
Synthetic Studies toward 5,6,7,3′,4′-Monomethoxytetrahydroxyflavones: Synthesis of Pedalitin
 , and
, and
Abstract
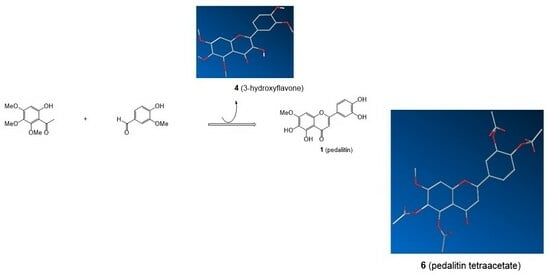
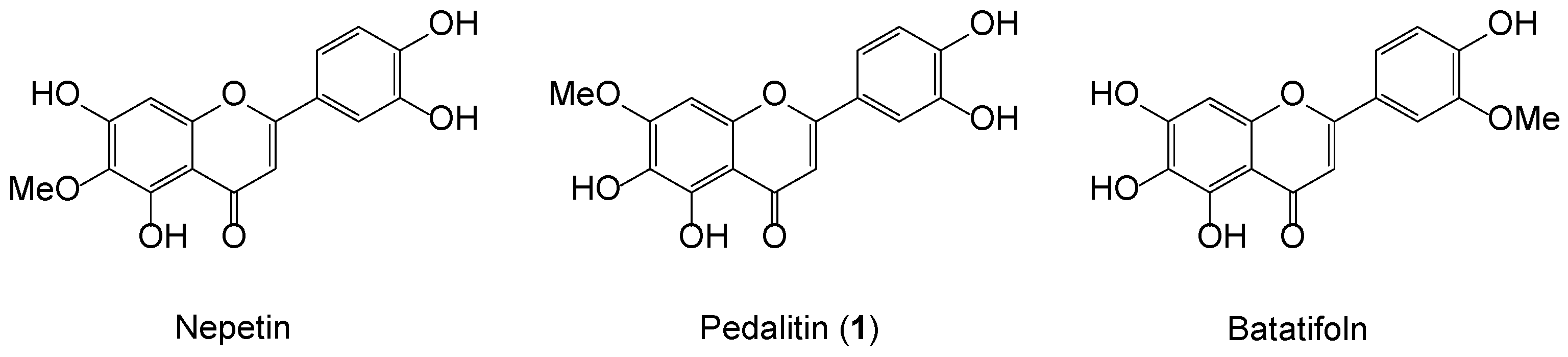
1. Introduction
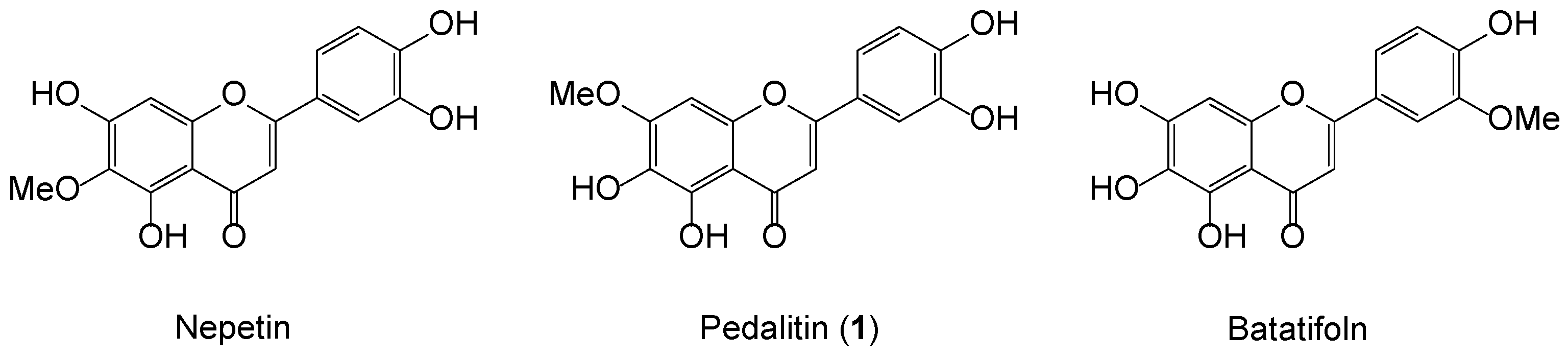
