, E.; Rubira, A.; Borges, O.; et al. Activity and cell-death pathway in Leishmania infantum induced by sugiol: Vectorization using yeast cell wall particles obtained from Saccharomyces cerevisiae. Front. Cell. Infect. Microbiol. 2019, 9, 208. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, X.; Han, S.; Dou, Y.; Liu, M.; Zhang, L.; Guo, J.; Shi, Q.; Gong, G.; Wang, R.; et al. Yeast microcapsule-mediated targeted delivery of diverse nanoparticles for imaging and therapy via the oral route. Nano Lett. 2017, 17, 1056–1064. [Google Scholar] [CrossRef]
- Shank, J.L. Encapsulation Process Utilizing Microorganisms and Products Produced Thereby. US4001480A, 13 April 1976. [Google Scholar]
- Sentosa, O.; Katherine, K.; Sugih, A. Effect of temperature in the encapsulation of temulawak extract with yeast Saccharomyces cereviceae. In Proceedings of the 2015 4th International Conference on Instrumentation, Communications, Information Technology, and Biomedical Engineering (ICICI-BME), Bandung, Indonesia, 2–3 November 2015; pp. 332–336. [Google Scholar]
- Dadkhodazade, E.; Mohammadi, A.; Shojaee-Aliabadi, S.; Mortazavian, A.M.; Mirmoghtadaie, L.; Hosseini, S.M. Yeast cell microcapsules as a novel carrier for cholecalciferol encapsulation: Development, characterization and release properties. Food Biophys. 2018, 13, 404–411. [Google Scholar] [CrossRef]
- Czerniak, A.; Kubiak, P.; Białas, W.; Jankowski, T. Improvement of oxidative stability of menhaden fish oil by microencapsulation within biocapsules formed of yeast cells. J. Food Eng. 2015, 167, 2–11. [Google Scholar] [CrossRef]
- Kavosi, M.; Mohammadi, A.; Shojaee-Aliabadi, S.; Khaksar, R.; Seyede, M.H. Characterization and oxidative stability of purslane seed oil microencapsulated in yeast cells biocapsules. J. Sci. Food Agric. 2017, 98, 2490–2497. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.P.; Nelson, G.; Lamb, J. Microencapsulation in yeast cells. J. Microencapsul. 1998, 15, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Miyamoto, A.; Hy, Q.L.; Tanaka, Y.; Fushimi, Y.; Yoshii, H. Microencapsulation of flavors by spray drying using Saccharomyces cerevisiae. J. Food Eng. 2017, 199, 36–41. [Google Scholar] [CrossRef]
- Errenst, C.; Petermann, M.; Kilzer, A. Encapsulation of limonene in yeast cells using the concentrated powder form technology. J. Supercrit. Fluids 2021, 168, 105076. [Google Scholar] [CrossRef]
- Pham-hoang, B.N.; Voilley, A.; Waché, Y. Molecule structural factors influencing the loading of flavoring compounds in a natural-preformed capsule: Yeast cells. Colloids Surf. B Biointerfaces 2016, 148, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Vulić, J.; Ranitovic, A.; Tumbas-Saponjac, V.; Cetkovic, G.; Canadanovic-Brunet, J.; Cvetkovic, D.; Jovic, B. Microencapsulation of beetroot pomace extractin cells of Saccharomyces cerevisiae. Chem. Ind. Chem. Eng. Q. 2019, 25, 321–327. [Google Scholar] [CrossRef]
- Lang, I.; Sassmann, S.; Schmidt, B.; Komis, G. Plasmolysis: Loss of turgor and beyond. Plants 2014, 3, 583–593. [Google Scholar] [CrossRef]
