

Catalytic Degradation of Lignin over Sulfonyl-Chloride-Modified Lignin-Based Porous Carbon-Supported Metal Phthalocyanine: Effect of Catalyst Concentrations

Abstract
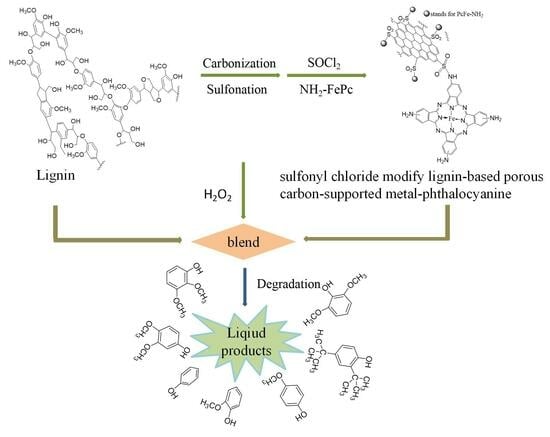
1. Introduction
2. Results and Discussions
2.1. Characterization of Catalyst
2.1.1. FTIR Analysis
2.1.2. XPS Analysis
2.1.3. BET Analysis
2.1.4. SEM Analysis
2.2. Optimizing the Reaction Conditions
2.3. Analysis of Liquid Products
2.3.1. GC-MS Analysis
2.3.2. FT-IR and 1H-NMR Analysis
2.3.3. Elemental Analysis
2.4. Characterization of the Solid Residue Fractions
2.4.1. FT-IR Analysis
2.4.2. 1H-NMR Analysis
2.4.3. GPC Analysis
2.4.4. SEM Analysis
3. Experimental
3.1. Materials
3.2. Experimental Procedure
3.2.1. Catalyst Preparation
Manufacture of Sulphonyl Chloride Derivative of Lignin-Base Carbon
The Synthesis of Iron Amino Phthalocyanine (FePc-NH2)
Synthesis of Sulfonyl-Chloride-Modified Lignin-Based Porous Carbon-Supported Iron Phthalocyanine
3.2.2. Treatment of Lignin with FePc@ SO2Cl-Clig
3.3. Characterizations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, X.; Duan, X.; Wei, W.; Wang, S.; Ni, B. Photocatalytic conversion of lignocellulosic biomass to valuable products. Green. Chem. 2019, 21, 4266–4289. [Google Scholar] [CrossRef]
- Hilde, V.H.; Tanja, B. Quantitative NMR analysis of the aqueous phase from hydrothermal liquefaction of lignin. J. Anal. Appl. Pyrolysis 2020, 151, 104919–104927. [Google Scholar]
- Zhang, Y.; Maryam, N. Lignin: A review on structure, properties, and applications as a light-colored UV absorber. ACS Sustain. Chem. Eng. 2021, 9, 1427–1442. [Google Scholar] [CrossRef]
- Mahdi, M.A.; Katalin, B.; Gregg, T.B.; Jeremy, S.L.; John, R.; Roberto, R.; Yuriy, R.L.; Samec, J.S.M.; Sels, B.F.; Wang, F. Guidelines for performing lignin-first biorefining. Energy Environ. Sci. 2021, 14, 262–292. [Google Scholar]
- Yao, L.; Lu, Y.C.; Hu, H.Q.; Xie, F.J.; Wei, X.Y.; Fan, X. Structural characterization of lignin and its degradation products with spectroscopic methods. J. Spectrosc. 2017, 2017, 8951658. [Google Scholar]
- Patryk, J.; Maurice, N.C.; Teofl, J.; Łukasz, K. The role of lignin and lignin-based materials in sustainable construction—A comprehensive review. Int. J. Biol. Macromol. 2021, 187, 624–650. [Google Scholar]
- Yokoyama, T. Revisiting the Mechanism of β-O-4 bond cleavage during acidolysis of lignin. Part 6: A review. J. Wood Chem. Technol. 2014, 35, 27–42. [Google Scholar] [CrossRef]
- Constant, S.; Basset, C.; Dumas, C.e; Di Renzo, F.; Robitzer, M.; Barakat, A.; Quignard, F. Reactive organosolv lignin extraction from wheat straw: Influence of lewis acid catalysts on structural and chemical properties of lignins. Ind. Crop. Prod. 2015, 65, 180–189. [Google Scholar] [CrossRef]