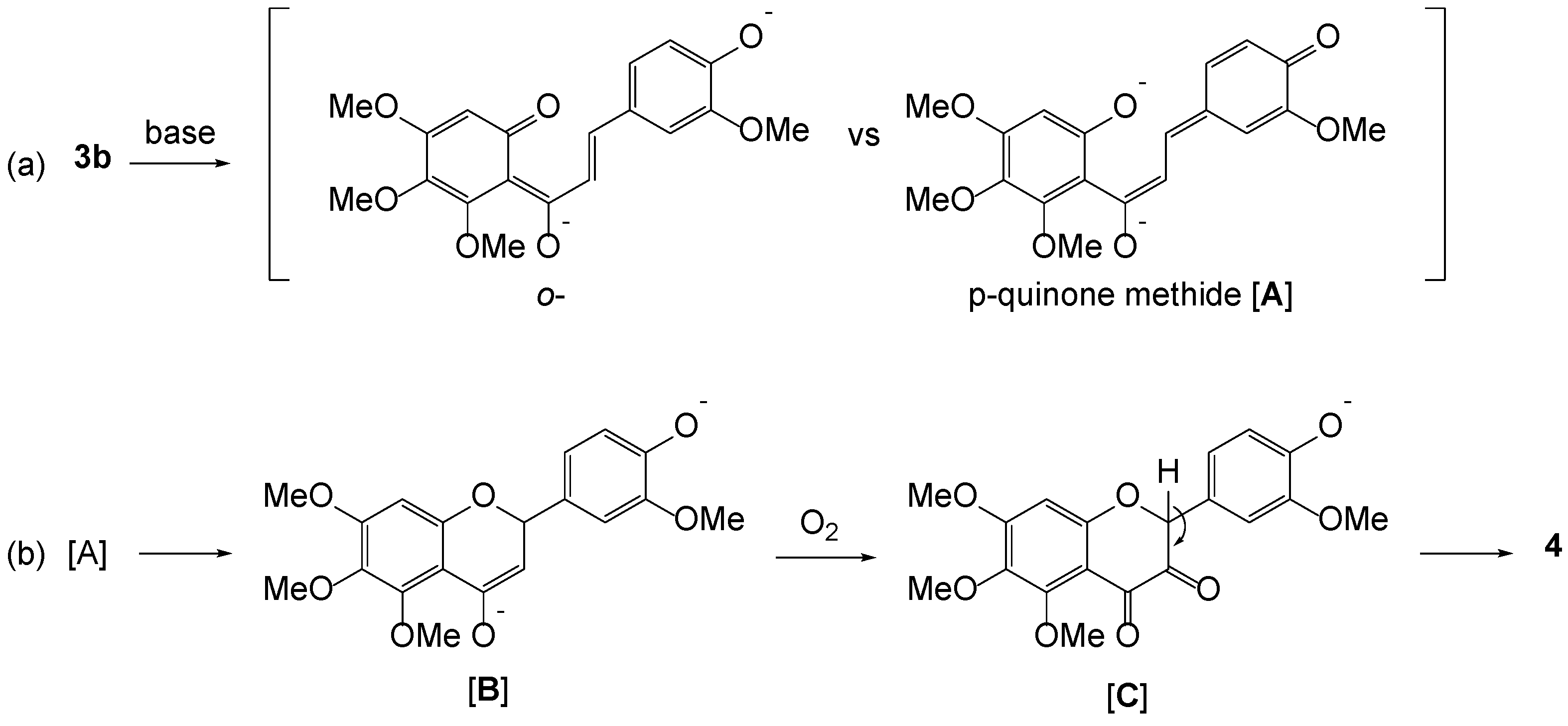
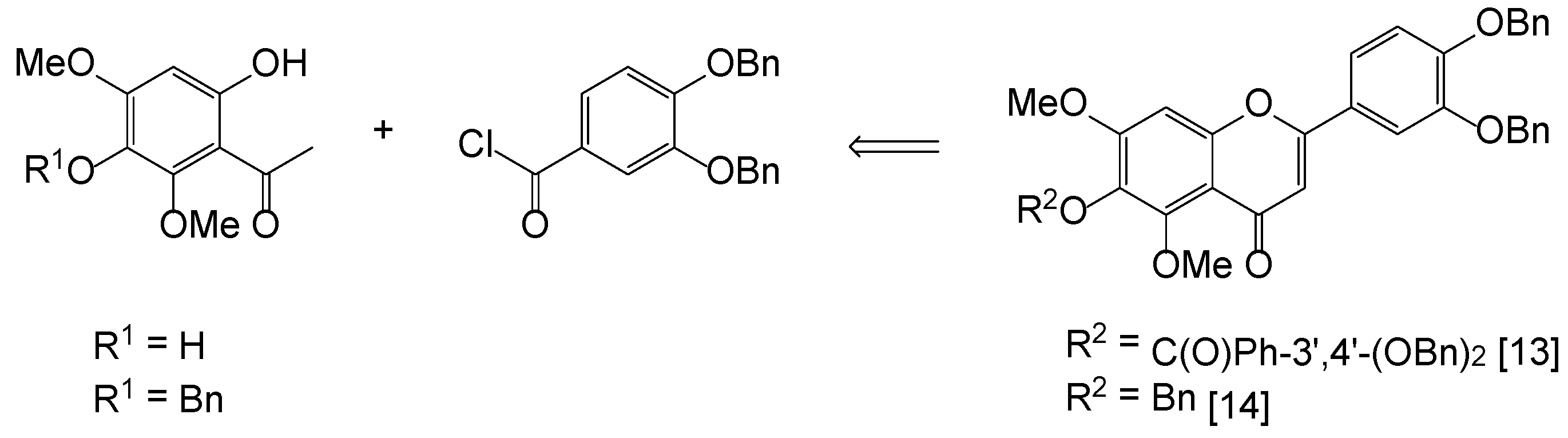
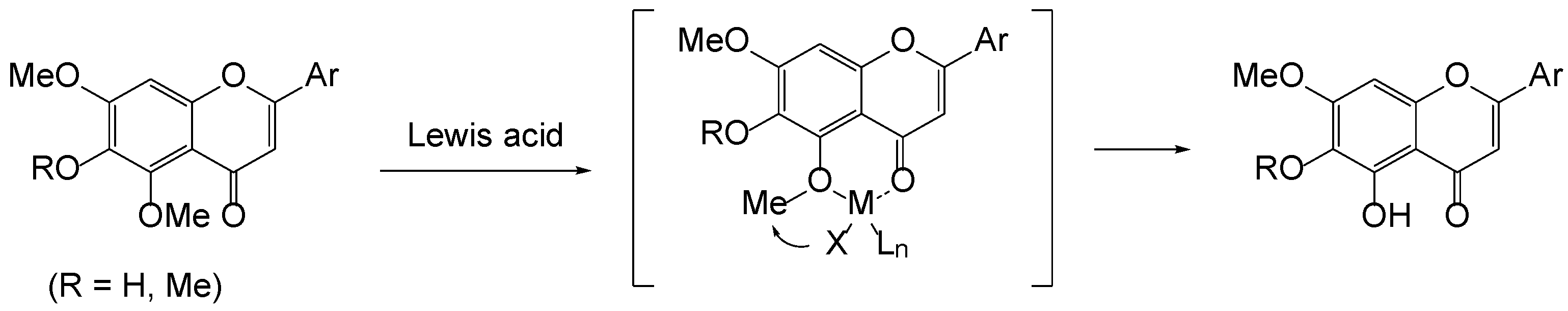
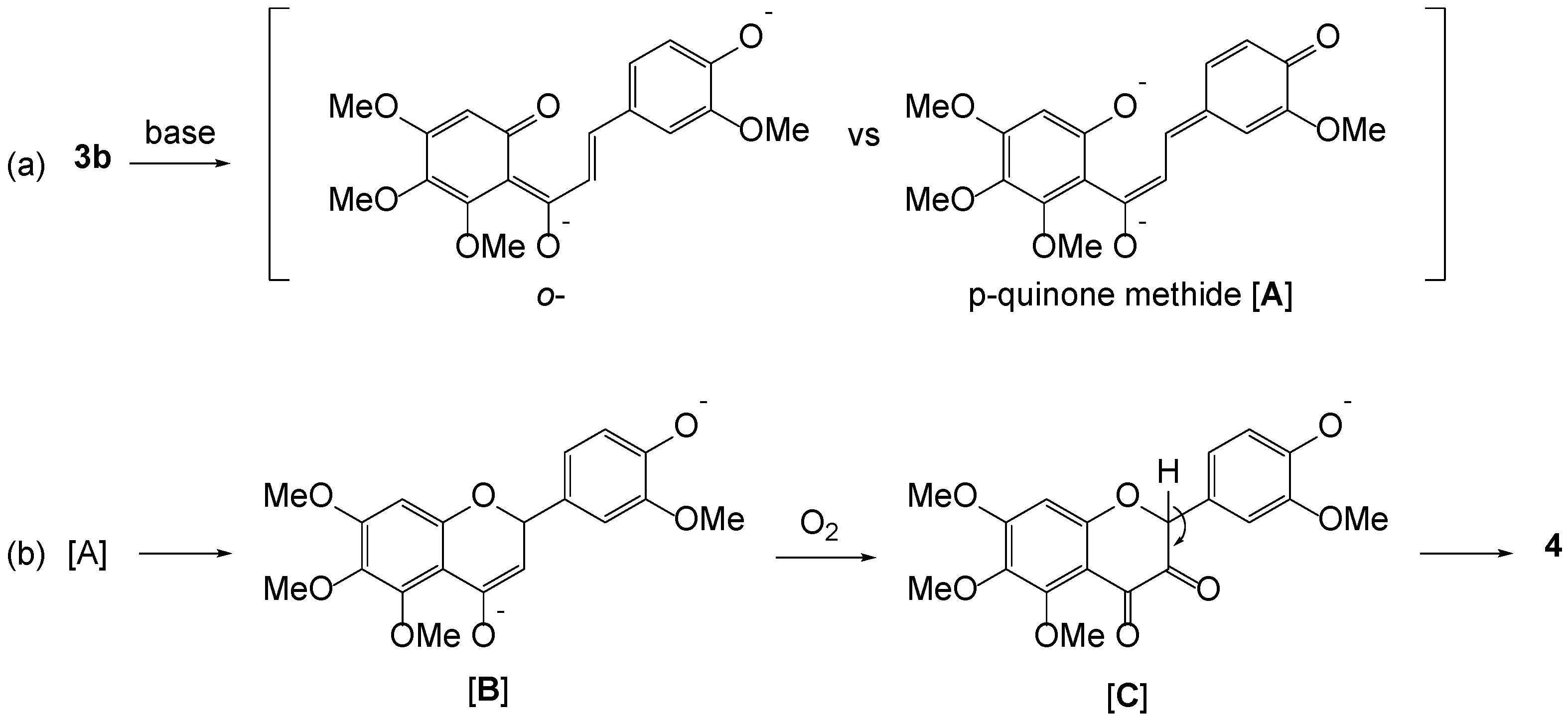
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Preparation of 3,6-Dihydroxy-2,4-dimethoxyacetophenone 