
Novel Flurbiprofen Derivatives as Antioxidant and Anti-Inflammatory Agents: Synthesis, In Silico, and In Vitro Biological Evaluation

 ,
,  ,
,  ,
,  , ,
, ,  , and
, and
Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis
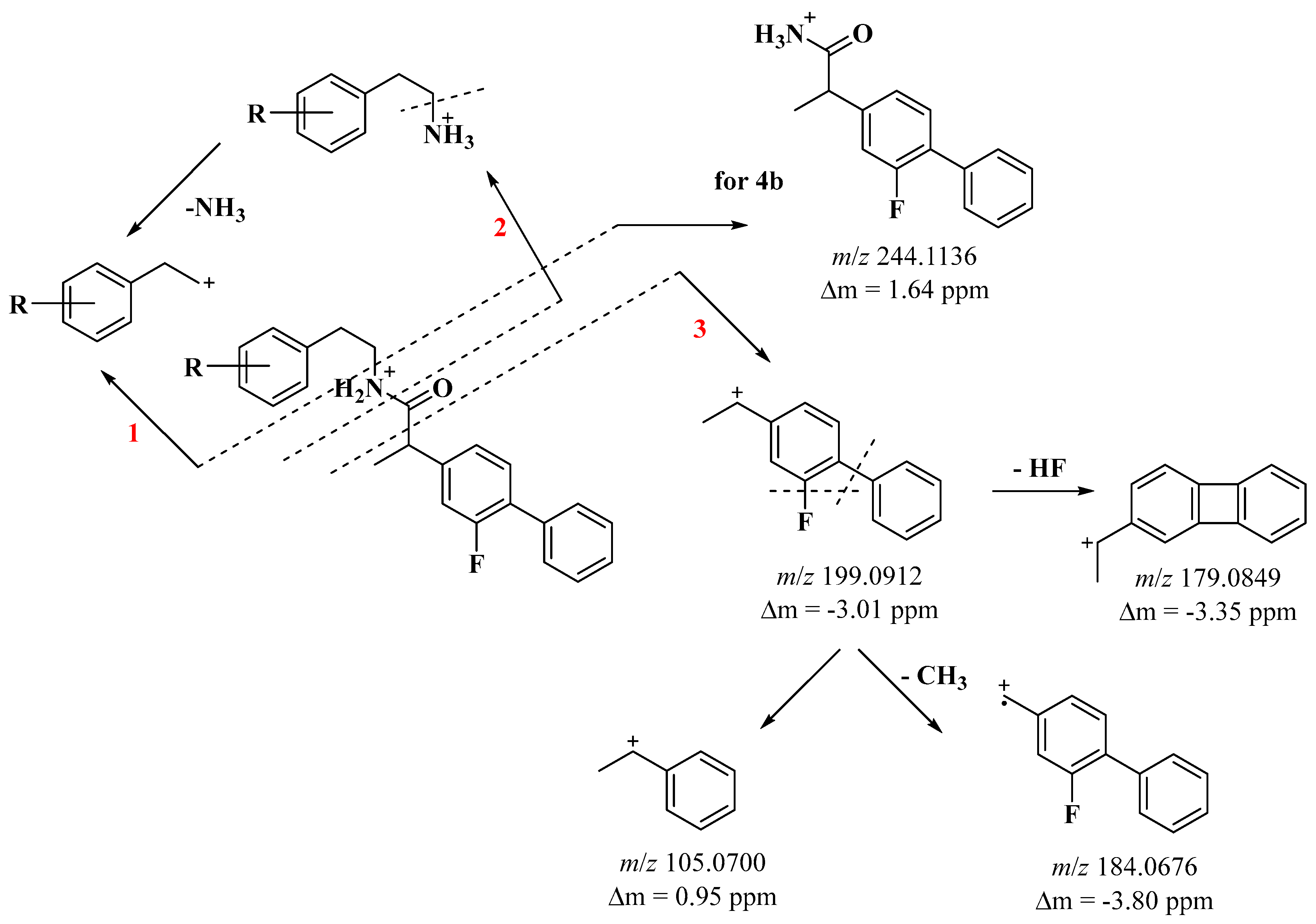
2.2. Mass Analysis
2.3. In Vitro Biological Assessment
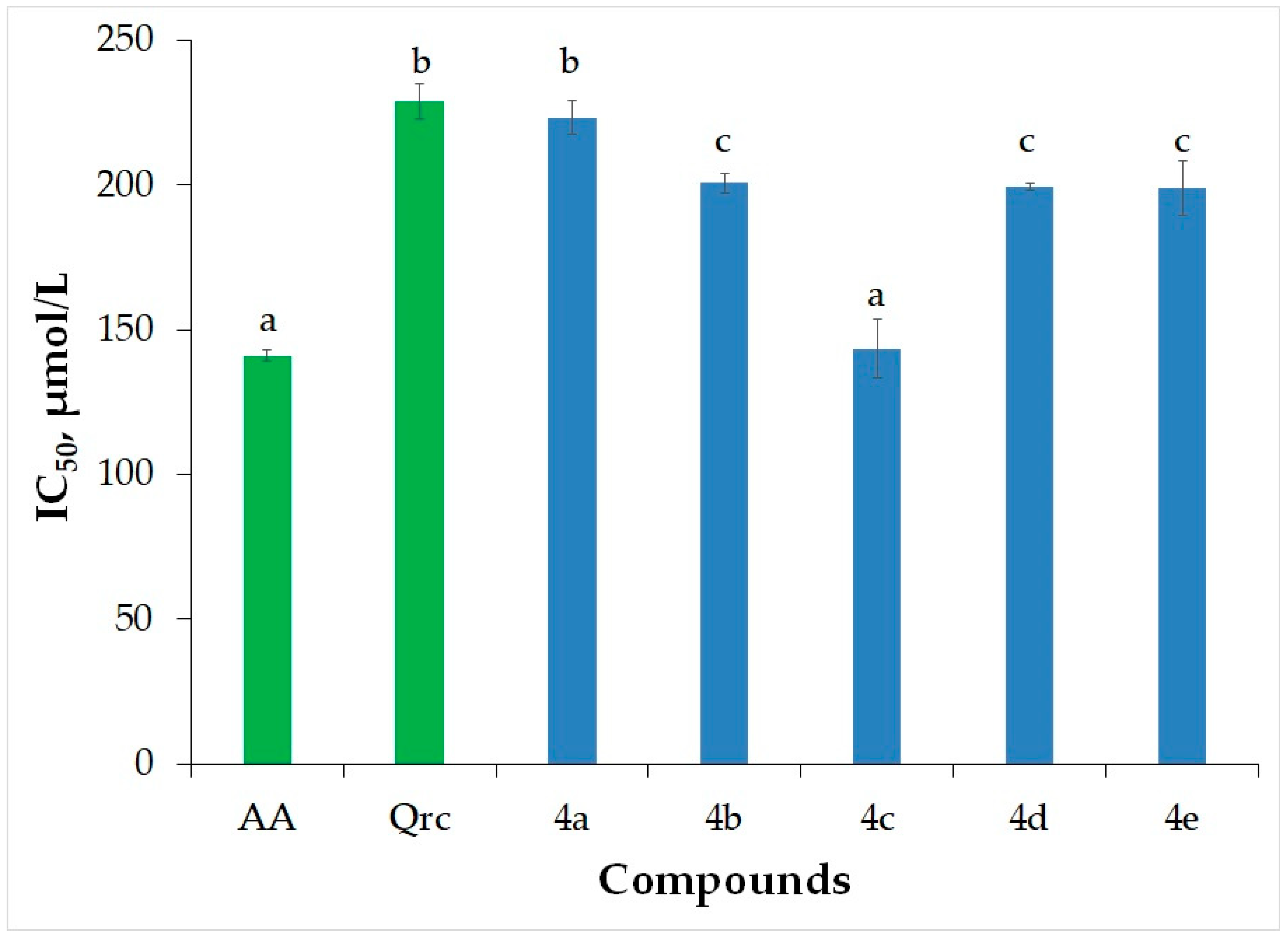
2.3.1. Hydrogen Peroxide Scavenging Activity (HPSA)
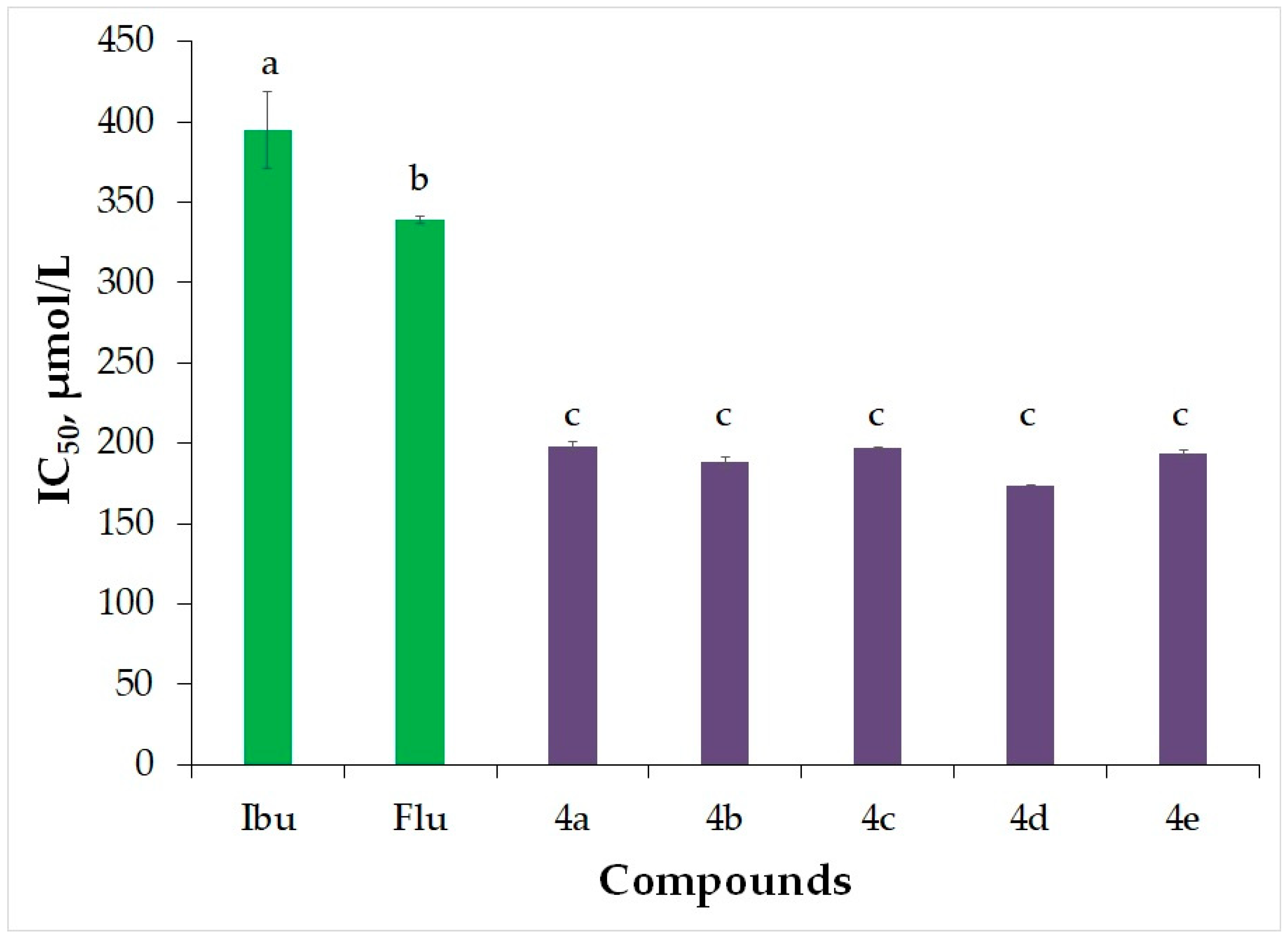
2.3.2. Inhibition of Albumin Denaturation (IAD)
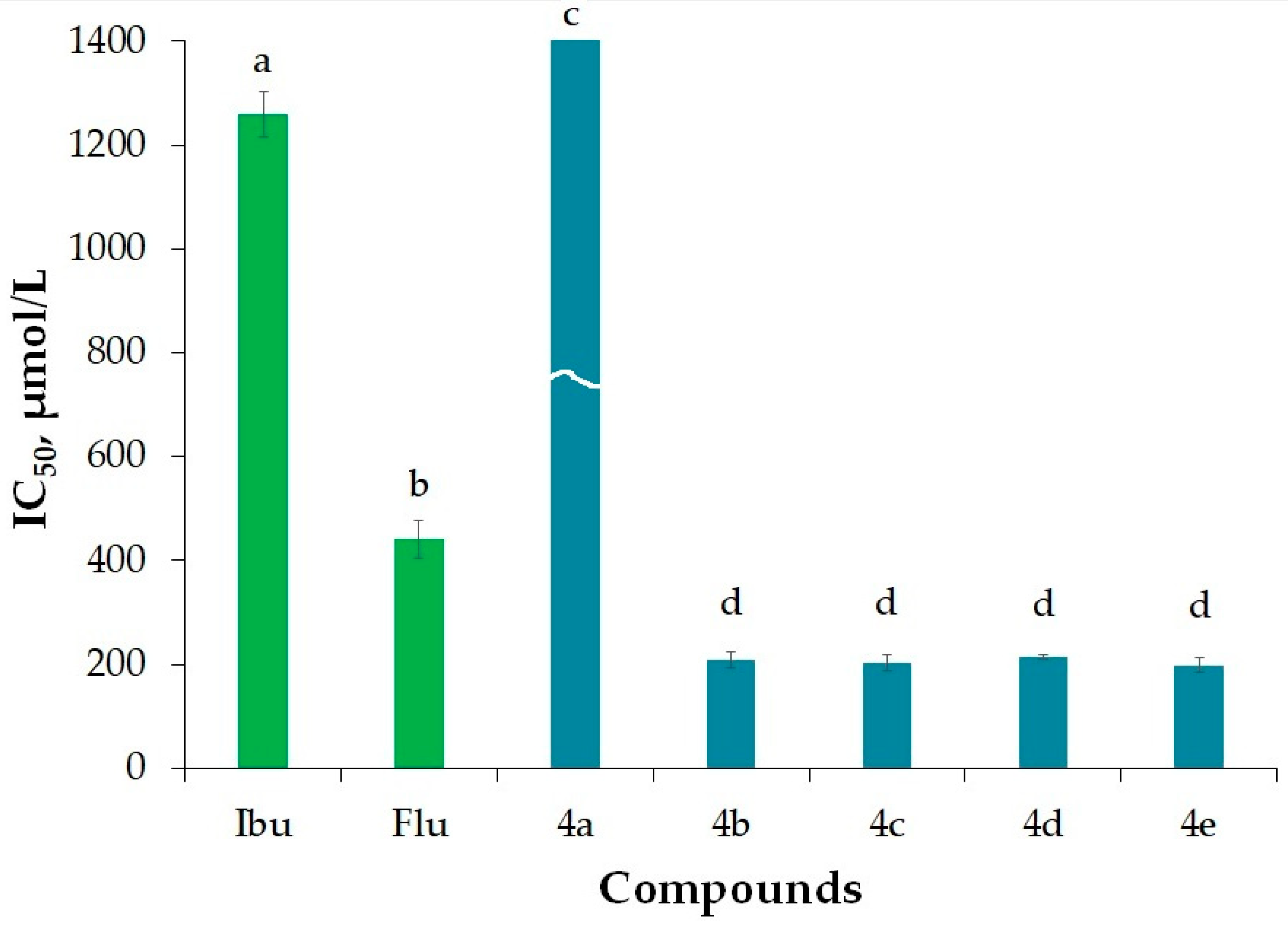
2.3.3. Antitryptic Activity (ATA)
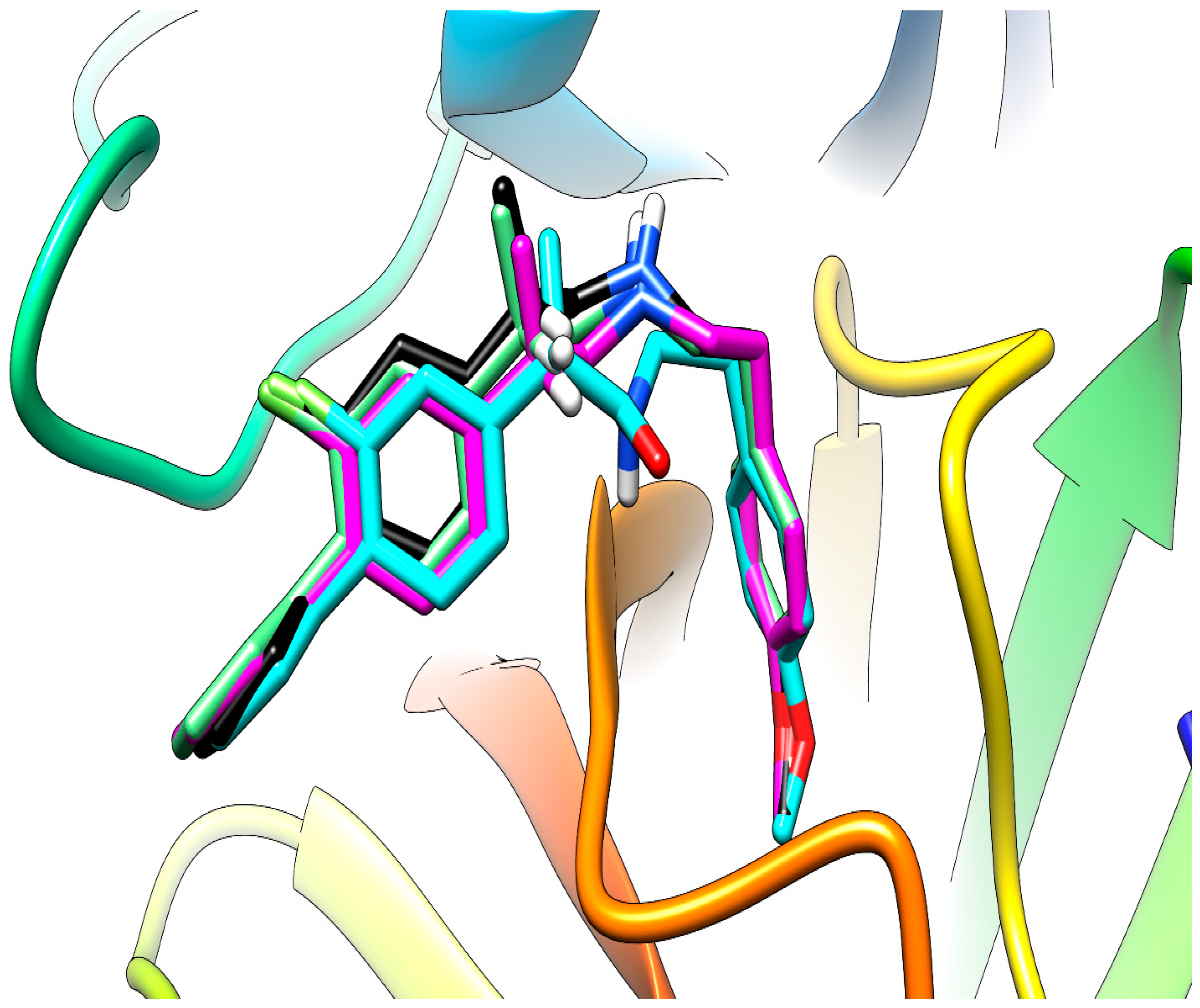
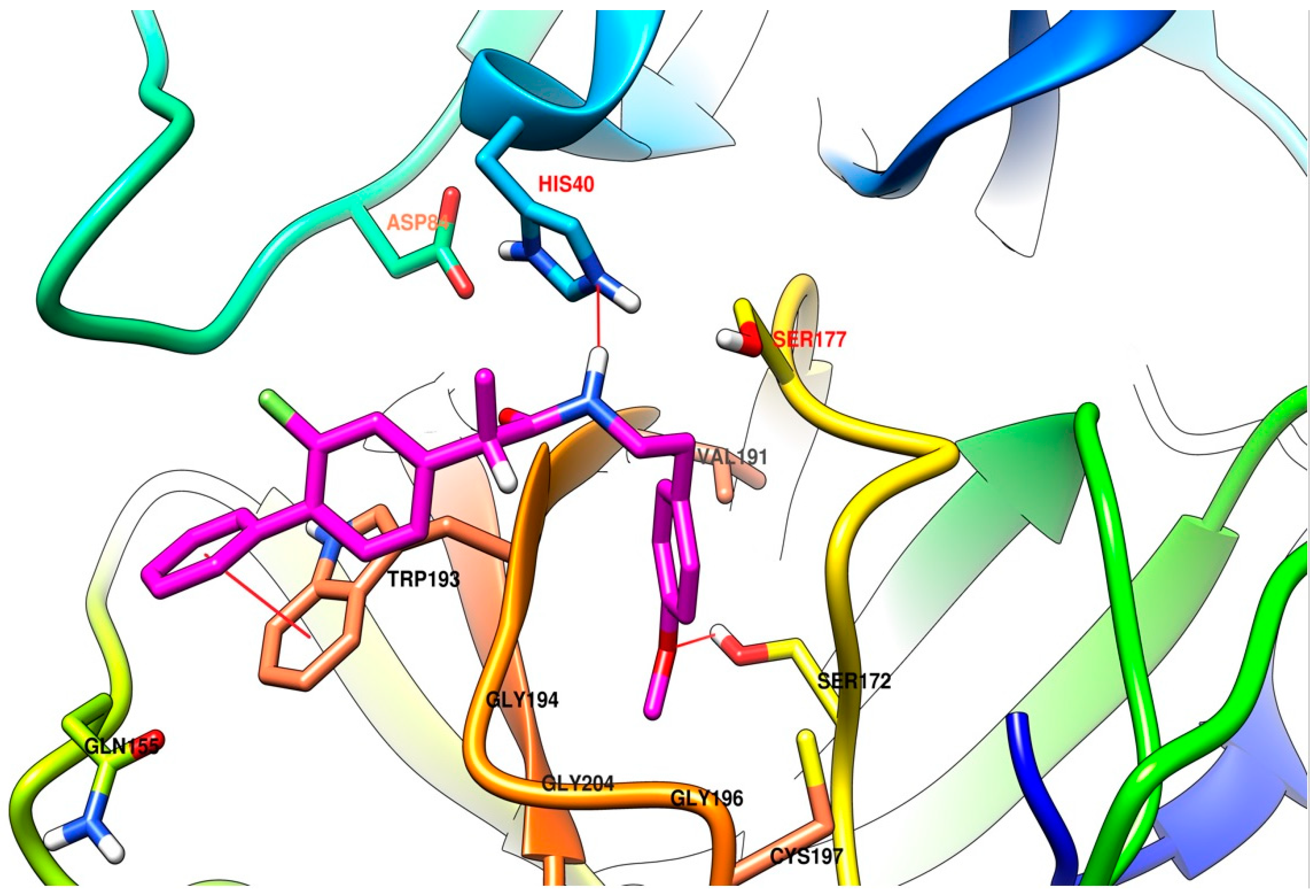
2.3.4. Molecular Docking
2.3.5. Experimental Determination of Lipophilicity (RM)
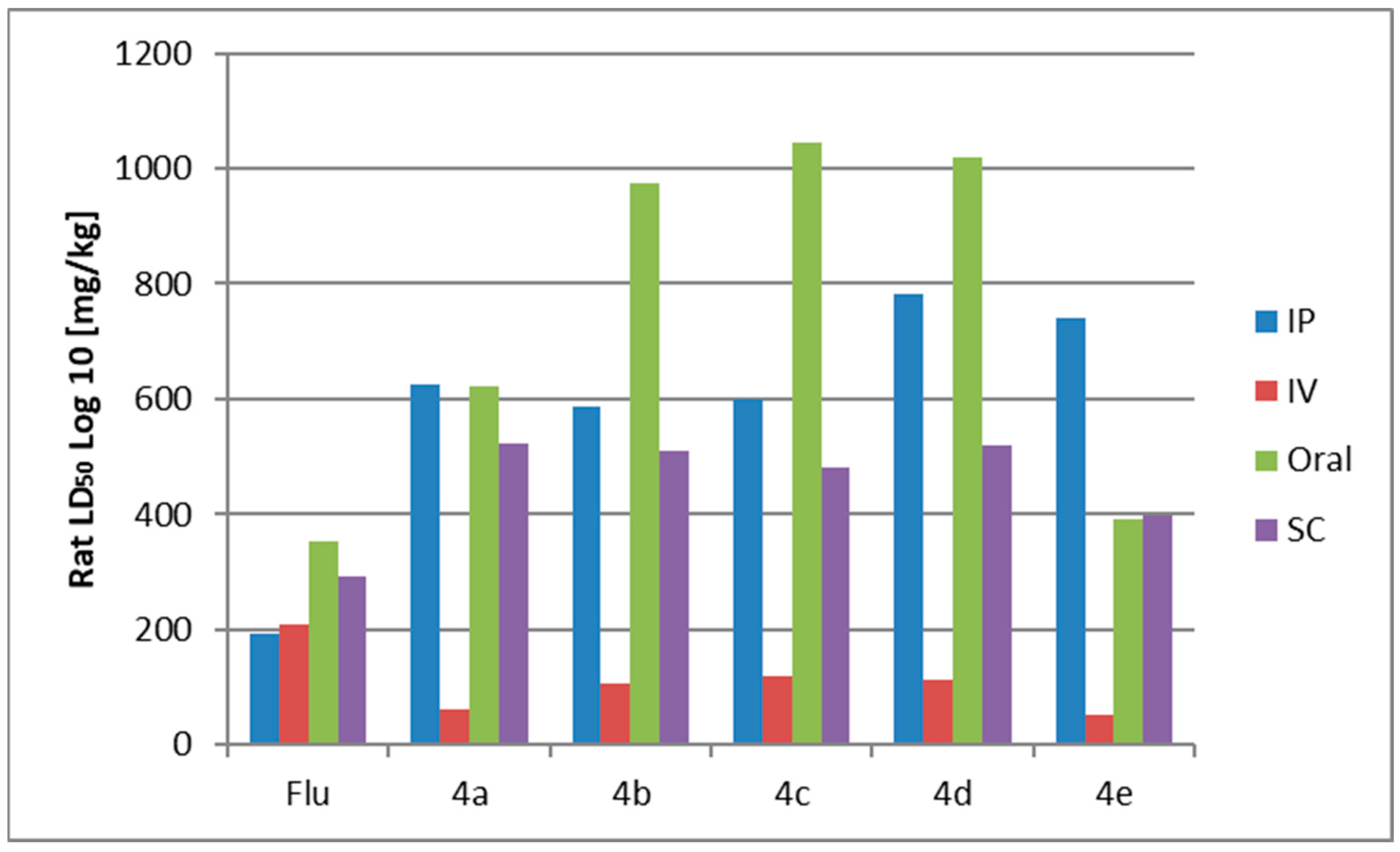
2.3.6. Prediction of Acute Rat Toxicity by Gusar Software
3. Materials and Methods
3.1. General
3.2. Synthesis
- 4a 2-(2-fluoro-[1,1′-biphenyl]-4-yl)-N-phenethylpropanamide
- 4b 2-(2-fluoro-[1,1′-biphenyl]-4-yl)-N-(3-methoxyphenethyl)propanamide
- 4c 2-(2-fluoro-[1,1′-biphenyl]-4-yl)-N-(4-methoxyphenethyl)propanamide
- 4d N-(3,4-dimethoxyphenethyl)-2-(2-fluoro-[1,1′-biphenyl]-4-yl)propanamide
- 4e N-(3-chlorophenethyl)-2-(2-fluoro-[1,1′-biphenyl]-4-yl)propanamide
3.3. HRMS Analysis
3.4. Biological Activity
3.4.1. Hydrogen Peroxide Scavenging Activity (HPSA)
3.4.2. Inhibition of Albumin Denaturation (IAD)
3.4.3. Antitryptic Activity (ATA)
3.4.4. Molecular Docking
3.4.5. Determination of Lipophilicity as cLogP
3.4.6. Experimental Determination of Lipophilicity (RM)