- Deepa, A.K.; Dhepe, P.L. Lignin depolymerization into aromatic monomers over solid acid catalysts. ACS Catal. 2015, 5, 365–379. [Google Scholar] [CrossRef]
- Deuss, P.J.; Scott, M.; Tran, F.; Westwood, N.J.; de Vries, J.G.; Barta, K. Aromatic monomers by in situ conversion of reactive intermediates in the acid-catalyzed depolymerization of lignin. J. Am. Chem. Soc. 2015, 137, 7456–7467. [Google Scholar] [CrossRef]
- Yang, X.J.; Feng, M.Q.; Choi, J.S.; Meyer III, H.M.; Yang, B. Depolymerization of corn stover lignin with bulk molybdenum carbide catalysts. Fuel 2019, 244, 528–535. [Google Scholar] [CrossRef]
- Chen, L.; Dou, J.; Ma, Q.; Li, N.; Wu, R.; Bian, H.; Yelle, D.J.; Vuorinen, T.; Fu, S.; Pan, X.; et al. Rapid and near-complete dissolution of wood lignin at </=80 degrees C by a recyclable acid hydrotrope. Sci. Adv. 2017, 3, e1701735. [Google Scholar]
- Miller, J.E.; Evans, L.; Littlewolf, A.; Trudell, D.E. Batch microreactor studies of lignin and lignin model compound depolymerization by bases in alcohol solvents. Fuel 1999, 78, 1363–1366. [Google Scholar] [CrossRef]
- Alfonso, C.; Fernando, B.; Rui, M.; Karina, H.; Íñigo, G.Y.; Maitane, M.; António, P.; Gandía, L.M.; Víctor, M.M. Production of aromatic compounds by catalytic depolymerization of technical and downstream biorefinery lignins. Biomolecules 2020, 10, 1338–1398. [Google Scholar]
- Bentivenga, G.; Bonini, C.; ĎAuria, M.; Bona, A.D. Degradation of steam-exploded lignin from beech by using Fenton’s reagent. Biomass Bioenerg. 2003, 24, 233–238. [Google Scholar] [CrossRef]
- Torrades, F.; Saiz, S.; García-Hortal, J.A. Using central composite experimental design to optimize the degradation of black liquor by Fenton reagent. Desalination 2011, 268, 97–102. [Google Scholar] [CrossRef]
- Phisit, S.; Ampin, K.; Arthitaya, K.; Charin, T.; Thanongsak, C.; Noppol, L. Improvement in efficiency of lignin degradation by Fenton reaction using synergistic catalytic action. Ecol. Eng. 2015, 85, 283–287. [Google Scholar]
- Lin, Y.Y.; Lu, S.Y. Selective and efficient cleavage of lignin model compound into value-added aromatic chemicals with CuFe2O4 nanoparticles decorated on partially reduced graphene oxides via sunlight-assisted heterogeneous Fenton processes. J. Taiwan Inst. Chem. Eng. 2019, 97, 264–271. [Google Scholar] [CrossRef]
- Pecci, L.; Montefoschi, G.; Cavallini, D. Some new details of the copperhydrogen peroxide interaction. Biochem. Biophys. Res. Commun. 1997, 235, 264–267. [Google Scholar] [CrossRef]
- Valdeir, A.; Adriane, M.F.M. The effect of a catecholate chelator as a redox agent in Fenton-based reactions on degradation of lignin-model substrates and on COD removal from effluent of an ECF kraft pulp mill. J. Hazard. Mater. 2007, 141, 273–279. [Google Scholar]