2a and 6-hydroxy-2,3,4-trimethoxyacetophenone 2b
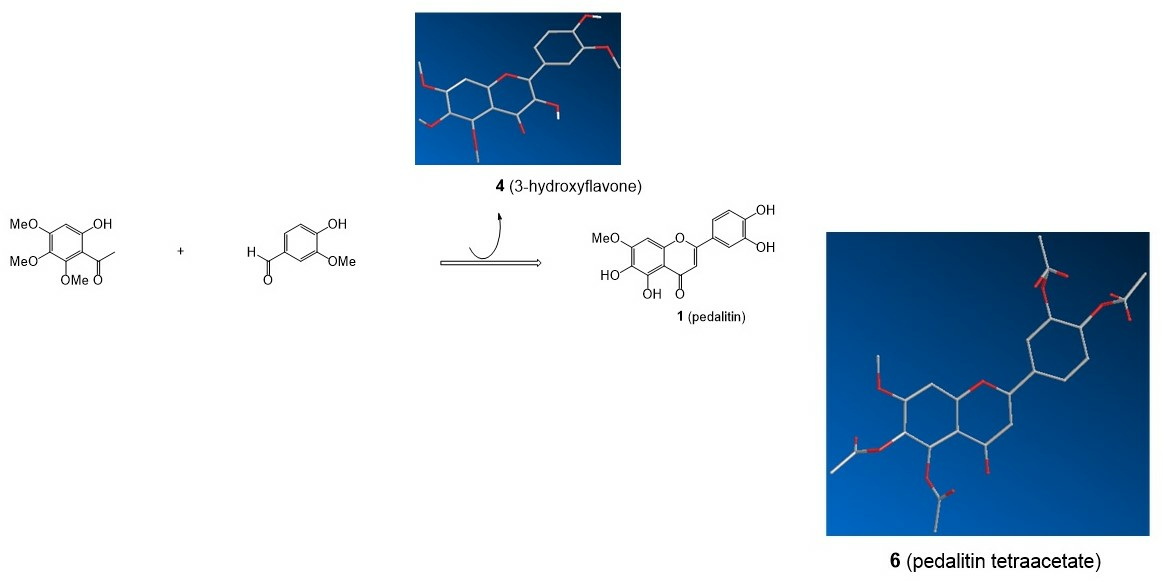
3.3. Aldol Condensation of 2a with Vanillin (Scheme 1)
3.4. Aldol Condensation of 2b with Vanillin (Entry 2, Table 1)
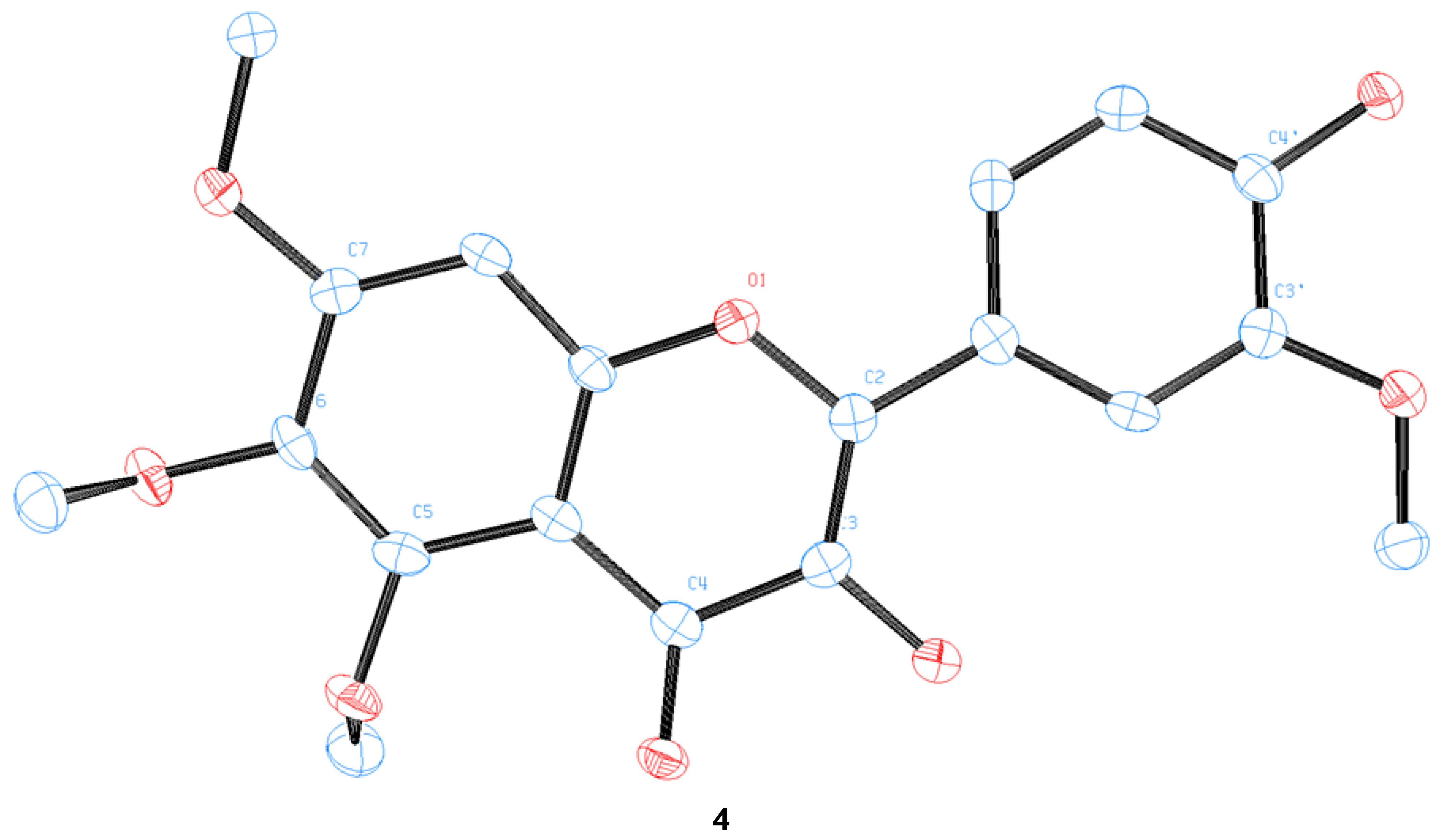