3.4.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dhanda, S.; Evans, A.; Roy, D.; Osborne, V.; Townsley, A.; Countinho, G.; Kulasekaran, A.; Shakir, S. A systematic review of flurbiprofen 8.75 mg dose and risk of haemorrhagic events. Front. Pharmacol. 2021, 12, 726141. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, T.-T.; Yin, L.; Huang, J.; Chen, Y.-J.; Xiong, L.-L.; Wang, T.-H. Analgesic effects of sufentanil in combination with flurbiprofen axetil and dexmedetomidine after open gastrointestinal tumor surgery: A retrospective study. BMC Anesthesiol. 2022, 22, 130. [Google Scholar] [CrossRef]
- Hofer, M.; Hoferová, Z.; Falk, M. Brief Story on Prostaglandins, Inhibitors of their Synthesis, Hematopoiesis, and Acute Radiation Syndrome. Molecules 2019, 24, 4019. [Google Scholar] [CrossRef]
- Phillips, W.; Currier, B. Analgesic pharmacology: II. Specific analgesics. J. Am. Acad. Orthop. Surg. 2004, 12, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Chaiamnuay, S.; Allison, J.J.; Curtis, J.R. Risks versus benefits of cyclooxygenase-2-selective nonsteroidal antiinflammatory drugs. Am. J. Health Syst. Pharm. 2006, 63, 1837–1851. [Google Scholar] [CrossRef]
- Pitzer, J.; Steiner, K. Amides in Nature and Biocatalysis. J. Biotechnol. 2016, 235, 32–46. [Google Scholar] [CrossRef]
- Kumari, S.; Carmona, A.; Tiwari, A.; Trippier, P. Amide Bond Bioisosteres: Strategies, Synthesis and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef]
- Ghosh, P.; Raj, N.; Verma, H.; Patel, M.; Chakraborti, S.; Khatri, B.; Doreswamy, C.; Anandakumar, S.; Seekallu, S.; Dinesh, M.; et al. An amide to thioamide substitution improves the permeability and bioavailability of macrocyclic peptides. Nat. Commun. 2023, 14, 6050. [Google Scholar] [CrossRef]