- Kim, H.E.; Lee, J.; Lee, H.; Lee, C. Synergistticc effects of TiO2 photo catalysis in combination with Fenton-like reaction oxideation of organic compounds at circumneutral pH. Appl. Catal. B Environ. 2012, 115–116, 219–224. [Google Scholar] [CrossRef]
- Martίn, R.; Navalon, S.; Alvaro, M.; Hermenegildo, G. Optimized water treatment by combining catalytic Fenton reaction using diamond supported gold and biological degradation. Appl. Catal. B Environ. 2011, 103, 246–252. [Google Scholar] [CrossRef]
- Colomban, C.; Kudrik, E.V.; Afanasiev, P.; Alexander, B.S. Degradation of chlorinated phenols in water in the presence of H2O2 and water-soluble μ-nitrido diiron phthalocyanine. Catal. Today 2014, 235, 14–19. [Google Scholar] [CrossRef]
- Tao, X.; Ma, W.; Zhang, T.; Zhao, J.C. A novel approach for the oxidative degradation of organic pollutants in aqueous solutions mediated by iron tetrasulfophthalocyanine under visible light radiation. Chem. Eur. J. 2002, 8, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, Y. Heterogeneous photo-Fenton degradation of methylene blue under visible irradiation by iron tetrasulphophthalocyanine immobilized layered double hydroxide at circumneutral pH. Dyes Pigments 2016, 134, 397–408. [Google Scholar] [CrossRef]
- Huang, L.; Denis, T.G.S.; Xuan, Y.; Huang, Y.Y.; Masamitsu, T.; Andrzej, Z.; Tadeusz, S.; Michael, R. HamblinParadoxical potentiation of methylene blue-mediated antimicrobial photodynamic inactivation by sodium azide: Role of ambient oxygen and azide radicals. Free Radic. Biol. Med. 2012, 53, 2062–2071. [Google Scholar] [CrossRef]
- Shi, M.; Chen, Z.M.; Guo, L.X.; Liang, X.H.; Zhang, J.L.; He, C.Y.; Wang, B.; Wu, Y.Q. A multiwalled carbon nanotube/tetra-bisoheptyloxyphthalocyanine cobalt(II) composite with high dispersibility for electrochemical detection of ascorbic acid. J. Mater. Chem. B 2014, 2, 4876–4882. [Google Scholar] [CrossRef]
- Yan, X.M.; Xu, X.; Zhong, Z.; Liu, J.J.; Tian, X.M.; Kang, L.T.; Yao, J.N. The effect of oxygen content of carbon nanotubes on the catalytic activity of carbon-based iron phthalocyanine for oxygen reduction reaction. Electrochim. Acta 2018, 281, 562–570. [Google Scholar] [CrossRef]
- Topacli, A.; Kesimli, B. Investigation on sulfanilamide and its interation eith some metals and lincomycin by infrared spectroscopy. Spectrosc. Lett. 2001, 34, 513–526. [Google Scholar] [CrossRef]
- AmalAl, A.; Sajjad, K. Cross-linked chitosan aerogel modifed with Pd(II)/phthalocyanine: Synthesis, characterization, and catalytic application. Sci. Rep. 2019, 9, 13849. [Google Scholar]