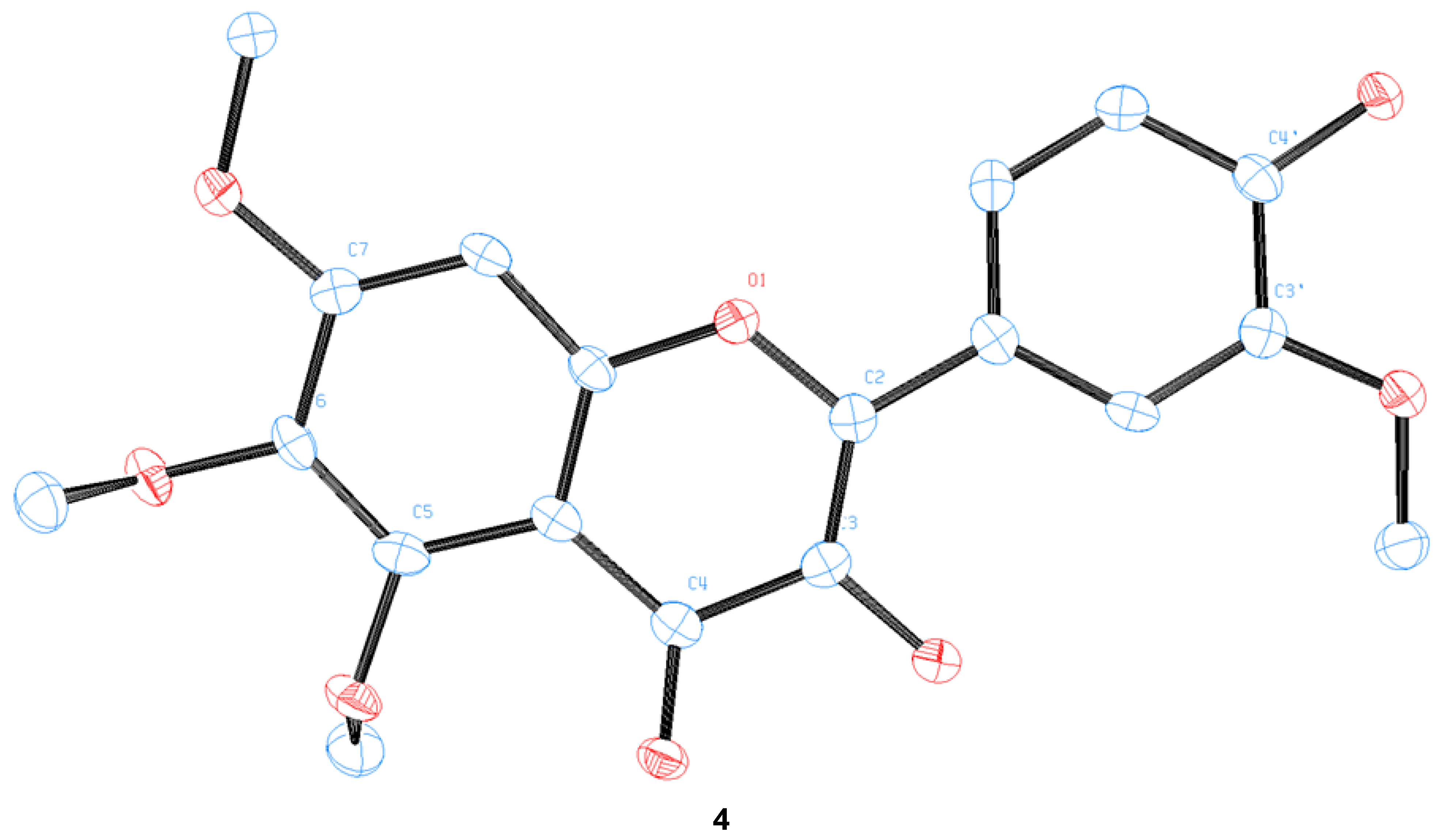
3.5. Preparation of a Single Crystal of 4
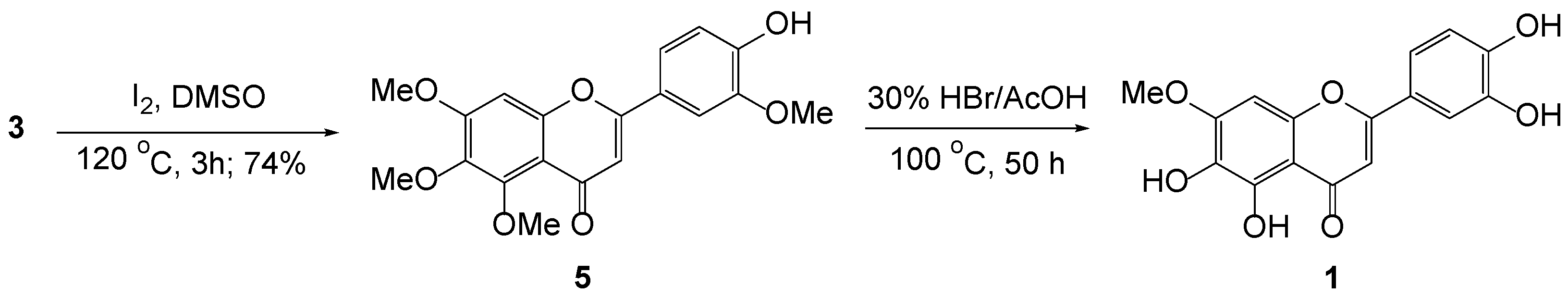
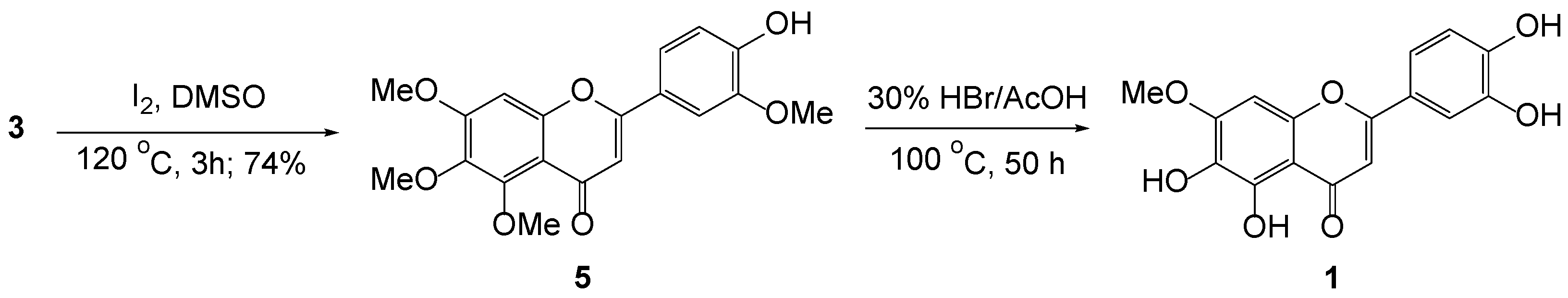
3.6. Intramolecular Cyclization of 3b to 5
3.7. Demethylation of 5 to Pedalitin 1
- (i)
- The reaction showed the consumption of starting material and multiple spots on TLC after 8 h;
- (ii)
- After 20 h, the reaction showed the consumption of multiple spots and enriched a major spot apparently;
- (iii)
- No further progress was observed after an additional 12 h.