- Wang, H.; Wei, D.; Wan, Z.; Du, Q.; Zhang, B.; Ling, M.; Liang, C. Epoxy and amide crosslinked polarity enhanced polysaccharides binder for silicon anode in lithium-ion batteries. Electrochim. Acta 2021, 368, 137580. [Google Scholar] [CrossRef]
- Tang, Z.; Li, S. A review of recent developments of friction modifiers for liquid lubricants (2007–present). Curr. Opin. 2014, 18, 119–139. [Google Scholar] [CrossRef]
- Alam, A.; Alandis, N. Corn oil based poly(ether amide urethane) coating material-Synthesis, characterization and coating properties. Ind. Crops Prod. 2014, 57, 17–28. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.; Golan, D. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef]
- Lal, S.; Snape, T. Aromatic amides and ureas as novel molecular probes for diagnosing disease. Med. Hypotheses 2014, 83, 751–754. [Google Scholar] [CrossRef]
- Thientunyakit, T.; Shiratori, S.; Ishi, K.; Gelovani, J. Molecular PET Imaging in Alzheimer’s Disease. J. Med. Biol. Eng. 2022, 42, 301–317. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Wang, Y.-T.; Sun, L.; Wang, S.-Q.; Chen, Z.-S. Synthesis and clinical application of new drugs approved by FDA in 2022. Mol. Biomed. 2023, 4, 26. [Google Scholar] [CrossRef]
- Aboelez, M.O.; Kamel, M.S.; Belal, A.; El Badry Abdel-Aziz, A.; Abourehab, M.A.; Abdel-Ghany, H.; El-Remaily, M.A.E.A.A. Microwave-assisted synthesis, spectroscopic characterization, and biological evaluation of fused thieno[2,3-d]pyrimidines as potential anti-cancer agents targeting EGFRWT and EGFRT790M. Mol. Divers. 2023, 27, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Abu-Dief, A.; Said, M.; Elhady, O.; Alzahrani, S.; Aljohani, F.; Eskander, T.; El-Ramaily, M. Design, structural inspection of some new metal chelates based on benzothiazol-pyrimidin-2-ylidene ligend: Biomedical studies and molecular docking approach. Inorg. Chem. Comm. 2023, 158, 111587. [Google Scholar] [CrossRef]