- Watkins, J.D.; Lawrence, R.; Taylor, J.E.; Bull, S.D.; Nelson, G.W.; Foord, J.S.; Wolverson, D.; Rassaei, L.; Evans, N.D.; Gascond, M.S.A.; et al. Carbon nanoparticle surface functionalisation: Converting negatively charged sulfonate to positively charged sulfonamide. Phys. Chem. Chem. Phys. 2010, 12, 4872–4878. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Yasin, G.; Korai, R.M.; Slimani, U.Y.; Muhammad, F.A.; Mohammad, T.; Nazir, M.T.; Nguyen, T.A. Boosting oxygen reduction reaction activity by incorporating the iron phthalocyanine nanoparticles on carbon nanotubes network. Inorg. Chem. Commun. 2020, 120, 108160. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Dou, M.L.; Ji, J.; Wang, F. Phthalocyanine tethered iron phthalocyanine on graphitized carbon black as superior electrocatalyst for oxygen reduction reaction. Nano Energy 2017, 34, 338–343. [Google Scholar] [CrossRef]
- Schick, G.A.; Sun, Z.Q. Spectroscopic characterization of sulfonyl chloride immobilization on silica. Langmuir 1994, 10, 3105–3110. [Google Scholar] [CrossRef]
- Exner, O.; Juška, T. X-Ray Photoelectron Spectroscopic Study of Sulphonamides: Charge distrbution and tautomerism. Collect. Czechoslov. Chern. Commun. 1984, 49, 51–57. [Google Scholar] [CrossRef]
- Catherine, D.C.; Haskouri, S.; Cachet, H. Study by XPS of the chlorination of proteins aggregated onto tin dioxide during electrochemical production of hypochlorous acid. Appl. Surf. Sci. 2007, 253, 5506–5510. [Google Scholar]
- Hou, L.J.; Zhang, J.L.; Pu, Y.F.; Li, W. Effects of nitrogen-dopants on Ru-supported catalysts for acetylene hydrochlorination. RSC Adv. 2016, 6, 18026–18032. [Google Scholar] [CrossRef]
- Wang, H.Q.; Hana, J.; Zhao, B.; Qin, L.B.; Wang, Y.; Yu, F. Non-thermal plasma enhanced dry reforming of CH4 with CO2 over activated carbon supported Ni catalysts. Mol. Catal. 2019, 475, 110486–110495. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, Y.S.; Liu, Z.M. Pyrolysis of iron phthalocyanine on activated carbon as highly efficient nonnoble metal oxygen reduction catalyst in microbial fuel cells. Chem. Eng. J. 2019, 361, 416–427. [Google Scholar] [CrossRef]
- Yang, T.; Li, K.; Pu, L.; Liu, Z.; Ge, B.; Pan, Y.; Liu, Y. Hollow-spherical Co/N-C nanoparticle as an efficient electrocatalyst used in air cathode microbial fuel cell. Biosens. Bioelectron. 2016, 86, 129–134. [Google Scholar] [CrossRef]
- Lappalainen, J.; Baudouin, D.; Hornung, U.; Schuler, J.; Melin, K.; Bjelíc, S.; Vogel, F.; Konttinen, J.; Joronen, T. Sub- and supercritical water liquefaction of kraft lignin and black liquor derived lignin. Energies 2020, 13, 3309. [Google Scholar] [CrossRef]
- Rangarajan, G.; Koh, J.M.; Farnood, R. Touch wood: Conversion of lignin to dicarboxylic acids using biochar-based solid-acid photo-Fenton catalysts. J. Clean. Prod. 2023, 415, 137816–137830. [Google Scholar] [CrossRef]