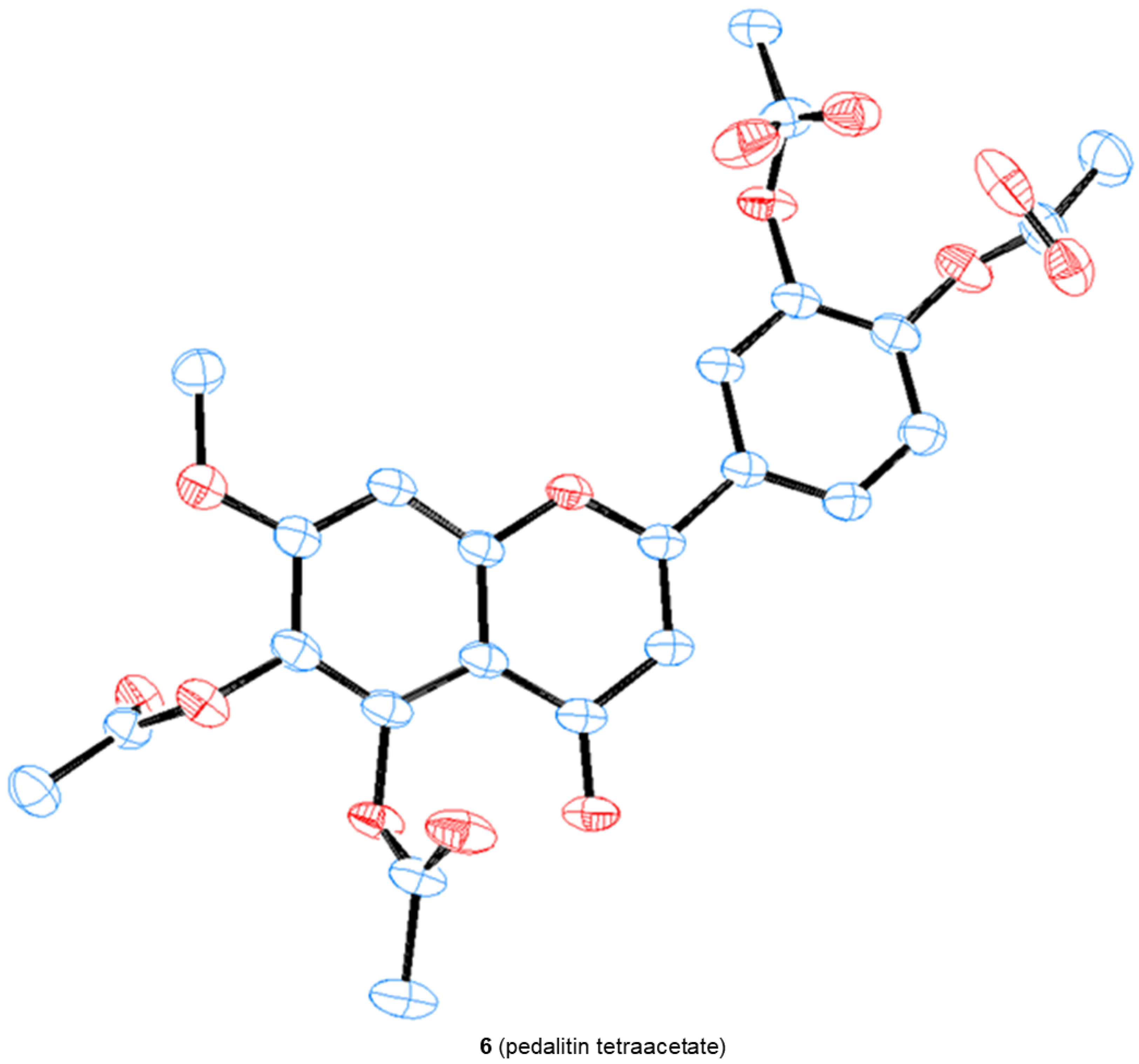
3.8. Crystal Structure Determination of 1 (as Tetraacetate 6)
3.9. Crystallographic Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, N.; Doseff, A.I.; Grotewold, E. Flavones: From Biosynthesis to Health Benefits. Plants 2016, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Ahn-Jarvis, J.H.; Parihar, A.; Doseff, A.I. Dietary Flavonoids for Immunoregulation and Cancer: Food Design for Targeting Disease. Antioxidants 2019, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lo, C.-Y.; Ho, C.-T. Hydroxylated Polymethoxyflavones and Methylated Flavonoids in Sweet Orange (Citrus sinensis) Peel. J. Agric. Food Chem. 2006, 54, 4176–4185. [Google Scholar] [CrossRef] [PubMed]
- Burapan, S.; Kim, M.; Han, J. Demethylation of polymethoxyflavones by human gut bacterium, Blautia sp. MRG-PMF1. J. Agric. Food Chem. 2017, 65, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tian, G.; Zhao, C.; Han, Y.; DiMarco-Crook, C.; Lu, C.; Bao, Y.; Li, C.; Xiao, H.; Zheng, J. Characterization of polymethoxyflavone demethylation during drying processes of citrus peels. Food Funct. 2019, 10, 5707–5717. [Google Scholar] [CrossRef]
- Wollenweber, E.; Dietz, V.H. Occurrence and distribution of free flavonoid aglycones in plants. Phytochemistry 1981, 20, 869–932. [Google Scholar] [CrossRef]
- Da, Q.; Shao, J.; Zhang, P.; Zhang, J.; Ma, H.; Jing, L. The design, synthesis, antioxidant, and antihypoxia activities of two new hydroxydaidzein derivatives. J. Chem. Res. 2022, 46, 17475198221131528. [Google Scholar] [CrossRef]
- Chen, D.Z.; Yang, J.; Yang, B.; Wu, Y.S.; Wu, T. Total synthesis of baicalein. J. Asian Nat. Prod. Res. 2010, 12, 124–128. [Google Scholar] [CrossRef]
- Alshammari, M.D.; Kucheryavy, P.V.; Ashpole, N.M.; Colby, D.A. Synthesis, biological evaluation, and NMR studies of 3-fluorinated derivatives of 3′,4′,5′-trihydroxyflavone and 3′,4′,5′-trimethoxyflavone. Bioorg. Med. Chem. Lett. 2021, 32, 127720. [Google Scholar] [CrossRef]
- Oyama, K.; Yoshida, K.; Kondo, T. Recent Progress in the Synthesis of Flavonoids: From Monomers to Supra-Complex Molecules. Curr. Org. Chem. 2011, 15, 2567–2607. [Google Scholar] [CrossRef]
- Lin, L.; Dong, Y.; Zhao, H.; Wen, L.; Yang, B.; Zhao, M. Comparative evaluation of rosmarinic acid, methyl rosmarinate and pedalitin isolated from Rabdosia serra (MAXIM.) HARA as inhibitors of tyrosinase and α-glucosidase. Food Chem. 2011, 129, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Herz, W.; Santhanam, P.S.; Wagner, H.; Höer, R.; Hörhammer, L.; Farkas, L. Isolierung, Struktur und Synthese von 5.6.7.4′-Tetrahydroxy-3′-methoxy-flavon (Batatifolin), einem neuen Flavon aus Mikania batatifolia D.C. Chem. Ber. 1970, 103, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Nakayama, M.; Matsui, T.; Masumura, M.; Horie, T. ペダリチンおよび関連化合物の合成. 日本化學雜誌 1969, 90, 1270. (In Japanese) [Google Scholar] [CrossRef]
- Munekazu, I.; Shin, M.; Toshiyuki, T. Synthetic Studies on Flavone Derivatives. XV. Isomerization of Chalcones into Flavanones in Methyl Cellosolve-Phosphoric Acid. Chem. Pharm. Bull. 1984, 32, 1472–1476. [Google Scholar]
- Zhang, W.; Liu, Y.; Chen, X.; Bergmeier, S.C. Novel inhibitors of basal glucose transport as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 2191–2194. [Google Scholar] [CrossRef]