- Manolov, S.; Ivanov, I.; Bojilov, D.; Nedialkov, P. Synthesis, In Vitro anti-inflammatory activity, and HRMS analysis of new amphetamine derivatives. Molecules 2023, 28, 151. [Google Scholar] [CrossRef]
- Bayir, H. Reactive oxygen species. Crit. Care Med. 2005, 33, S498–S501. [Google Scholar] [CrossRef]
- Galano, A.; Macías-Ruvalcaba, N.A.; Campos, O.N.M.; Pedraza-Chaverri, J. Mechanism of the OH radical scavenging activity of nordihydroguaiaretic acid: A combined theoretical and experimental study. J. Phys. Chem. B 2010, 114, 6625–6635. [Google Scholar] [CrossRef] [PubMed]
- Manolov, S.; Ivanov, I.; Bojilov, D. Synthesis of new 1,2,3,4-tetrahydroquinoline hybrid of ibuprofen and its biological evaluation. Molbank 2022, 2022, M1350. [Google Scholar] [CrossRef]
- Ebrahimzadeh, M.; Nabavi, S.; Nabavi, S.; Bahramian, F.; Bekhradnia, A. Antioxidant and free radical scavenging activity of H. officinalis L. var. angustifolius, V. odorata, B. hyrcana and C. speciosum. Pak. J. Pharm. Sci. 2010, 23, 29–34. [Google Scholar]
- Vane, J.R.; Botting, R.M. New insights into the mode of action of anti-inflammatory drugs. Inflamm. Res. 1995, 44, 1–10. Available online: http://link.springer.com/10.1007/BF01630479 (accessed on 9 January 2024). [CrossRef] [PubMed]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef]
- Insel, P.A. Analgesic-antipyretic and anti-inflammatory agents and drugs employed in the treatment of gout. In The Pharmacological Basics of Therapeutics, 9th ed.; Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W., Gilman, A., Eds.; 1325 6th Avenie; McGraw Hill: New York, NY, USA, 1996; pp. 617–657. [Google Scholar]