- Peng, F.; Peng, P.; Xu, F.; Sun, R.C. Fractional purification and bioconversion of hemicelluloses. Biotechnol Adv. 2012, 30, 879–903. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.T.; Chen, B.Y.; Li, B.Y.; Tseng, M.C.; Han, C.C.; Shyu, S.G. Efficient pretreatment of lignocellulosic biomass with high recovery of solid lignin and fermentable sugars using Fenton reaction in a mixed solvent. Biotechnol. Biofuels 2018, 11, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Schrank, S.G.; Santos, J.N.R.; Souza, D.S.; Souza, E.E.S. Decolourisation effects of Vat Green 01 textile dye and textile wastewater using H2O2/UV process. J. Photochem. Photobiol. Chem. 2007, 186, 125–129. [Google Scholar] [CrossRef]
- Xu, F.; Lu, Q.W.; Li, K.P.; Zhu, T.T.; Wang, W.K.; Hu, Z.H. Green synthesis of magnetic mesoporous carbon from waste-lignin and its application as an efficient heterogeneous Fenton catalyst. J. Clean. Prod. 2021, 285, 125363–125372. [Google Scholar] [CrossRef]
- Wang, Y.S.; Tang, Z.Y.; Chen, M.Q.; Zhang, J.H.; Shi, J.J.; Wang, C.S.; Yang, Z.L.; Wang, J. Effect of Mo content in Mo/Sepiolite catalyst on catalytic depolymerization of Kraft lignin under supercritical ethanol. Energy Convers. Manag. 2020, 222, 113227–113240. [Google Scholar] [CrossRef]
- Leybo, D.V.; Konopatsky, A.S.; Fang, X.S.; Shtansky, D.V. Photocatalytic phenol oxidation over ball milled hexagonal boron nitride. J. Water Process. Eng. 2023, 51, 103367–103377. [Google Scholar] [CrossRef]
- Huang, X.M.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 2015, 17, 4941–4950. [Google Scholar] [CrossRef]
- Yan, L.; Wang, Y.; Li, J.; Zhang, Y.; Ma, L.; Fu, F.; Chen, B.; Liu, H.J. Hydrothermal liquefaction of Ulva prolifera macroalgae and the influence of base catalysts on products. Bioresour. Technol. 2019, 292, 121286. [Google Scholar] [CrossRef]
- Song, Y.; Motagamwale, A.H.; Karlen, S.D.; Dumesic, J.A.; Ralph, J.; Mobley, J.K.; Crocker, M. A comparative study of secondary depolymerization methods on oxidized lignin. Green Chem. 2019, 21, 3904–3947. [Google Scholar] [CrossRef]
- Zhu, Y.F.; Song, W.Q.; Yao, R.J.; Zhao, Y.Z.; Xu, G.Y. In-situ catalytic hydropyrolysis of lignin for the production of aromatic rich bio-oil. J. Energy Inst. 2022, 101, 187–193. [Google Scholar] [CrossRef]
- Biswas, B.; Kumar, A.A.; Bisht, Y.; Singh, R.; Kumar, J.; Bhaskar, T. Effects of temperature and solvents on hydrothermal liquefaction of Sargassum tenerrimum algae. Bioresour. Technol. 2017, 242, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Biswasa, B.; Kumara, A.; Fernandesc, A.C.; Sainia, K.; Negia, S.; Muraleedharanc, U.D.; Bhaskar, T. Solid base catalytic hydrothermal liquefaction of macroalgae: Effects of process parameter on product yield and characterization. Bioresour. Technol. 2020, 307, 123232–123239. [Google Scholar] [CrossRef] [PubMed]
- Raikova, S.; Allen, M.J.; Chuck, C.J. Hydrothermal liquefaction of macroalgae for the production of renewable biofuels. Biofuels Bioprod. Biof. 2019, 13, 1483–1504. [Google Scholar] [CrossRef]