- Asakawa, T.; Sagara, H.; Kanakogi, M.; Hiza, A.; Tsukaguchi, Y.; Ogawa, T.; Nakayama, M.; Ouchi, H.; Inai, M.; Kan, T. Practical Synthesis of Polymethylated Flavones: Nobiletin and Its Desmethyl Derivatives. Org. Process Res. Dev. 2019, 23, 595–602. [Google Scholar] [CrossRef]
- Horie, T.; Tominaga, H.; Kawamura, Y.; Yamada, T. Studies of the selective O-alkylation and dealkylation of flavonoids. 13. An improved method for synthesizing 5,6,7-trihydroxyflavones from 6-hydroxy-5,7-dimethoxyflavones. J. Org. Chem. 1992, 57, 3343–3347. [Google Scholar] [CrossRef]
- Martin-Benlloch, X.; Elhabiri, M.; Lanfranchi, D.A.; Davioud-Charvet, E. A Practical and Economical High-Yielding, Six-Step Sequence Synthesis of a Flavone: Application to the Multigram-Scale Synthesis of Ladanein. Org. Process Res. Dev. 2014, 18, 613–617. [Google Scholar] [CrossRef]
- Li, S.; Pan, M.H.; Lai, C.S.; Lo, C.Y.; Dushenkov, S.; Ho, C.T. Isolation and syntheses of polymethoxyflavones and hydroxylated polymethoxyflavones as inhibitors of HL-60 cell lines. Bioorg. Med. Chem. 2007, 15, 3381–3389. [Google Scholar] [CrossRef]
- Wang, X.; Li, D.; Cao, Y.; Ho, C.T.; Huang, Q. Biotransformation and Quantification of Sinensetin and Its Metabolites in Plasma, Urine, and Feces of Rats. J. Agric. Food Chem. 2021, 69, 14143–14150. [Google Scholar] [CrossRef] [PubMed]
- Waghmode, S.B.; Mahale, G.; Patil, V.P.; Renalson, K.; Singh, D. Efficient Method for Demethylation of Aryl Methyl Ether Using Aliquat-336. Synth. Commun. 2013, 43, 3272–3280. [Google Scholar] [CrossRef]
- Righi, G.; Antonioletti, R.; Silvestri, I.P.; D’Antona, N.; Lambusta, D.; Bovicelli, P. Convergent synthesis of mosloflavone, negletein and baicalein from crysin. Tetrahedron 2010, 66, 1294–1298. [Google Scholar] [CrossRef]
- Wei, G.-J.; Sheen, J.-F.; Lu, W.-C.; Hwang, L.S.; Ho, C.-T.; Lin, C.-I. Identification of Sinensetin Metabolites in Rat Urine by an Isotope-Labeling Method and Ultrahigh-Performance Liquid Chromatography–Electrospray Ionization Mass Spectrometry. J. Agric. Food. Chem. 2013, 61, 5016–5021. [Google Scholar] [CrossRef] [PubMed]
- Ichino, K.; Tanaka, H.; Ito, K.; Tanaka, T.; Mizuno, M. Synthesis of helilandin B, pashanone, and their isomers. J. Nat. Prod. 1988, 51, 906–914. [Google Scholar] [CrossRef]
- Bulut, O.; Yilmaz, M.D. Concise synthesis of quercetagetin (3,3′,4′,5,6,7-hexahydroxyflavone) with antioxidant and antibacterial activities. Results Chem. 2021, 3, 100255. [Google Scholar] [CrossRef]
- Hierold, J.; Baek, S.; Rieger, R.; Lim, T.G.; Zakpur, S.; Arciniega, M.; Lee, K.W.; Huber, R.; Tietze, L.F. Design, synthesis, and biological evaluation of quercetagetin analogues as JNK1 inhibitors. Chem. Eur. J. 2015, 21, 16887–16894. [Google Scholar] [CrossRef]
- Pereira, A.M.; Cidade, H.; Tiritan, M.E. Stereoselective Synthesis of Flavonoids: A Brief Overview. Molecules 2023, 28, 426. [Google Scholar] [CrossRef]
- Xiong, W.; Wang, X.; Shen, X.; Hu, C.; Wang, X.; Wang, F.; Zhang, G.; Wang, C. Synthesis of flavonols via pyrrolidine catalysis: Origins of the selectivity for flavonol versus aurone. J. Org. Chem. 2020, 85, 13160–13176. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Recent developments in 1,6-addition reactions of para-quinone methides (p-QMs). Org. Chem. Front. 2020, 7, 1743–1778. [Google Scholar] [CrossRef]