- Oparka, K.J. Plasmolysis: New insights into an old process. New Phytol. 1994, 126, 571–591. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, T.; Wang, Q.; Ren, T. Microorganism-based monodisperse microcapsules: Encapsulation of the fungicide tebuconazole and its controlled release properties. RSC Adv. 2015, 5, 25164–25170. [Google Scholar] [CrossRef]
- Ren, T.; Gou, J.; Sun, W.; Tao, X.; Tan, X.; Wang, P.; Zhang, Y.; He, H.; Yin, T.; Tang, X. Entrapping of nanoparticles in yeast cell wall microparticles for macrophage-targeted oral delivery of cabazitaxel. Mol. Pharm. 2018, 15, 2870–2882. [Google Scholar] [CrossRef]
- Paramera, E.; Konteles, S.; Karathanos, V. Microencapsulation of curcumin in cells of Saccharomyces cerevisiae. Food Chem. 2011, 125, 892–902. [Google Scholar] [CrossRef]
- Bitsikas, V.; Karachaliou, M.; Gournas, C.; Diallinas, G. Hypertonic conditions trigger transient plasmolysis, growth arrest and blockage of transporter endocytosis in Aspergillus nidulans and Saccharomyces cerevisiae. Mol. Membr. Biol. 2011, 28, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Inoue, C.; Ishiguro, M.; Ishiwaki, N.; Yamada, K. A Process for the Preparation of Microcapsules. EP0453316A1, 23 October 1991. [Google Scholar]
- Coradello, G.; Tirelli, N. Yeast cells in microencapsulation. General features and controlling factors of the encapsulation process. Molecules 2021, 26, 3123. [Google Scholar] [CrossRef] [PubMed]
- Pham-Hoang, B.N.; Romero-Guido, C.; Phan-Thi, H.; Waché, Y. Encapsulation in a natural, preformed, multi-component and complex capsule: Yeast cells. Appl. Microbiol. Biotechnol. 2013, 97, 6635–6645. [Google Scholar] [CrossRef] [PubMed]
- Sangwai, M.; Vavia, P. Effect of decisive formulation variables on bioencapsulation efficiency and integrity of yeast biocapsules for oral itraconazole delivery. J. Microencapsul. 2011, 28, 311–322. [Google Scholar] [CrossRef]
- Greenspan, P.; Fowler, S.D. Spectrofluorometric studies of the lipid probe, Nile Red. J. Lipid Res. 1985, 26, 781–789. [Google Scholar] [CrossRef]
- Rostron, K.A.; Lawrence, C.L. Nile Red staining of neutral lipids in yeast. In Histochemistry of Single Molecules: Methods and Protocols; Pellicciari, C., Biggiogera, M., Eds.; Springer: Dordrecht, The Netherlands, 2017; Volume 1560, pp. 219–229. [Google Scholar]
- Siloto, R.M.P.; Truksa, M.; He, X.H.; McKeon, T.; Weselake, R.J. Simple methods to detect triacylglycerol biosynthesis in a yeast-based recombinant system. Lipids 2009, 44, 963–973. [Google Scholar] [CrossRef]
- Simonin, H.; Beney, L.; Gervais, P. Controlling the membrane fluidity of yeasts during coupled thermal and osmotic treatments. Biotechnol. Bioeng. 2008, 100, 325–333. [Google Scholar] [CrossRef]
- Malmberg, C.G.; Maryott, A.A. Dielectric constant of water from 0 to 100 °C. J. Res. Nat. Bur. Stand. 1956, 56, 2641–2649. [Google Scholar] [CrossRef]
- Wyman, J. The dielectric constant of mixtures of ethyl alcohol and water from −5 to 40°. J. Am. Chem. Soc. 1931, 53, 3292–3301. [Google Scholar] [CrossRef]
- Akerlof, G. Dielectric constants of some organic solvent-water mixtures at various temperatures. J. Am. Chem. Soc. 1932, 54, 4125–4139. [Google Scholar] [CrossRef]
- Dardelle, G.; Normand, V.; Steenhoudt, M.; Bouquerand, P.-E.; Chevalier, M.; Baumgartner, P. Flavour-encapsulation and flavour-release performances of a commercial yeast-based delivery system. Food Hydrocoll. 2007, 21, 953–960. [Google Scholar] [CrossRef]