- Yang, Z.X.; Lei, H.W.; Zhang, Y.Y.; Qian, K.Z.; Villot, E.; Qian, M.; Yadavalli, G.; Sun, H. Production of renewable alkyl-phenols from catalytic pyrolysis of Douglas fr sawdust over biomass-derived activated carbons. Appl Energy 2018, 220, 426–436. [Google Scholar] [CrossRef]
- Chen, H.T.; He, Z.X.; Zhang, B.; Feng, H.; Kandasamy, S.; Wang, B. Efects of the aqueous phase recycling on bio-oil yield in hydrothermal liquefaction of Spirulina Platensis, a-cellulose, and lignin. Energy 2019, 179, 1103–1113. [Google Scholar] [CrossRef]
- Liu, H.M.; Li, M.F.; Yang, S.; Sun, R.C. Understanding the mechanism of cypress liquefaction in hot-compressed water through characterization of solid residues. Energies 2013, 6, 1590–1603. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Qin, L.; Ge, Y. Enhancing antioxidant performance of lignin by enzymatic treatment with laccase. ACS Sustain. Chem. Eng. 2018, 6, 2591–2595. [Google Scholar] [CrossRef]
- Ramires, E.C.; Megiatto, J.D.; Gardrat, J.C.; Castellan, A.; Frollini, E. Valorization of an industrial organosolv-sugarcane bagasse lignin: Characterization and use as a matrix in biobased composites reinforced with sisal fibers. Biotechnol. Bioeng. 2010, 107, 612–621. [Google Scholar] [CrossRef]
- Wang, W.Y.; Qin, Z.; Liu, H.M.; Wang, X.D.; Gao, J.H.; Qin, G.Y. Structural changes in milled wood lignin (MWL) of chinese quince (Chaenomeles sinensis) fruit subjected to subcritical water treatment. Molecules 2021, 26, 398. [Google Scholar] [CrossRef] [PubMed]
- Glasser, W.G.; Kaar, W.E.; Jain, R.K.; Sealey, J.E. Isolation options for non-cellulosic heteropolysaccharides (HetPS). Cellulose 2000, 7, 299–317. [Google Scholar] [CrossRef]
- Zhu, J.H.; Li, Y.X.; Chen, Y.; Wang, J.; Zhang, B.; Zhang, J.J.; Werner, J.B. Graphene oxide covalently functionalized with zinc phthalocyanine for broadband optical limiting. Carbon 2011, 49, 1900–1905. [Google Scholar] [CrossRef]
- Nemykin, V.N.; Polshyna, A.E.; Borisenkova, S.A.; Strelko, V.V. Preparation, characterization, and catalytic activity of synthetic carbon-supported (phthalocyaninato)cobalt-containing complexes in dodecane-1-thiol oxidation reaction. J. Mol. Catal A Chem. 2007, 264, 103–109. [Google Scholar] [CrossRef]
- Achar, B.N.; Lokesh, K.S. Studies on tetra-amine phthalocyanines. J. Organomet. Chem. 2004, 689, 3357–3361. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET (m2/g) | SMic (m2/g) | Vtotal (cm3/g) | VMic (cm3/g) |
|---|---|---|---|---|
| SO2Cl-Clig | 643.67 | 540.79 | 0.337 | 0.210 |
| FePc@ SO2Cl-Clig | 638.98 | 603.57 | 0.291 | 0.246 |
| Retention Time/s | Compound Name | Catalyst Concentrations (wt%) Quantitative Content (Area %) | ||||
|---|---|---|---|---|---|---|
| 0.5 | 1.5 | 2.5 | 3.5 | 4.5 | ||
| 5.712 | Propanoic acid, 3-mercapto-, methyl ester | - | 0.14 | - | - | 0.35 |
| 5.958 | Propanoic acid, ethyl ester | 5.54 | 17.42 | 65.18 | 51.59 | 59.06 |