- Marliyah, M.; Ananthu, T. In Vitro anti-inflamatory activity of seed extract of Zea mays (L.). J. Glob. Biosci. 2015, 4, 2168–2173. Available online: https://www.mutagens.co.in/jgb/vol.04/5/09.pdf (accessed on 9 January 2024).
- Sen, S.; Chakrabotru, R.; Maramsa, N.; Basak, M.; Deka, S.; Dey, B.K. In Vitro anti-inflamatory activity of Amaranthus caudatus L. leaves. Indian J. Nat. Prod. Resour. 2015, 6, 326–329. Available online: https://nopr.niscpr.res.in/bitstream/123456789/33666/1/IJNPR6%284%29326-329.pdf (accessed on 15 July 2023).
- Sangeetha, G.; Vidhya, R. In Vitro anti-inflammatory activity of different parts of Pedalium murex (L.). Int. J. Herb. Med. 2016, 4, 31–36. [Google Scholar]
- Opie, E.L. On the relation of necrosis and inflammation to denaturation of proteins. J. Exp. Med. 1962, 115, 597–608. [Google Scholar] [CrossRef]
- Oyedapo, O.O.; Famurewa, A.J. Antiprotease and membrane stabilizing activities of extracts of fagara zanthoxyloides, olax subscorpioides and tetrapleura tetraptera. Int. J. Pharmacogn. 1995, 33, 65–69. [Google Scholar] [CrossRef]
- Jayashree, V.; Bagyalakshmi, S.; Manjula Devi, K.; Richard Daniel, D. In Vitro anti-inflamatory activity of 4-benzylpiperidine. Asian J. Pharm. Clin. Res. 2016, 9, 108–110. [Google Scholar] [CrossRef]
- Pontiki, E.; Hadjipavlou-Litina, D. Synthesis and pharmacochemical evaluation of novel aryl-acetic acid inhibitors of lipoxygenase, antioxidants, and anti-inflammatory agents. Bioorg. Med. Chem. 2007, 15, 5819–5827. [Google Scholar] [CrossRef] [PubMed]
- Raies, A.; Bajic, V. In silico toxicology: Computational methods for the prediction of chemical toxicity. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef]