- Kilcher, G.; Delneri, D.; Duckham, C.; Tirelli, N. Probing (macro)molecular transport through cell walls. Faraday Discuss. 2008, 139, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Duša, F.; Witos, J.; Ruokonen, S.-K.; Wiedmer, S.K. Determination of the main phase transition temperature of phospholipids by nanoplasmonic sensing. Sci. Rep. 2018, 8, 14815. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Perdue, E.M.; Pavlostathis, S.G.; Araujo, R. Physicochemical properties of selected monoterpenes. Environ. Int. 1998, 24, 353–358. [Google Scholar] [CrossRef]
- Griffin, S.; Wyllie, S.G.; Markham, J. Determination of octanol-water partition coefficient for terpenoids using reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1999, 864, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.A.; Hawkins, J.E. Physical and Thermodynamic Properties of Terpenes. IV. The Dielectric Constant, Refractive Index and Density of Some Terpenes. J. Am. Chem. Soc. 1954, 76, 4856–4858. [Google Scholar] [CrossRef]
- Ramirez-Castrillon, M.; Jaramillo-Garcia, V.P.; Barros, H.L.; Henriques, J.A.P.; Stefani, V.; Valente, P. Nile Red incubation time before reading fluorescence greatly influences the yeast neutral lipids quantification. Front. Microbiol. 2021, 12, 619313. [Google Scholar] [CrossRef]
- Hicks, R.H.; Chuck, C.J.; Scott, R.J.; Leak, D.J.; Henk, D.A. Comparison of Nile Red and cell size analysis for high-throughput lipid estimation within oleaginous yeast. Eur. J. Lipid Sci. Technol. 2019, 121, 1800355. [Google Scholar] [CrossRef]
- Sitepu, I.R.; Ignatia, L.; Franz, A.K.; Wong, D.M.; Faulina, S.A.; Tsui, M.; Kanti, A.; Boundy-Mills, K. An improved high-throughput Nile red fluorescence assay for estimating intracellular lipids in a variety of yeast species. J. Microbiol. Methods 2012, 91, 321–328. [Google Scholar] [CrossRef]
- Zhao, C.; Luo, M.T.; Huang, C.; Chen, X.F.; Xiong, L.; Li, H.L.; Chen, X.D. Determining intracellular lipid content of different oleaginous yeasts by one simple and accurate Nile Red fluorescent method. Prep. Biochem. Biotechnol. 2019, 49, 597–605. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coradello, G.; Setti, C.; Donno, R.; Ghibaudi, M.; Catalano, F.; Tirelli, N. A Quantitative Re-Assessment of Microencapsulation in (Pre-Treated) Yeast. Molecules 2024, 29, 539. https://doi.org/10.3390/molecules29020539
Coradello G, Setti C, Donno R, Ghibaudi M, Catalano F, Tirelli N. A Quantitative Re-Assessment of Microencapsulation in (Pre-Treated) Yeast. Molecules. 2024; 29(2):539. https://doi.org/10.3390/molecules29020539
Chicago/Turabian StyleCoradello, Giulia, Chiara Setti, Roberto Donno, Matilde Ghibaudi, Federico Catalano, and Nicola Tirelli. 2024. "A Quantitative Re-Assessment of Microencapsulation in (Pre-Treated) Yeast" Molecules 29, no. 2: 539. https://doi.org/10.3390/molecules29020539
APA StyleCoradello, G., Setti, C., Donno, R., Ghibaudi, M., Catalano, F., & Tirelli, N. (2024). A Quantitative Re-Assessment of Microencapsulation in (Pre-Treated) Yeast. Molecules, 29(2), 539. https://doi.org/10.3390/molecules29020539