| 8.344 | Acetic acid, butyl ester | 0.33 | 0.92 | 3.28 | 2.65 | 2.92 |
| 8.489 | Acetosyringone | 0.82 | 0.84 | 1.49 | 1.38 | 1.88 |
| 10.753 | Cyclohexanone | - | 0.17 | 0.11 | 0.08 | - |
| 12.449 | 2-Anthracenamine | 0.08 | - | - | - | - |
| 13.431 | Phenol | 0.16 | 0.24 | 3.11 | 4.22 | 3.70 |
| 13.734 | 1,2,4-trimethoxybenzene | - | 0.14 | 0.46 | 0.41 | 0.49 |
| 15.611 | Eicosyl nonyl ether | - | 0.09 | - | - | - |
| 16.818 | Phenol, 2-methoxy- | - | 0.61 | 0.72 | 1.56 | 0.61 |
| 16.923 | Mequinol | 0.13 | - | - | - | - |
| 18.42 | formic acid | - | - | 0.16 | 0.10 | 0.14 |
| 19.095 | 2-Isopropylpyrazine | - | - | 0.08 | - | - |
| 19.765 | Dodecane | - | 0.13 | - | - | - |
| 20.154 | Formic Acid, 4-methoxyphenyl ester | - | 0.08 | - | - | - |
| 20.408 | Benzofuran, 2,3-dihydro- | 13.72 | 48.16 | - | - | - |
| 20.44 | Benzaldehyde, 3,4-dimethyl- | - | - | 0.74 | 1.23 | 0.24 |
| 21.202 | Benzonitrile, 2-(4-methylphenyl)- | - | - | - | - | 0.11 |
| 21.375 | Benzene, 1,3-bis(1,1-dimethylethyl)- | 0.16 | 0.94 | 0.27 | 0.16 | 0.40 |
| 21.655 | Naphthalene, 2,6-bis(1,1-dimethylethyl)- | 0.27 | 1.05 | 2.56 | 5.78 | 5.41 |
| 21.985 | (S)-(−)-2-Amino-3-phenyl-1 -propanol | - | - | - | 0.09 | - |
| 21.991 | Dodecane, 1-iodo- | - | 0.40 | - | - | 0.11 |
| 22.004 | 1,4-Benzenediamine, N,N′-bis(1-methylethyl)- | 0.18 | - | - | - | - |
| 22.363 | Octane, 2,3,6,7-tetramethyl- | - | 0.07 | - | - | - |
| 22.666 | Ethanone, 2-(2-benzothiazolylthio)-1-(3,5-dimethylpyrazolyl)- | - | - | - | - | 0.09 |
| 22.737 | Acetaldehyde, bis(1-methylethyl)hydrazone | 0.11 | 0.27 | - | - | - |
| 22.809 | Benzaldehyde, 2,4,5-trimethyl- | - | - | - | - | 0.07 |
| 23.198 | Linoleic acid, ethyl ester | - | - | 0.18 | 0.17 | - |
| 23.204 | Undecanoic acid | - | - | - | 0.18 | |
| 23.21 | Hexadecane, 1-iodo- | - | - | -- | - | |
| 24.119 | Phenol, 2,6-dimethoxy- | - | - | 2.15 | 10.78 | 0.90 |
| 25.186 | Tetradecane | 0.12 | 0.68 | - | - | - |
| 25.556 | [1,2,4]Oxadiazole, 5-(4-tert-butylphenoxymethyl)-3-(thiophen-2-yl)- | - | - | - | - | 0.224 |
| 25.632 | Benzaldehyde, 4-hydroxy- | 0.29 | - | - | - | - |
| 25.798 | Benzaldehyde, 3-hydroxy-4-methoxy- | 0.47 | 5.38 | - | - | - |
| 26.872 | desaspinidol | - | - | 0.52 | 0.84 | 0.69 |
| 27.049 | 3-Ethyl-2,6,10- trimethylundecane | - | 0.33 | - | - | - |
| 27.072 | Dimethyl phthalate | - | - | 0.34 | 0.20 | 0.12 |
| 27.472 | 2-Heptanone, 3-propylidene- | - | - | - | 0.12 | - |
| 27.753 | Tetradecane, 2,6,10-trimethyl- | - | 0.25 | - | - | - |
| 27.982 | Octacosane, 1-iodo- | 0.362 | 1.62 | - | - | - |
| 27.993 | Hexacosane | - | - | - | - | 0.18 |
| 27.999 | 2,3-Dimethoxyphenol | - | - | - | 0.08 | - |
| 28.056 | Phenol, 3,4-dimethoxy- | - | - | - | 0.18 | - |
| 28.062 | Pentadecane | - | 0.69 | - | - | - |