- Lagunin, A.; Zakharov, A.; Filimonov, D.; Poroikov, V. QSAR Modelling of rat acute toxicity on the basis of PASS prediction. Mol. Inf. 2011, 30, 241–250. [Google Scholar] [CrossRef]
- Tang, S.; Chen, R.; Lin, M.; Lin, Q.; Zhu, Y.; Ding, J.; Hu, H.; Ling, M.; Wu, J. Accelerating AutoDock Vina with GPUs. Molecules 2022, 27, 3041. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Iyaguchi, D.; Kawano, S.; Takada, K.; Toyota, E. Structural basis for the design of novel Schiff base metal chelate inhibitors of trypsin. Bioorg. Med. Chem. 2010, 18, 2076–2080. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Ayeni, C.; Breuer, S.; Torbett, B.E. Virtual Screening for HIV Protease Inhibitors: A Comparison of AutoDock 4 and Vina. PLoS ONE 2010, 5, e11955. [Google Scholar] [CrossRef]
- Makeneni, S.; Thieker, D.F.; Woods, R.J. Applying Pose Clustering and MD Simulations To Eliminate False Positives in Molecular Docking. J. Chem. Inf. Model. 2018, 58, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Stoica, C.I.; Marc, G.; Pîrnău, A.; Vlase, L.; Araniciu, C.; Oniga, S.; Palage, M.; Oniga, O. Thiazolyl-oxadiazole derivatives targeting lanosterol 14α-demethylase as potential antifungal agents: Design, synthesis and molecular docking studies. Farmacia 2016, 64, 390–397. [Google Scholar]
- Toma, C.; Imre, S.; Farczadi, L.; Ion, V.; Marc, G. Enantioselective binding of carvedilol to human serum albumin and alpha-1-acid glycoprotein. Chirality 2023, 35, 779–792. [Google Scholar] [CrossRef]