- Nagayoshi, H.; Murayama, N.; Kakimoto, K.; Tsujino, M.; Takenaka, S.; Katahira, J.; Lim, Y.-R.; Kim, D.; Yamazaki, H.; Komori, M.; et al. Oxidation of Flavone, 5-Hydroxyflavone, and 5,7-Dihydroxyflavone to Mono-, Di-, and Tri-Hydroxyflavones by Human Cytochrome P450 Enzymes. Chem. Res. Toxicol. 2019, 32, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhang, Q.; Wang, J.-R.; Mei, X. Cocrystals of Baicalein with Higher Solubility and Enhanced Bioavailability. Cryst. Growth Des. 2017, 17, 1893–1901. [Google Scholar] [CrossRef]
- Xu, J.; Shi, Q.; Wang, Y.; Wang, Y.; Xin, J.; Cheng, J.; Li, F. Recent Advances in Pharmaceutical Cocrystals: A Focused Review of Flavonoid Cocrystals. Molecules 2023, 28, 613. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.; Bekkouch, K.; Thozet, A. Structure of 2-hydroxybiphenyl. Acta Cryst. 1987, C43, 980–982. [Google Scholar] [CrossRef]
- Grein, F. Twist Angles and Rotational Energy Barriers of Biphenyl and Substituted Biphenyls. J. Phys. Chem. A 2002, 106, 3823–3827. [Google Scholar] [CrossRef]
- Huang, W.-H.; Chien, P.-Y.; Yang, C.-H.; Lee, A.-R. Novel synthesis of flavonoids of Scutellaria baicalensis GEORGI. Chem. Pharm. Bull. 2003, 51, 339–340. [Google Scholar] [CrossRef]
- Pham, T.-A.; Che, H.; Phan, P.-T.; Lee, J.-W.; Kim, S.-S.; Park, H. Oroxylin A analogs exhibited strong inhibitory activities against iNOS-mediated nitric oxide (NO) production. Bioorg. Med. Chem. Lett. 2012, 22, 2534–2535. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
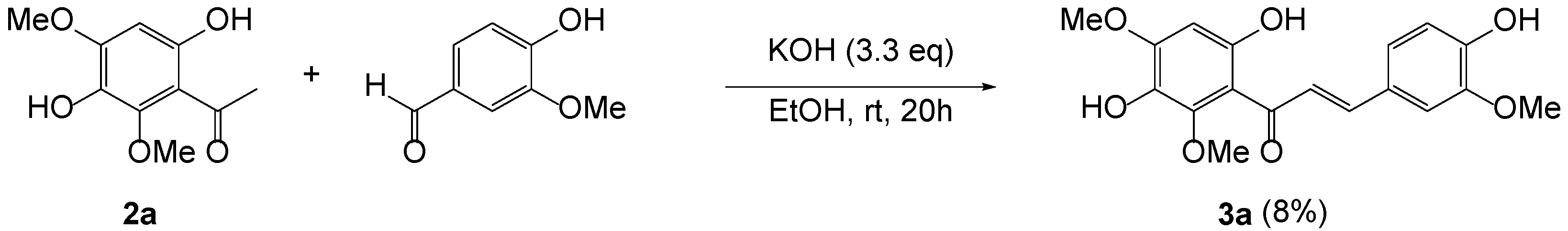{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry a | Conditions | Yield (%) of 3b/4 |
| 1 | KOH (3.3 eq), EtOH/H2O (5/2), rt, 20 h | 15/10 |
| 2 | KOH (3.0 eq), EtOH, reflux, 20 h | 28/20 |
| 3 | KOBut (3.0 eq), THF, rt, 12 h | 12/5 |
| 4 | NaH (3.0 eq), THF, rt, 12 h | 15/8 |
| 5 | LiHMDS (3.5 eq), THF, −78 °C to rt, 20 h | 18/3 |
| 6 | THF; freeze-pump-thaw (3 cycles); LiHMDS (3.5 eq), −78 °C to rt, 20 h; under Ar | 23/- |
| Geometric Data | 4 | 6 |
|---|---|---|
| Empirical formula | C19H18O8 | C24H20O11 |
| Formula weight | 374.33 | 484.40 |
| Crystal system | Orthorhombic | triclinic |
| Space group | Pna21 | P-1 |
| Unit cell dimensions | a = 8.2312(19) Å | a = 8.94440(10) Å |
| b = 16.306(3) Å | b = 10.9710(2) Å | |
| c = 12.099(3) Å | c = 13.8699(2) Å | |
| Volume | 1623.9(6) Å3 | 1164.11(4) Å3 |
| Density (calculated) | 1.531 g/cm3 | 1.382 g/cm3 |
| Reflections collected | 22,072 | 35,274 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0492, wR2 = 0.1038 | R1 = 0.0543, wR2 = 0.1661 |
| R indices (all data) | R1 = 0.0552, wR2 = 0.1065 | R1 = 0.0630, wR2 = 0.1754 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamma, K.R.; Cho, J.; Won, H.J.; Nam, S.-Y.; Le, N.H.; Jung, J.H.; Lee, K.-I. Synthetic Studies toward 5,6,7,3′,4′-Monomethoxytetrahydroxyflavones: Synthesis of Pedalitin. Molecules 2024, 29, 513. https://doi.org/10.3390/molecules29020513
Kamma KR, Cho J, Won HJ, Nam S-Y, Le NH, Jung JH, Lee K-I. Synthetic Studies toward 5,6,7,3′,4′-Monomethoxytetrahydroxyflavones: Synthesis of Pedalitin. Molecules. 2024; 29(2):513. https://doi.org/10.3390/molecules29020513
Chicago/Turabian StyleKamma, Koteswara Rao, Joungmo Cho, Hyo Jun Won, So-Yeon Nam, Ngan Hong Le, Je Hyeong Jung, and Kee-In Lee. 2024. "Synthetic Studies toward 5,6,7,3′,4′-Monomethoxytetrahydroxyflavones: Synthesis of Pedalitin" Molecules 29, no. 2: 513. https://doi.org/10.3390/molecules29020513
APA StyleKamma, K. R., Cho, J., Won, H. J., Nam, S.-Y., Le, N. H., Jung, J. H., & Lee, K.-I. (2024). Synthetic Studies toward 5,6,7,3′,4′-Monomethoxytetrahydroxyflavones: Synthesis of Pedalitin. Molecules, 29(2), 513. https://doi.org/10.3390/molecules29020513