| 28.073 | Nonahexacontanoic acid | - | - | - | - | 0.20 |
| 28.216 | Vanillin | - | - | - | - | - |
| 28.545 | 2,4-Di-tert-butylphenol | 3.01 | 10.12 | 15.73 | 14.84 | 17.08 |
| 29.075 | Eicosane | - | 1.11 | - | - | 0.07 |
| 29.083 | Decane, 3,8-dimethyl- | 0.29 | - | - | - | - |
| 29.311 | Vanillin | 0.11 | 0.42 | - | - | - |
| 30.351 | Hexadecane | 0.21 | 0.54 | - | - | - |
| 30.362 | Pyrrolidine, 1-(1-cyclohexen-1-yl)- | -- | - | - | - | 0.11 |
| 30.366 | Benzaldehyde, 4-(methylthio)- | - | 0.20 | - | 0.07 | - |
| 31.089 | Eicosane | - | 0.42 | - | - | - |
| 31.118 | 3-Methoxy-4-[3-oxo-3-(pyrrolidin-1-yl)propoxy]benzaldehyde | - | - | - | - | 0.28 |
| 31.93 | Dotriacontane | - | 0.51 | - | - | - |
| 31.966 | Tetracosane, 1-iodo- | 0.19 | - | - | - | - |
| 32.073 | Octadecane | - | 0.55 | - | - | - |
| 32.104 | Docosane, 1-iodo- | 0.14 | - | - | - | - |
| 32.514 | Heneicosane | 0.78 | 3.09 | - | - | - |
| 32.554 | Hexanoic acid, 4-tridecyl ester | - | - | - | 0.83 | 1.04 |
| Samples | Elemental Composition (wt%) | HHV (MJ/kg) | |||
|---|---|---|---|---|---|
| C | H | O | N | ||
| Lignin | 61.3 | 6.4 | 29.1 | 1.7 | 20.5 |
| 0.5 wt% catalyst | 63.2 | 6.9 | 26.7 | 0.9 | 23.3 |
| 1.5 wt% catalyst | 65.5 | 6.8 | 25.2 | 0.7 | 24.9 |
| 2.5 wt% catalyst | 66.9 | 6.8 | 24.7 | 0.6 | 27.1 |
| 3.5 wt% catalyst | 69.2 | 7.0 | 21.8 | 0.7 | 30.2 |
| 4.5 wt% catalyst | 68.6 | 7.2 | 22.4 | 0.6 | 29.0 |
| Samples | Mw (g/mol) | Mn (g/mol) | Mw/Mn |
|---|---|---|---|
| Lignin | 66,671 | 36,726 | 1.81 |
| Residues (40 min) | 62,256 | 36,208 | 1.72 |
| Residues (80 min) | 41,063 | 27,202 | 1.51 |
| Residues (120 min) | 31,421 | 25,633 | 1.22 |
| Residues (150 min) | 33,468 | 14,597 | 2.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, F.; Xian, X.; Tang, P.; Li, Y. Catalytic Degradation of Lignin over Sulfonyl-Chloride-Modified Lignin-Based Porous Carbon-Supported Metal Phthalocyanine: Effect of Catalyst Concentrations. Molecules 2024, 29, 347. https://doi.org/10.3390/molecules29020347
Du F, Xian X, Tang P, Li Y. Catalytic Degradation of Lignin over Sulfonyl-Chloride-Modified Lignin-Based Porous Carbon-Supported Metal Phthalocyanine: Effect of Catalyst Concentrations. Molecules. 2024; 29(2):347. https://doi.org/10.3390/molecules29020347
Chicago/Turabian StyleDu, Fangli, Xuequan Xian, Peiduo Tang, and Yanming Li. 2024. "Catalytic Degradation of Lignin over Sulfonyl-Chloride-Modified Lignin-Based Porous Carbon-Supported Metal Phthalocyanine: Effect of Catalyst Concentrations" Molecules 29, no. 2: 347. https://doi.org/10.3390/molecules29020347
APA StyleDu, F., Xian, X., Tang, P., & Li, Y. (2024). Catalytic Degradation of Lignin over Sulfonyl-Chloride-Modified Lignin-Based Porous Carbon-Supported Metal Phthalocyanine: Effect of Catalyst Concentrations. Molecules, 29(2), 347. https://doi.org/10.3390/molecules29020347