- Borlan, R.; Stoia, D.; Gaina, L.; Campu, A.; Marc, G.; Perde-Schrepler, M.; Silion, M.; Maniu, D.; Focsan, M.; Astilean, S. Fluorescent Phthalocyanine-Encapsulated Bovine Serum Albumin Nanoparticles: Their Deployment as Therapeutic Agents in the NIR Region. Molecules 2021, 26, 4679. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | HPSA | IAD | ATA | RM ± SD | cLogP |
|---|---|---|---|---|---|
| IC50 ± SD (µmol/L) | |||||
| AA | 141.0 ± 2.0 | - | - | - | |
| Qrc | 229 ± 6 | - | - | - | |
| Ibu | 618 ± 25 | 395 ± 24 | 1260 ± 44 | 1.11 ± 0.01 | 3.72 |
| Flu | 1158 ± 64 | 339.3 ± 2.3 | 441 ± 36 | 1.37 ± 0.02 | 3.57 |
| 4a | 223 ± 6 | 198.4 ± 2.3 | 2261 ± 73 | 1.24 ± 0.02 | 4.95 |
| 4b | 200.8± 3.3 | 188.3 ± 3.0 | 208 ± 15 | 1.13 ± 0.02 | 5.04 |
| 4c | 144 ± 10 | 196.9 ± 0.5 | 203 ± 14 | 1.21 ± 0.02 | 5.07 |
| 4d | 199.5 ± 1.3 | 173.74 ± 0.13 | 215 ± 5 | 1.33 ± 0.02 | 4.90 |
| 4e | 199 ± 9 | 193.5 ± 2.2 | 198 ± 14 | 1.41 ± 0.03 | 5.44 |
| Compound | Enantiomer | AutoDock 4.2 | AutoDock Vina 1.1.2 Rigid | AutoDock Vina 1.1.2 Flexible | Vina-GPU 1.0 |
|---|---|---|---|---|---|
| 4a | R | −6.7 | −7.4 | −7.5 | −7.3 |
| S | −6.9 | −7.7 | −7.7 | −7.5 | |
| 4b | R | −7.3 | −7.6 | −7.6 | −7.6 |
| S | −7.4 | −8.0 | −8.0 | −8.0 | |
| 4c | R | −7.5 | −8.1 | −8.3 | −8.0 |
| S | −7.8 | −8.2 | −8.5 | −8.2 | |
| 4d | R | −7.4 | −7.5 | −7.7 | −7.3 |
| S | −7.5 | −7.9 | −7.9 | −7.9 | |
| 4e | R | −7.6 | −7.8 | −8.0 | −7.7 |
| S | −7.7 | −8.0 | −8.3 | −8.0 |
| AutoDock 4.2 | AutoDock Vina 1.1.2 Rigid | AutoDock Vina 1.1.2 Flexible | Vina-GPU 1.0 | |
|---|---|---|---|---|
| AutoDock 4.2 | - | 0.75 | 0.82 | 0.76 |
| AutoDock Vina 1.1.2 rigid | 0.75 | - | 0.94 | 0.98 |
| AutoDock Vina 1.1.2 flexible | 0.82 | 0.94 | - | 0.90 |
| Vina-GPU 1.0 | 0.76 | 0.98 | 0.90 | - |
| Rat LD50 Log10 (mg/kg) | ||||
|---|---|---|---|---|
| IP | IV | Oral | SC | |
| Flu | 191.5 4a | 209.9 4a | 353.1 4a | 293.5 4a |
| 4a | 626.3 5a | 62.29 4a | 621.3 4a | 521.3 4a |
| 4b | 587.5 5a | 106.2 4a | 973.6 4a | 511.1 4a |
| 4c | 598.4 5a | 119.5 4a | 1045 4a | 479.9 4a |
| 4d | 781.4 5a | 114.2 4a | 1020 4a | 521.1 4a |
| 4e | 739.3 5a | 51.4 4a | 392.4 4a | 398.5 4a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanov, I.; Manolov, S.; Bojilov, D.; Marc, G.; Dimitrova, D.; Oniga, S.; Oniga, O.; Nedialkov, P.; Stoyanova, M. Novel Flurbiprofen Derivatives as Antioxidant and Anti-Inflammatory Agents: Synthesis, In Silico, and In Vitro Biological Evaluation. Molecules 2024, 29, 385. https://doi.org/10.3390/molecules29020385
Ivanov I, Manolov S, Bojilov D, Marc G, Dimitrova D, Oniga S, Oniga O, Nedialkov P, Stoyanova M. Novel Flurbiprofen Derivatives as Antioxidant and Anti-Inflammatory Agents: Synthesis, In Silico, and In Vitro Biological Evaluation. Molecules. 2024; 29(2):385. https://doi.org/10.3390/molecules29020385
Chicago/Turabian StyleIvanov, Iliyan, Stanimir Manolov, Dimitar Bojilov, Gabriel Marc, Diyana Dimitrova, Smaranda Oniga, Ovidiu Oniga, Paraskev Nedialkov, and Maria Stoyanova. 2024. "Novel Flurbiprofen Derivatives as Antioxidant and Anti-Inflammatory Agents: Synthesis, In Silico, and In Vitro Biological Evaluation" Molecules 29, no. 2: 385. https://doi.org/10.3390/molecules29020385
APA StyleIvanov, I., Manolov, S., Bojilov, D., Marc, G., Dimitrova, D., Oniga, S., Oniga, O., Nedialkov, P., & Stoyanova, M. (2024). Novel Flurbiprofen Derivatives as Antioxidant and Anti-Inflammatory Agents: Synthesis, In Silico, and In Vitro Biological Evaluation. Molecules, 29(2), 385. https://doi.org/10.3390/molecules29020385